
Effects of (Oxy-)Fluorination on Various High-Performance Yarns




Abstract
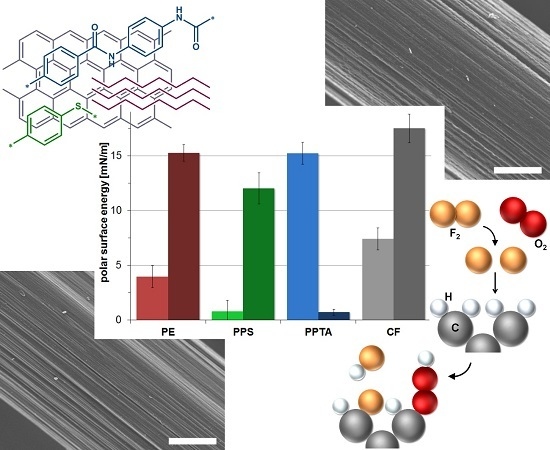
:
1. Introduction
2. Results and Discussion
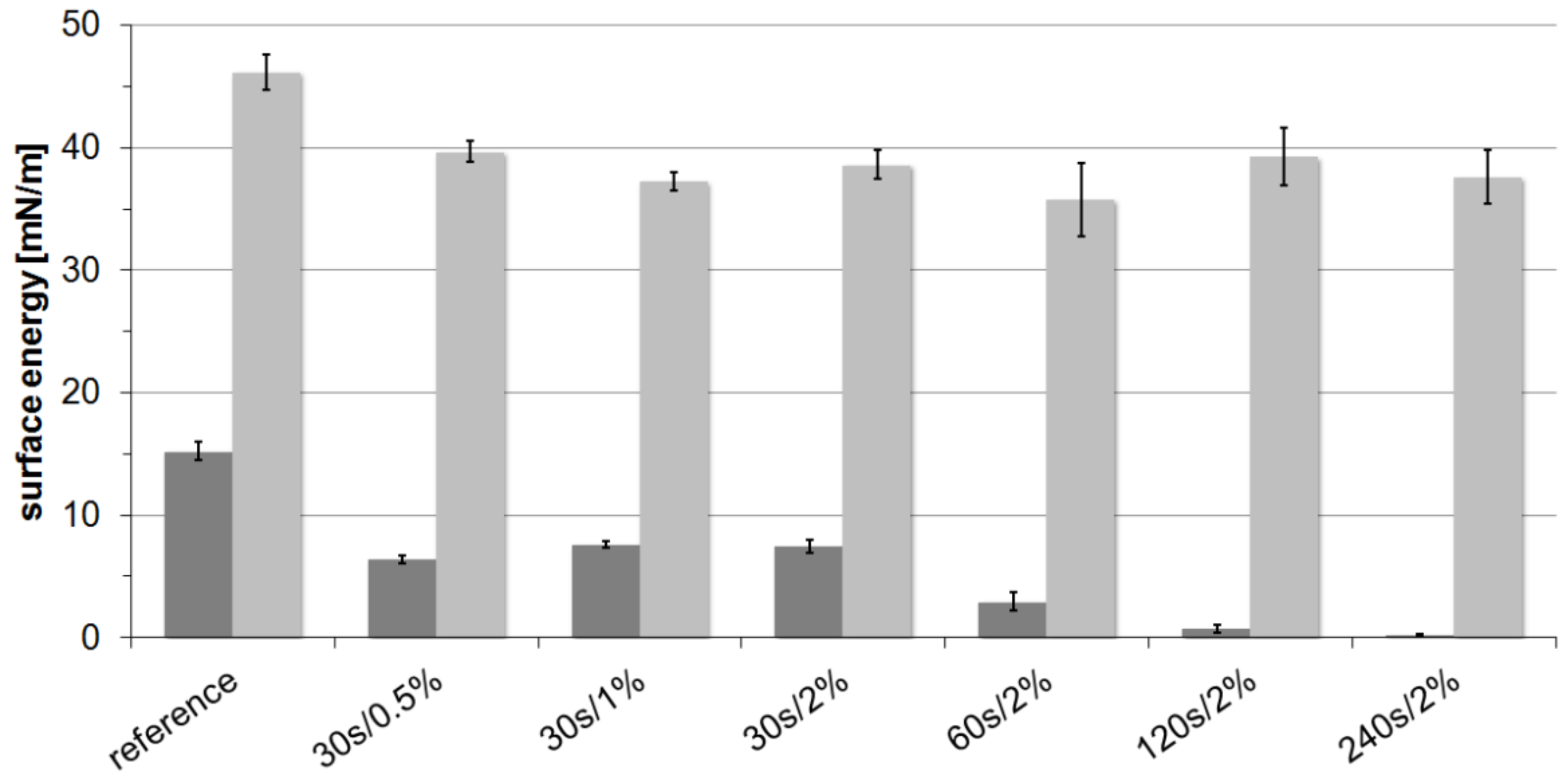
2.1. Contact Angles and Surface Energies
2.2. Scanning Electron Microscopy
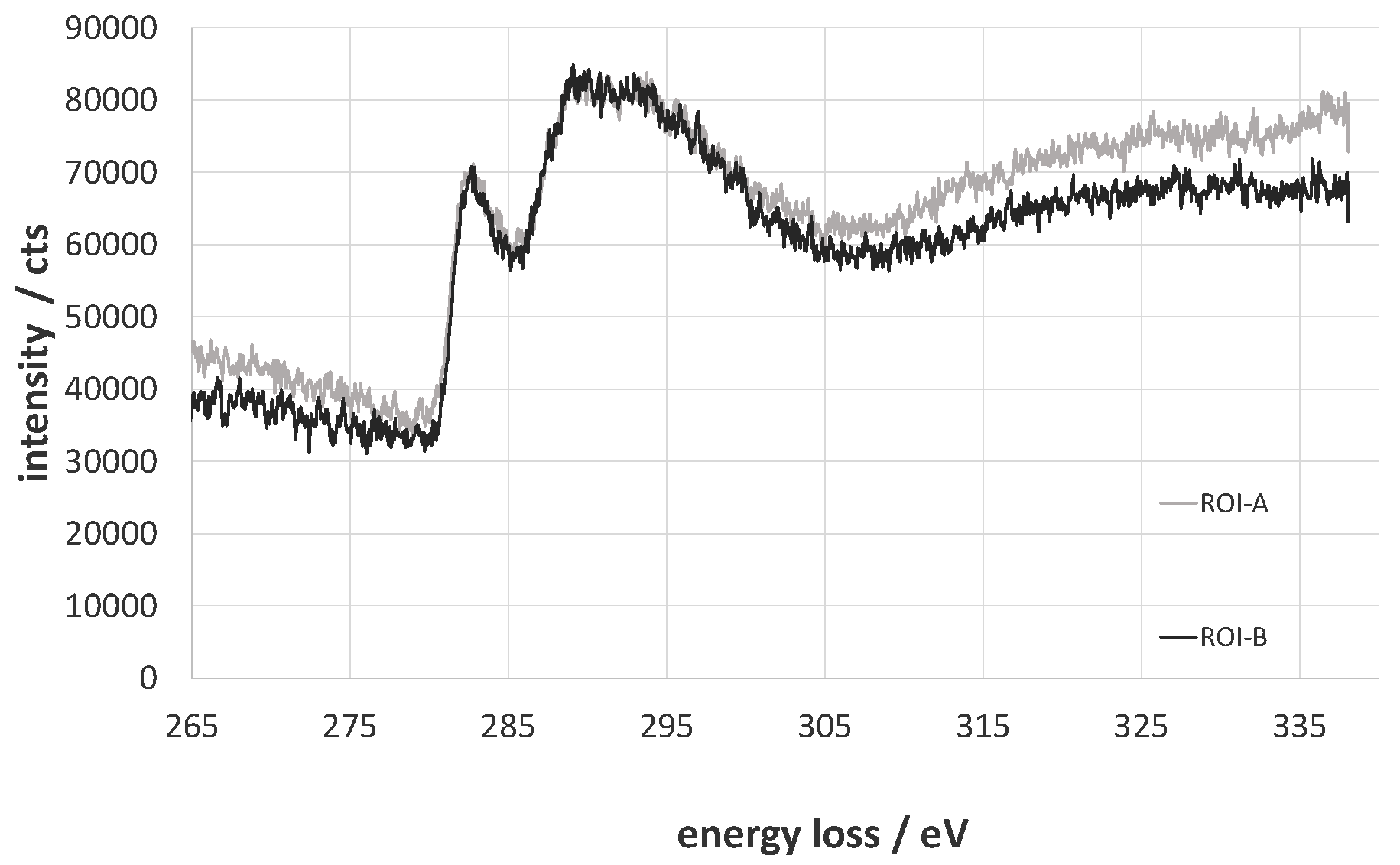
2.3. X-Ray Photoelectron Spectroscopy
2.4. Tensile Strength Tests
3. Materials and Methods
3.1. Investigated High-Performance Fibers
3.2. (Oxy-)Fluorination
3.3. Contact Angle Measurements and Surface Energy Calculation
3.4. Scanning Electron Microscopy
3.5. Transmission Electron Microscopy, Electron Energy-Loss Spectroscopy
3.6. X-Ray Photoelectron Spectroscopy
3.7. Tensile Strength Tests
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CF | carbon fiber |
| EELS | electron energy-loss spectroscopy |
| ELNES | electron energy-loss near edge structure |
| FIB | focussed ionic beam |
| PEEK | polyether ether ketone |
| PPS | poly(p-phenylene sulfide) |
| PPTA | poly(p-phenylene terephthalamide) |
| SEM | scanning electron microscopy |
| (S)TEM) | scanning electron microscopy |
| UHMW PE | ultra-high-molecular-weight polyethylene |
| XPS | X-ray photoelectron spectroscopy |
References
- Cherif, C. (Ed.) Textile Materials for Lightweight Constructions, 1st ed.; Springer-Verlag: Berlin, Germany; Heidelberg, Germany, 2016; pp. 1–6.
- Rudolph, H.; Sheeting, P.; Schmalz, E.; Frenzel, W.; Heidenreich, R. Resistances of high-performance fibers depending on environmental. Tech. Text. 2013, 2013, 19–22. [Google Scholar]
- Bartusch, M.; Hund, R.-D.; Hund, H.; Cherif, C. Surface functionalisation of UHMW polyethylene textile with atmospheric pressure plasma. Fibers Polym. 2014, 15, 736–743. [Google Scholar] [CrossRef]
