
Small Molecule-Photoactive Yellow Protein Labeling Technology in Live Cell Imaging

Abstract
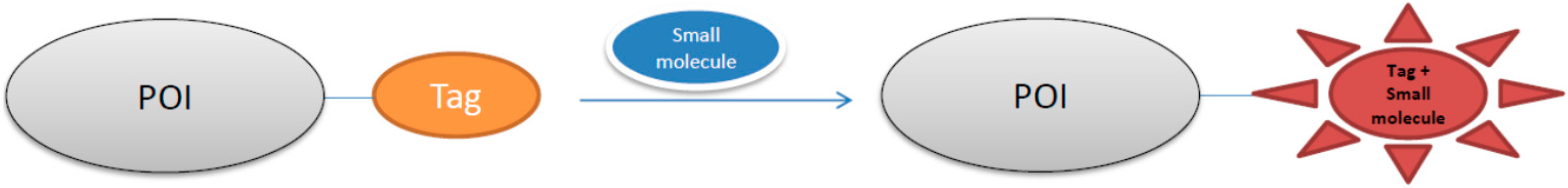
:1. Introduction
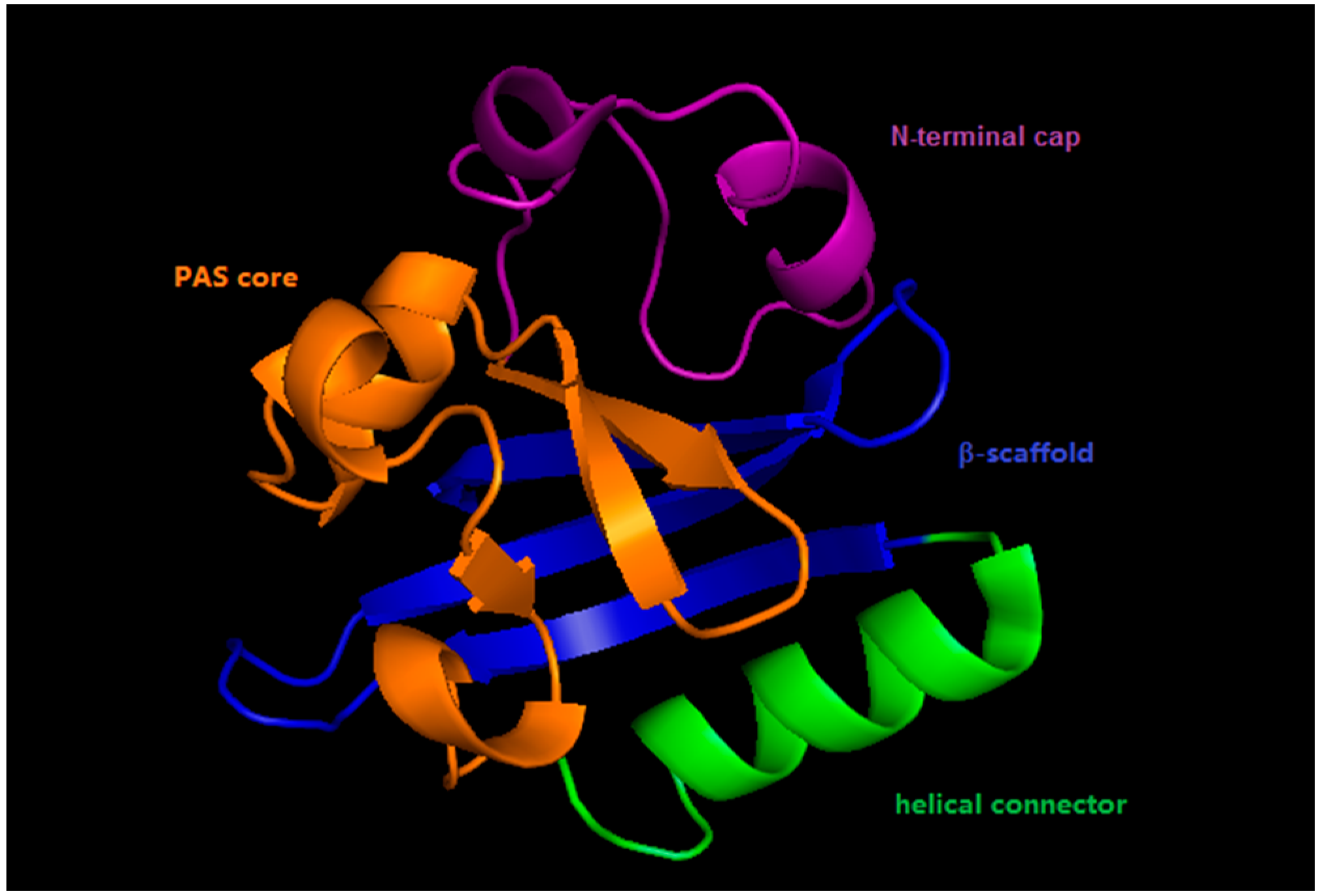
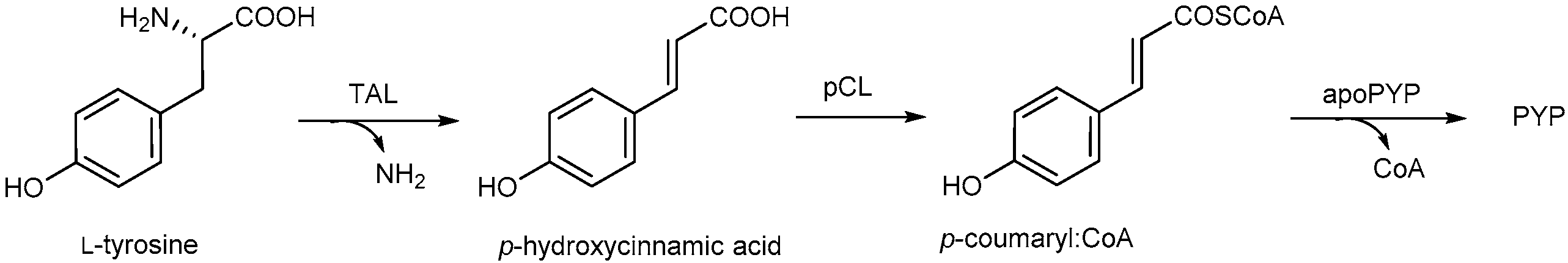
2. Photoactive Yellow Protein
3. Small Molecule-Photoactive Yellow Protein Labeling Technology in Live-Cell Imaging
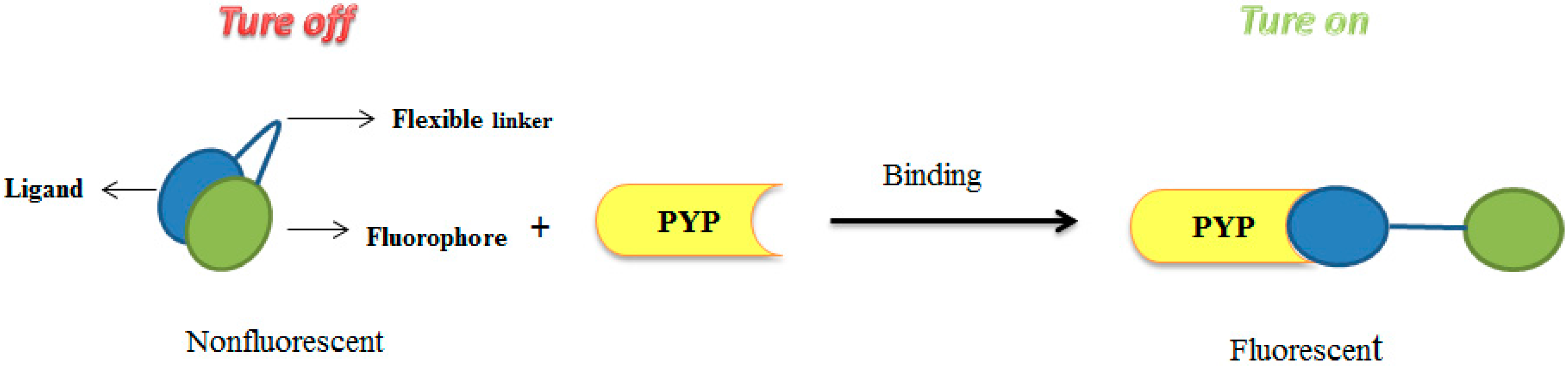
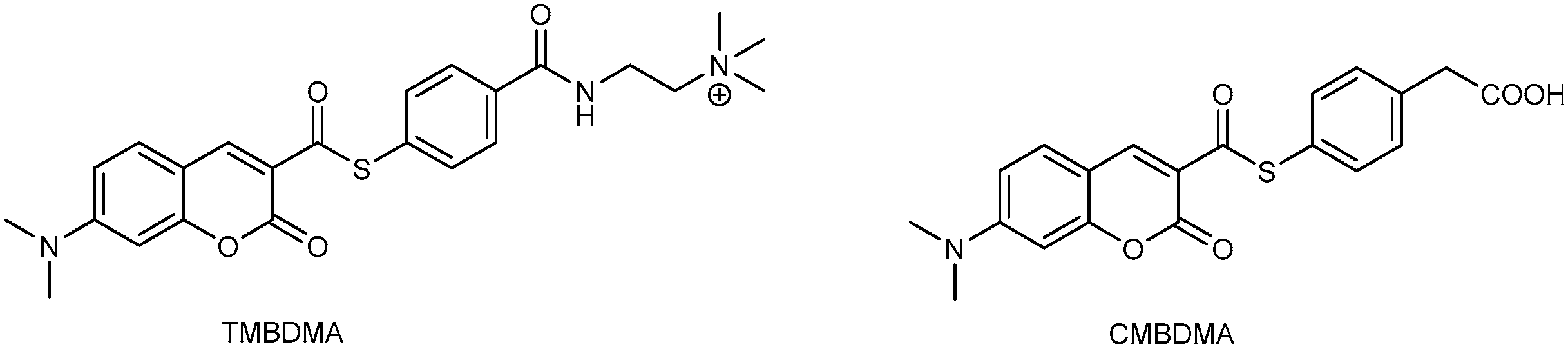
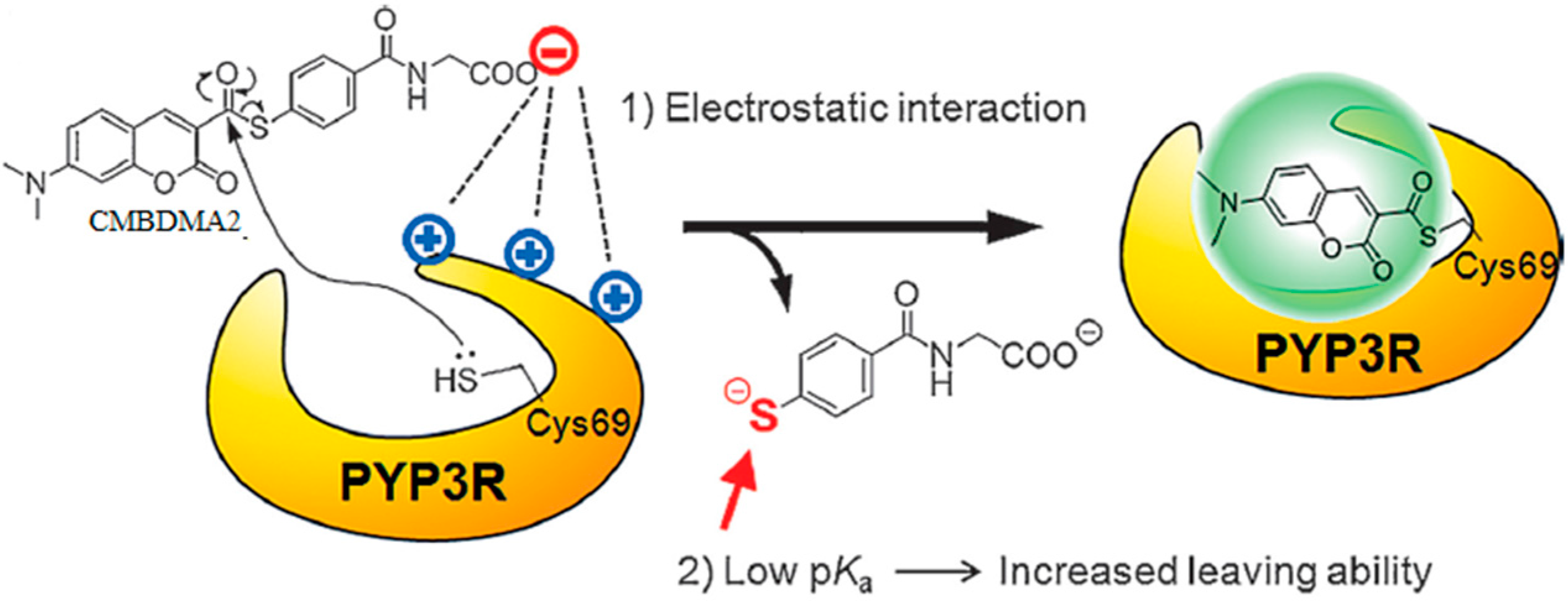
3.1. Development of Turn-On Fluorescent Protein Labeling System Based on Photoactive Yellow Protein
3.2. Development of Protein-Labeling Probes for No-Wash Live Cell Imaging
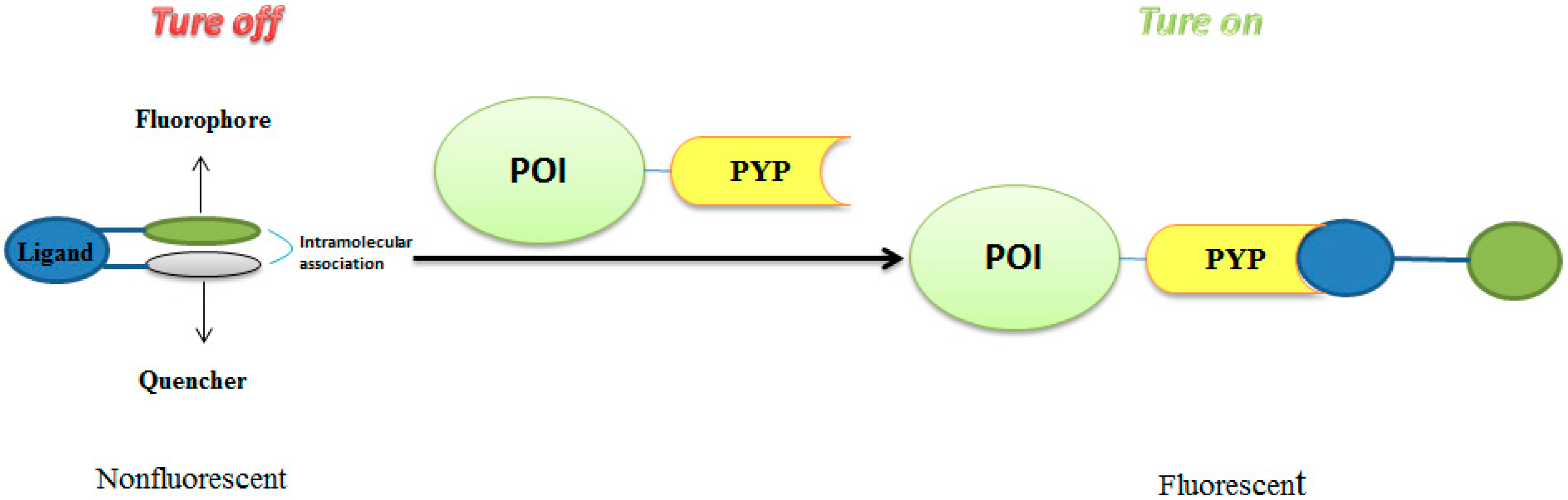
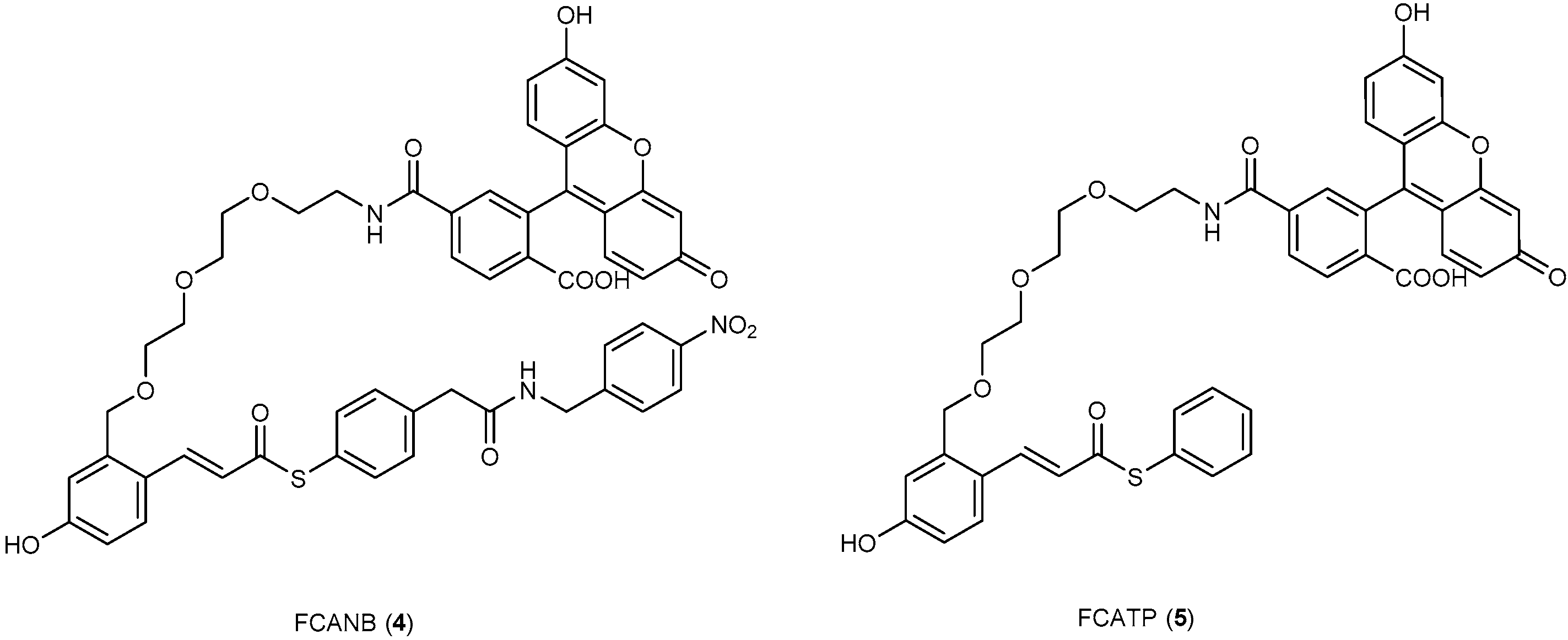
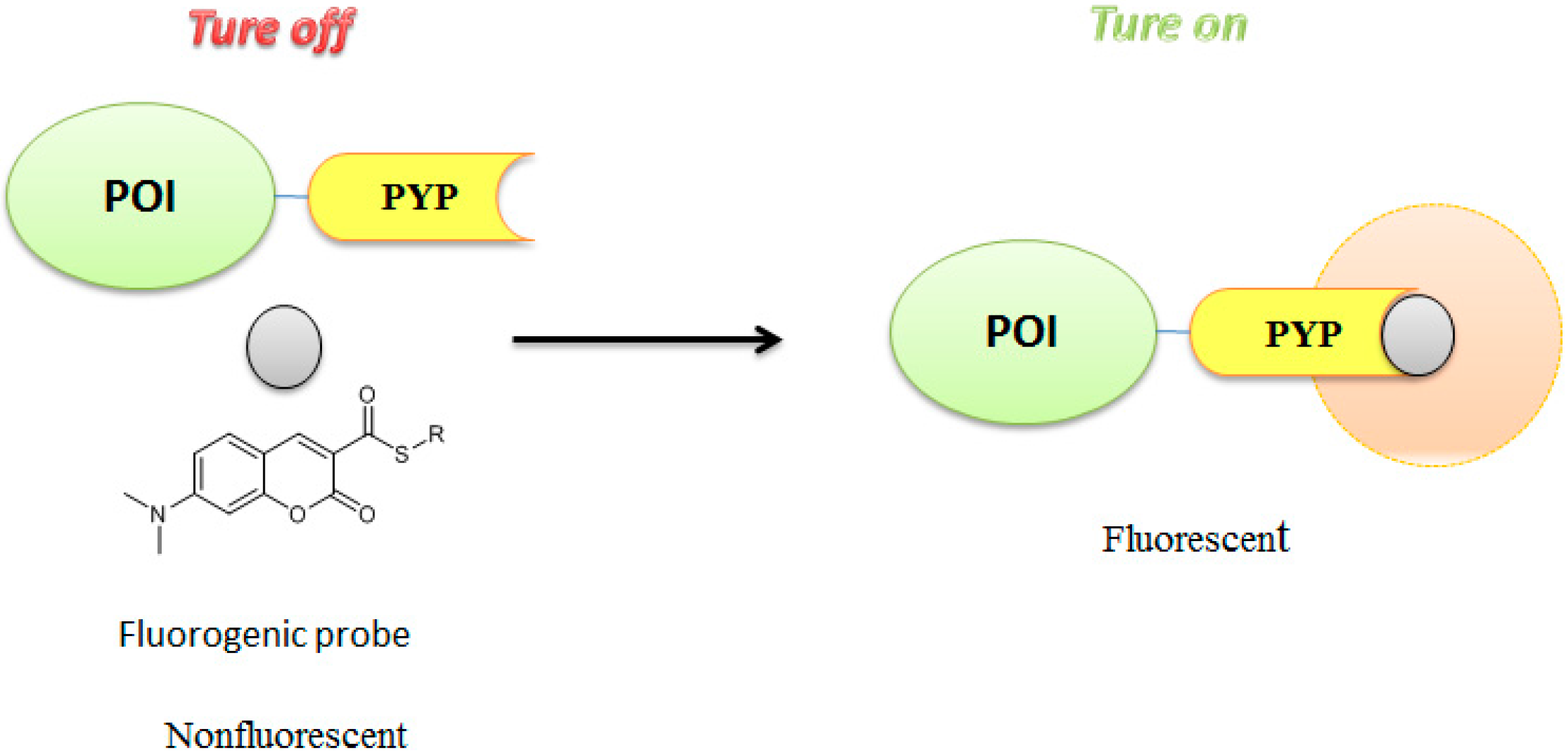
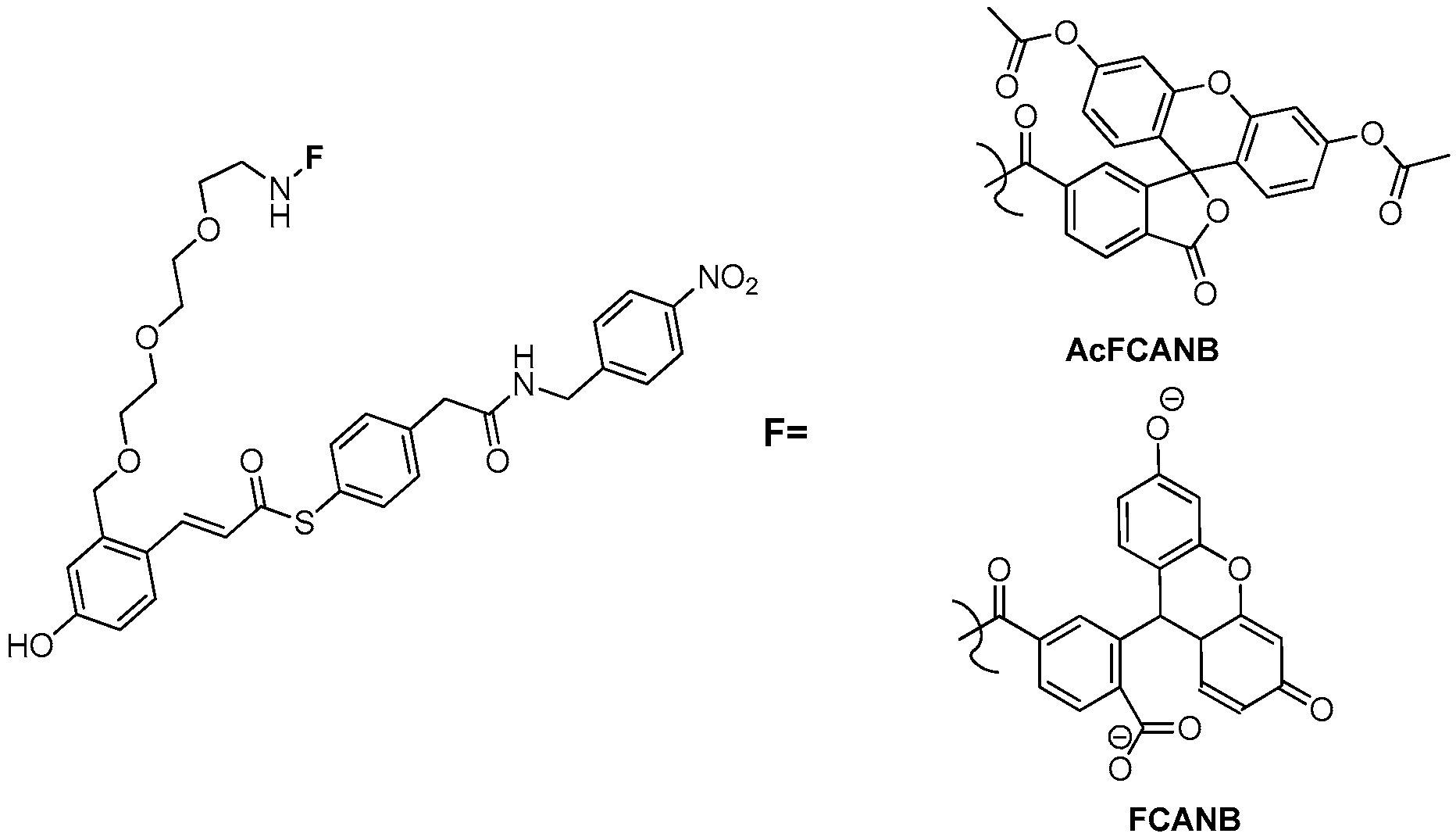
3.3. Development of Flurogenic Probes for Imaging of Intracellular Proteins in Living Cells
3.4. Turn-On Fluorescent Protein Labeling System Based on Mutant PYP-tag
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Grammel, M.; Hang, H.C. Chemical reporters for biological discovery. Nat. Chem. Biol. 2013, 9, 475–484. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, H.M.; Johnsson, K.; Gautier, A. Chemical probes shed light on protein function. Curr. Opin. Struct. Biol. 2007, 17, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, Y.; Ojida, A.; Hamachi, I. Protein organic chemistry and applications for labeling and engineering in live-cell systems. Angew. Chem. Int. Ed. 2013, 52, 4088–4106. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, A. Proteins on the move: Insights gained from fluorescent protein technologies. Nat. Rev. Mol. Cell Biol. 2011, 12, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Bruchez, M.P. Advances in chemical labeling of proteins in living cells. Cell Tissue Res. 2015, 360, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Chandrudu, S.; Simerska, P.; Toth, I. Chemical methods for peptide and protein production. Molecules 2013, 18, 4373–4388. [Google Scholar] [CrossRef] [PubMed]
- Van Engelenburg, S.B.; Palmer, A.E. Fluorescent biosensors of protein function. Curr. Opin. Chem. Biol. 2008, 12, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Shui, W.; Carlson, B.L.; Hu, N.; Rabuka, D.; Lee, J.; Bertozzi, C.R. Site-specific chemical modification of recombinant proteins produced in mammalian cells by using the genetically encoded aldehyde tag. Proc. Natl. Acad. Sci. USA 2009, 106, 3000–3005. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.; Ting, A.Y. Site-specific labeling of proteins with small molecules in live cells. Curr. Opin. Chem. Biol. 2005, 16, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.R.; Campbell, R.E.; Gross, L.A.; Martin, B.R.; Walkup, G.K.; Yao, Y.; Llopis, J.; Tsien, R.Y. New biarsenical Ligands and tetracysteine motifs for protein labeling in vitro and in vivo: Synthesis and biological applications. J. Am. Chem. Soc. 2002, 124, 6063–6076. [Google Scholar] [CrossRef] [PubMed]