- Brown, J.R.; Chappell, P.J.C.; Mathys, Z. Plasma surface modification of advanced organic fibers Part II. J. Mater. Sci. 1992, 27, 3167–3172. [Google Scholar] [CrossRef]
- Teodoru, S.; Kusano, Y.; Rozlosnik, N.; Michelsen, P.K. Continuous Plasma Treatment of Ultra-High- Molecular-Weight Polyethylene (UHMWPE) Fibers for Adhesion Improvement. Plasma Process. Polym. 2009, 6, S375–S381. [Google Scholar] [CrossRef]
- Väänänen, R.; Heikkilä, P.; Tuominen, M.; Kuusipalo, J.; Harlin, A. Fast and efficient surface treatment for nonwoven materials by atmospheric pressure plasma. Int. J. Adhes. Adhes. 2010, 10, 8–13. [Google Scholar]
- Biswas, M.K.; Shayed, M.A.; Hund, R.-D.; Cherif, C. Surface modification of Twaron aramid fiber by the atmospheric air plasma technique. Technol. Res. J. 2012, 83, 406–417. [Google Scholar] [CrossRef]
- Plawky, U.; Londschien, M.; Michaeli, W. Surface modification of an aramid fiber treated in a low-temperature microwave plasma. J. Mater. Sci. 1996, 31, 6043–6053. [Google Scholar] [CrossRef]
- Li, Y.; Sun, Y.; Deng, X.; Yang, Q.; Bai, Z.; Xu, Z. Graft polymerization of acrylic acid onto polyphenylene sulfide nonwoven initiated by low temperature plasma. J. Appl. Polym. Sci. 2006, 102, 5884–5889. [Google Scholar] [CrossRef]
- Anagreh, N.; Dorn, L.; Bilke-Krause, C. Low-pressure plasma pretreatment of polyphenylene sulfide (PPS) surfaces for adhesive bonding. Int. J. Adhes. Adhes. 2008, 28, 16–22. [Google Scholar] [CrossRef]
- Käppler, I.; Hund, R.-D.; Cherif, C. Surface modification of carbon fibers using plasma technique. Autex Res. J. 2014, 14, 12–16. [Google Scholar] [CrossRef]
- Bian, X.S.; Ambrosio, L.; Kenny, J.M.; Nicolais, L.; Occhiello, E.; Morra, M. The effects of surface treatments of fibers on the interfacial properties in single-fiber composites. Part I. Single carbon fiber/epoxy resin composite system. J. Adhes. Sci. Technol. 1991, 5, 377–388. [Google Scholar] [CrossRef]
- Marcandalli, B.; Riccardi, C. Plasma Treatments of Fibers and Textiles; Shishoo, R., Ed.; Woodhead Publishing Limited: Cambridge, UK, 2007; pp. 282–300. [Google Scholar]
- Murakami, T.; Kuroda, S.; Osawa, Z. Dynamics of polymeric solid surfaces treated with oxygen plasma: Effect of aging media after plasma treatment. J. Colloid Interface Sci. 1998, 202, 37–44. [Google Scholar] [CrossRef]
- Kim, Y.J.; Lee, H.J.; Lee, S.W.; Cho, B.W.; Park, C.R. Effects of sulfuric acid treatment on the microstructure and electrochemical performance of a polyacrylonitrile (PAN)-based carbon anode. Carbon 2005, 43, 163–169. [Google Scholar] [CrossRef]
- Pittman, C.U.; He, G.-R.; Wu, B.; Gardner, S.D. Chemical modification of carbon fiber surfaces by nitric acid oxidation followed by reaction with tetraethylenepentamine. Carbon 1997, 35, 317–331. [Google Scholar] [CrossRef]
- Wu, G.M.; Hung, C.H.; You, J.H.; Liu, S.J. Surface modification of reinforcement fibers for composites by acid treatments. J. Polym. Res. 2004, 11, 31–36. [Google Scholar] [CrossRef]
- Zhang, G.; Sun, S.; Yang, D.; Dodelet, J.P.; Sacher, E. The surface analytical characterization of carbon fibers functionalized by H2SO4/HNO3 treatment. Carbon 2008, 46, 196–205. [Google Scholar] [CrossRef]