- Griffin, B.A.; Adams, S.R.; Tsien, R.Y. Specific covalent labeling of recombinant protein molecules inside live cells. Science 1998, 281, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Gautier, A.; Juillerat, A.; Heinis, C.; Corrêa, I.R., Jr.; Kindermann, M.; Beaufils, F.; Johnsson, K. An engineered protein tag for multiprotein labeling in living cells. Chem. Biol. 2008, 15, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Keppler, A.; Gendreizig, S.; Gronemeyer, T.; Pick, H.; Vogel, H.; Johnsson, K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 2003, 21, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Los, G.V.; Encell, L.P.; McDougall, M.G.; Hartzell, D.D.; Karassina, N.; Zimprich, C.; Wood, M.G.; Learish, R.; Ohana, R.F.; Urh, M.; et al. HatoTag: A novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 2008, 3, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.L.; Lord, S.J.; Iwanaga, S.; Zhan, K.; Xie, H.; Williams, J.C.; Wang, H.; Bowman, G.R.; Goley, E.D.; Shapiro, L.; et al. Superresolution Imaging of Targeted Proteins in Fixed and Living Cells Using Photoactivatable Organic Fluorophores. J. Am. Chem. Soc. 2010, 132, 15099–15101. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Jing, C.; Gallagher, S.S.; Sheetz, M.P.; Cornish, V.W. Second-Generation Covalent TMP-Tag for Live Cell Imaging. J. Am. Chem. Soc. 2012, 134, 13692–13699. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.Z.; Uttamapinant, C.; Poloukhtine, A.; Baskin, J.M.; Codelli, J.A.; Sletten, E.M.; Bertozzi, C.R.; Popik, V.V.; Ting, A.Y. Fluorophore Targeting to Cellular Proteins via Enzyme-Mediated Azide Ligation and Strain-Promoted Cycloaddition. J. Am. Chem. Soc. 2012, 134, 3720–3728. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, H.; Fujishima, S.H.; Uchinomiya, S.H.; Ojida, A.; Hamachi, I. Selective Covalent Labeling of Tag-Fused GPCR Proteins on Live Cell Surface with a Synthetic Probe for Their Functional Analysis. J. Am. Chem. Soc. 2010, 132, 9301–9309. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, S.; Yamamoto, T.; Yoshimura, A.; Watanabe, S.; Kikuchi, K. Covalent Protein Labeling with a Lanthanide Complex and Its Application to Photoluminescence Lifetime-Based Multicolor Bioimaging. Angew. Chem. Int. Ed. 2011, 50, 8750–8752. [Google Scholar] [CrossRef] [PubMed]
- Roberti, M.J.; Bertoncini, C.W.; Klement, R.; Jares-Erijman, E.A.; Jovin, T.M. Fluorescence imaging of amyloid formation in living cells by a functional, tetracysteine-tagged alpha-synuclein. Nat. Methods 2007, 4, 345–351. [Google Scholar] [PubMed]
- Sadhu, K.K.; Mizukami, S.; Watanabe, S.; Kikuchi, K. Turn-on fluorescence switch involving aggregation and elimination processes for beta-lactamase-tag. Chem. Commun. 2010, 46, 7403–7405. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, S.; Watanabe, S.; Hori, Y.; Kikuchi, K. Covalent Protein Labeling Based on Noncatalytic beta-Lactamase and a Designed FRET Substrate. J. Am. Chem. Soc. 2009, 131, 5016–5017. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, T.; Johnsson, K.; Okuno, H.; Bito, H.; Inoue, T.; Nagano, T.; Urano, Y. Real-Time Measurements of Protein Dynamics Using FluorescenceActivation-Coupled Protein Labeling Method. J. Am. Chem. Soc. 2011, 133, 6745–6751. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.L.; Zhang, A.; Baker, B.; Sun, L.; Howard, A.; Buswell, J.; Maurel, D.; Masharina, A.; Johnsson, K.; Noren, C.J.; et al. Development of SNAP-Tag Fluorogenic Probes for Wash-Free Fluorescence Imaging. ChemBioChem 2011, 12, 2217–2226. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.J.; Li, L.; Chen, G.Y.; Xu, Q.H.; Yao, S.Q. One- and Two-Photon Live Cell Imaging Using a Mutant SNAP-Tag Protein and Its FRET Substrate Pairs. Org. Lett. 2011, 13, 4160–4163. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.E. Isolation and Characterization of Soluble Cytochromes, Ferredoxins and Other Chromophoric Proteins from the Halophilic Phototrophic Bacterium Ectothiorhodospira-Halophila. Biochim. Biophys. Acta 1985, 806, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Cusanovich, M.A.; Meyer, T.E. Photoactive yellow protein: A prototypic PAS domain sensory protein and development of a common signaling mechanism. Biochemistry 2003, 42, 4759–4770. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.E.; Yakali, E.; Cusanovich, M.A.; Tollin, G. Properties of a Water-Soluble, Yellow Protein Isolated from a Halophilic Phototrophic Bacterium That Has Photochemical Activity Analogous to Sensory Rhodopsin. Biochemistry 1987, 26, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Borgstahl, G.E.O.; Williams, D.R.; Getzoff, E.D. 