- Yue, Z.R.; Jiang, W.; Wang, L.; Gardner, S.D.; Pittman, C.U. Surface characterization of electrochemically oxidized carbon fibers. Carbon 1999, 37, 1785–1796. [Google Scholar] [CrossRef]
- Pittman, C.U.; Jiang, W.; Yue, Z.R.; Gardner, S.; Wang, L.; Toghiani, H. Surface properties of electrochemically oxidized carbon fibers. Carbon 1999, 37, 1797–1807. [Google Scholar] [CrossRef]
- Rashkovan, I.A.; Korabel’nikov, Y.G. The effect of fiber surface treatment on its strength and adhesion to the matrix. Compos. Sci. Technol. 1997, 57, 1017–1022. [Google Scholar] [CrossRef]
- Park, S.-J.; Kim, M.-H. Effect of acidic anode treatment on carbon fibers for increasing fiber-matrix adhesion and its relationship to interlaminar shear strength of composites. J. Mater. Sci. 2000, 35, 1901–1905. [Google Scholar] [CrossRef]
- Alexander, M.R.; Jones, F.R. Effect of electrolytic oxidation upon the surface chemistry of type A carbon fibers—Part II, analysis of derivatised surface functionalities by XPS, and TOF SIMS. Carbon 1995, 33, 569–580. [Google Scholar] [CrossRef]
- Chan, C.-M.; Ko, T.-M.; Hiraoka, H. Polymer surface modification by plasmas and photons. Surf. Sci. Rep. 1996, 24, 1–54. [Google Scholar] [CrossRef]
- Yip, J.; Chan, K.; Sin, K.M.; Lau, K.S. Study of physico-chemical surface treatments on dyeing properties of polyamides. Part 2: Effect of UV excimer laser irradiation. Color. Technol. 2002, 118, 31–34. [Google Scholar] [CrossRef]
- Knittel, D.; Schollmeyer, E. Surface structuring of synthetic polymers by UV-laser irradiation. Part IV. Applications of excimer laser induced surface modification of textile materials. Polym. Int. 1998, 45, 110–117. [Google Scholar] [CrossRef]
- Joffre, S.P. Impermeable Polyethylene Film and Containers and Process of Making Same. U.S. Patent 2811468, 29 October 1957. [Google Scholar]
- Kruse, A.; Krüger, G.; Baalmann, A.; Hennemann, O.-D. Surface pretreatment of plastics for adhesive bonding. J. Adhes. Sci. Technol. 1995, 9, 1611–1621. [Google Scholar] [CrossRef]
- Friedrich, J.F.; Wigan, L.; Unger, W.; Lippitz, A.; Erdmann, J.; Gorsler, H.-V. Barrier properties of plasma and chemically fluorinated polypropylene and polyethyleneterephthalate. Surf. Coat. Technol. 1995, 74–75, 910–918. [Google Scholar] [CrossRef]
- Kharitonov, A.P.; Taege, R.; Ferrier, G.; Teplyakov, V.V.; Syrtsova, D.A.; Koops, G.-H. Direct fluorination—Useful tool to enhance commercial properties of polymer articles. J. Fluor. Chem. 2005, 126, 251–263. [Google Scholar] [CrossRef]
- Park, S.J.; Seo, M.K.; Rhee, K.Y. Studies on mechanical interfacial properties of oxy-fluorinated carbon fibers-reinforced composites. Mater. Sci. Eng. A 2003, 356, 219–226. [Google Scholar] [CrossRef]
- Park, S.-J.; Seo, M.-K.; Lee, J.-R. Relationship between surface characteristics and interlaminar shear strength of oxyfluorinated carbon fibers in a composite system. J. Colloid Interface Sci. 2003, 268, 127–132. [Google Scholar] [CrossRef]
- Seo, M.-K.; Park, S.-J. Surface characteristics of carbon fibers modified by direct oxyfluorination. J. Colloid Interface Sci. 2009, 330, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.-J.; Kim, J.W.; Im, J.S.; Park, S.-J.; Lee, Y.-S. Nitrogen and hydrogen adsorption of activated carbon fibers modified by fluorination. J. Ind. Eng. Chem. 2009, 15, 410–414. [Google Scholar] [CrossRef]