1.4 Angstrom Structure of Photoactive Yellow Protein, a Cytosolic Photoreceptor—Unusual Fold, Active-Site, and Chromophore. Biochemistry 1995, 34, 6278–6287. [Google Scholar] [CrossRef] [PubMed]
- Düx, P.; Rubinstenn, G.; Vuister, G.W.; Boelens, R.; Mulder, F.A.; Hård, K.; Hoff, W.D.; Kroon, A.R.; Crielaard, W.; Hellingwerf, K.J.; et al. Solution structure and backbone dynamics of the photoactive yellow protein. Biochemistry 1998, 37, 12689–12699. [Google Scholar] [CrossRef] [PubMed]
- Imamoto, Y.; Kataoka, M. Structure and photoreaction of photoactive yellow protein, a structural prototype of the PAS domain superfamily. Photochem. Photobiol. 2007, 83, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Pellequer, J.L.; Wager-Smith, K.A.; Kay, S.A.; Getzoff, E.D. Photoactive yellow protein: A structural prototype for the three-dimensional fold of the PAS domain superfamily. Proc. Natl. Acad. Sci. USA 1998, 95, 5884–5890. [Google Scholar] [CrossRef] [PubMed]
- Van der Horst, M.A.; Arents, J.C.; Kort, R.; Hellingwerf, K.J. Binding, tuning and mechanical function of the 4-hydroxy-cinnamic acid chromophore in photoactive yellow protein. Photochem. Photobiol. 2007, 6, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Kort, R.; Vonk, H.; Xu, X.; Hoff, W.D.; Crielaard, W.; Hellingwerf, K.J. Evidence for trans-cis isomerization of the p-coumaric acid chromophore as the photochemical basis of the photocycle of photoactive yellow protein. FEBS Lett. 1996, 382, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Xie, A.; Hoff, W.D.; Kroon, A.R.; Hellingwerf, K.J. Glu46 donates a proton to the 4-hydroxycinnamate anion chromophore during the photocycle of photoactive yellow protein. Biochemistry 1996, 35, 14671–14678. [Google Scholar] [CrossRef] [PubMed]
- Kyndt, J.A.; Meyer, T.E.; Cusanovich, M.A.; van Beeumen, J.J. Characterization of a bacterial tyrosine ammonia lyase, a biosynthetic enzyme for the photoactive yellow protein. FEBS Lett. 2002, 512, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Kyndt, J.A.; Vanrobaeys, F.; Fitch, J.C.; Devreese, B.V.; Meyer, T.E.; Cusanovich, M.A.; van Beeumen, J.J. Heterologous production of Halorhodospirahalophilaholo-photoactive yellow protein through tandem expression of the postulated biosynthetic genes. Biochemistry 2003, 42, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Imamoto, Y.; Ito, T.; Kataoka, M.; Tokunaga, F. Reconstitution Photoactive Yellow Protein from Apoprotein and P-CoumaricAcid-Derivatives. FEBS Lett. 1995, 374, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Genick, U.K.; Devanathan, S.; Meyer, T.E.; Canestrelli, I.L.; Williams, E.; Cusanovich, M.A.; Tollin, G.; Getzoff, E.D. Active site mutants implicate key residues for control of color and light cycle kinetics of photoactive yellow protein. Biochemistry 1997, 36, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.; Ueno, H.; Mizukami, S.; Kikuchi, K. Photoactive Yellow Protein-Based Protein Labeling System with Turn-On Fluorescence Intensity. J. Am. Chem. Soc. 2009, 131, 16610–16611. [Google Scholar] [CrossRef] [PubMed]