- Vargha, V.; Chetty, A.; Sulyok, Z.; Mihály, J.; Keresztes, Z.; Tòth, A.; Sajó, I.; Korecz, L.; Anandjiwala, R.; Boguslavsky, L. Functionalisation of polypropylene non-woven fabrics (NWFs) -F unctionalisation by oxyfluorination as a first step for graft polymerisation. J. Therm. Anal. Calorim. 2012, 109, 1019–1032. [Google Scholar] [CrossRef]
- Kharitonov, A.P.; Simbirtseva, G.V.; Bouznik, V.M.; Chebzubov, M.G.; Dubois, M.; Guérin, K.; Hamwi, A.; Kharbache, H.; Masin, F. Modification of ultra-high-molecular weight polyethylene by various fluorinating routes. J. Polym. Sci. Part A Polym. Chem. 2011, 49, 3559–3573. [Google Scholar] [CrossRef]
- Gao, J.; Dai, Y.; Wang, X.; Huang, J.; Yao, J.; Yang, J. Effects of different fluorination routes on aramid fiber surface structures and interlaminar shear strength of its composites. Appl. Surf. Sci. 2013, 270, 627–633. [Google Scholar] [CrossRef]
- Maity, J.; Jacob, C.; Das, C.K.; Kharitonov, A.P.; Singh, R.P.; Alam, S. Fluorinated aramid fiber reinforced polypropylene composites and their characterization. Polym. Compos. 2007, 28, 462–469. [Google Scholar] [CrossRef]
- Kaul, A.; Udipi, K. ESCA study of the surface oxidation of poly(phenylene sulfide) powder by heterogeneous reactions. Macromolecules 1989, 22, 1201–1207. [Google Scholar] [CrossRef]
- Dubois, M.; Guérin, K.; Giraudet, J.; Pilichowski, J.-F.; Thomas, P.; Delbé, K.; Mansot, J.-L.; Hamwi, A. Direct fluorination of poly(p-phenylene). J. Electron Spectrosc. Relat. Phenom. 2005, 46, 6736–6745. [Google Scholar] [CrossRef]
- Sherwood, P.M.A. Surface analysis of carbon and carbon fibers for composites. J. Electron Spectrosc. Relat. Phenom. 1996, 81, 319–342. [Google Scholar] [CrossRef]
- Bismarck, A.; Tahhan, R.; Springer, J.; Schulz, A.; Klapötke, T.M.; Zeil, H. Influence of fluorination on the properties of carbon fibers. J. Fluor. Chem. 1997, 87, 127–134. [Google Scholar] [CrossRef]
- Hamwi, A. Fluorine reactivity with graphite and fullerenes. Fluoride derivatives and some practical electrochemical applications. J. Phys. Chem. Solids 1995, 57, 677–688. [Google Scholar] [CrossRef]
- Schmid, H.K. Phase identification in Carbon and BN systems by EELS. Microsc. Microanal. Microstruct. 1995, 6, 99–111. [Google Scholar] [CrossRef]
- Langenhorst, F.; Solozhenko, V.L. ATEM-EELS study of a new diamond-like phases in B-C-N system. Phys. Chem. Chem. Phys. 2002, 4, 5183–5188. [Google Scholar] [CrossRef]
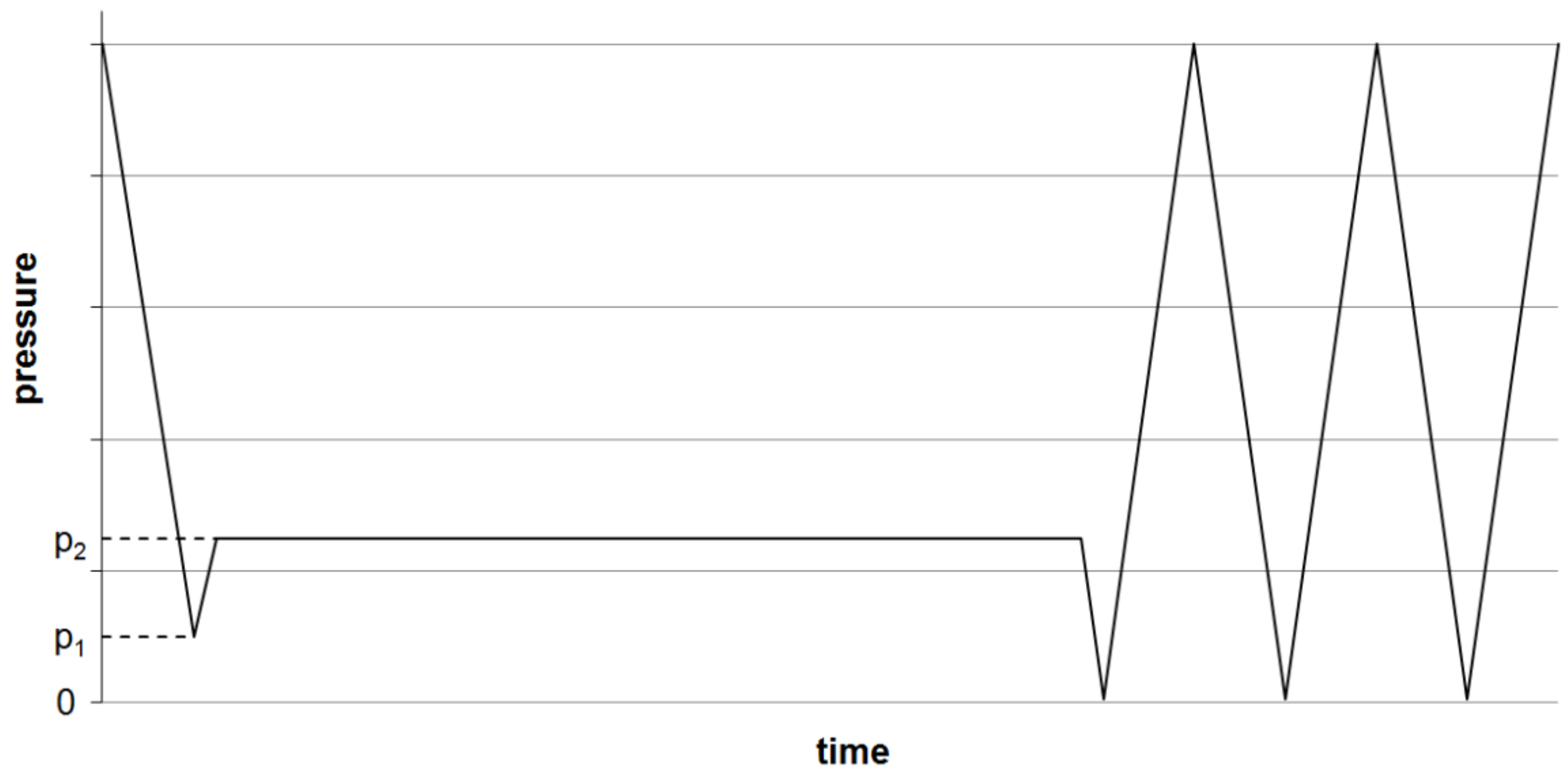
- Bartusch, M.; Hund, R.-D.; Cherif, C. Potential errors in the measurement of contact angles by means of single fiber tensiometry of fibers. Tech. Text. 2014, 4, 133–135. [Google Scholar]
- Kaelble, D.H.; Uy, K.C. A Reinterpretation of organic liquid-polytetrafluoroethylene surface interactions. J. Adhes. 1970, 2, 50–60. [Google Scholar] [CrossRef]
- Owens, D.K.; Wendt, R.C. Estimation of the surface free energy of polymers. J. Appl. Polym. Sci. 1969, 13, 1741–1747. [Google Scholar] [CrossRef]
- Rabel, W. Liquid interfaces in theory and applied technology. Phys. Bl. 1977, 33, 151–156. [Google Scholar] [CrossRef]
- Mühle, U.; Standke, Y.; Zschech, E.; Meinl, J.; Michaelis, A.; Kirsten, M.; Cherif, C. Structural characterization of carbon fibers along the fabrication process using SEM/FIB and TEM. In Proceedings of the 16th AUTEX World Textile Conference, Ljubljana, Slovenia, 8–10 June 2016; pp. 1–8.
- Sample Availability: Samples of the compounds are not available from the authors.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UHMW PE | PPS | PPTA | CF | |||||
|---|---|---|---|---|---|---|---|---|
| Reference | Sample | Reference | Sample | Reference | Sample | Reference | Sample | |
| CA | 84.9 (1.6) | 58.7 (2.8) | 87.2 (1.5) | 71.8 (4.9) | 50.4 (3.1) | 91.7 (5.3) | 68.9 (5.8) | 50.2 (5.1) |
| CA | 58.2 (2.7) | 49.7 (2.9) | 24.7 (3.6) | 67.6 (2.7) | 24.8 (5.8) | 40.4 (5.5) | 36.0 (7.1) | 37.8 (6.1) |
| (mN/m) | 4.0 (0.3) | 15.2 (0.8) | 0.8 (0.1) | 12.0 (1.4) | 15.2 (0.7) | 0.7 (0.3) | 7.4 (1.0) | 17.5 (1.3) |
| (mN/m) | 29.6 (0.9) | 34.4 (1.2) | 46.2 (1.1) | 24.2 (2.4) | 46.2 (1.4) | 39.3 (2.4) | 41.3 (2.7) | 40.5 (2.3) |
| (mN/m) | 33.6 (1.2) | 49.7 (1.9) | 47.0 (1.3) | 36.2 (3.8) | 61.4 (2.2) | 40.0 (2.7) | 48.7 (3.7) | 58.0 (3.6) |
| Sample | C:O | C:N | C:F | C:S |
|---|---|---|---|---|
| PE reference | 0.027 | 0.000 | 0.000 | 0.000 |
| PE treated | 0.176 | 0.000 | 0.157 | 0.000 |
| PPS reference | 0.115 | 0.000 | 0.000 | 0.040 |
| PPS treated | 0.215 | 0.010 | 0.198 | 0.033 |
| PPTA reference | 0.199 | 0.098 | 0.000 | 0.000 |
| PPTA treated | 0.251 | 0.042 | 0.241 | 0.000 |
| CF reference | 0.166 | 0.037 | 0.000 | 0.000 |
| CF treated | 0.175 | 0.027 | 0.050 | 0.000 |
| Sample | E Modulus (GPa) | Breaking Force (N) | Tensile Strength (MPa) | Elongation at Break (%) |
|---|---|---|---|---|
| PE reference | 82.4 (1.6) | 504 (13) | 2328 (61) | 3.19 (0.09) |
| PE treated | 80.0 (5.0) | 440 (33) | 2032 (151) | 2.59 (0.13) |
| PPS reference | 6.4 (0.1) | 39.8 (1.1) | 491 (16) | 23.73 (0.80) |
| PPS treated | 6.3 (0.1) | 38.9 (1.5) | 479 (15) | 23.71 (0.76) |
| PPTA reference | 66.4 (2.7) | 601 (31) | 2545 (131) | 3.33 (0.15) |
| PPTA treated | 64.7 (3.1) | 600 (27) | 2541 (116) | 3.34 (0.12) |
| CF reference | 136 (15.6) | 592 (13) | 1295 (28) | 1.19 (0.16) |
| CF treated | 124 (12.7) | 683 (26) | 1494 (57) | 1.34 (0.07) |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kruppke, I.; Bartusch, M.; Hickmann, R.; Hund, R.-D.; Cherif, C. Effects of (Oxy-)Fluorination on Various High-Performance Yarns. Molecules 2016, 21, 1127. https://doi.org/10.3390/molecules21091127
Kruppke I, Bartusch M, Hickmann R, Hund R-D, Cherif C. Effects of (Oxy-)Fluorination on Various High-Performance Yarns. Molecules. 2016; 21(9):1127. https://doi.org/10.3390/molecules21091127
Chicago/Turabian StyleKruppke, Iris, Matthias Bartusch, Rico Hickmann, Rolf-Dieter Hund, and Chokri Cherif. 2016. "Effects of (Oxy-)Fluorination on Various High-Performance Yarns" Molecules 21, no. 9: 1127. https://doi.org/10.3390/molecules21091127
APA StyleKruppke, I., Bartusch, M., Hickmann, R., Hund, R. -D., & Cherif, C. (2016). Effects of (Oxy-)Fluorination on Various High-Performance Yarns. Molecules, 21(9), 1127. https://doi.org/10.3390/molecules21091127