- Takakusa, H.; Kikuchi, K.; Urano, Y.; Higuchi, T.; Nagano, T. Intramolecular fluorescence resonance energy transfer system with coumarin donor included in beta-cyclodextrin. Anal. Chem. 2001, 73, 939–942. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Crosson, S.; Moffat, K. Short hydrogen bonds in photoactive yellow protein. Acta Crystallogr. D 2004, 60, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Fang, Y.Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [PubMed]
- Hori, Y.; Nakaki, K.; Sato, M.; Mizukami, S.; Kikuchi, K. Development of protein-labeling probes with a redesigned fluorogenic switch based on intramolecular association for no-wash live-cell imaging. Angew. Chem. Int. Ed. 2012, 51, 5611–5614. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.P.; Blumenthal, D.K.; Herron, J.N. Antibody-Mediated Fluorescence Enhancement Based on Shifting the Intramolecular Dimer-Reversible-Arrow-Monomer Equilibrium of Fluorescent Dyes. Anal. Chem. 1994, 66, 1500–1506. [Google Scholar] [CrossRef] [PubMed]
- Packard, B.Z.; Toptygin, D.D.; Komoriya, A.; Brand, L. Profluorescent protease substrates: Intramolecular dimers described by the exciton model. Proc. Natl. Acad. Sci. USA 1996, 93, 11640–11645. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Jackson, W.R.; Kanoktanaporn, S.; Halpern, A.M. Solvent Effects on Photophysical Parameters for Coumarin Laser-Dyes. Opt. Commun. 1980, 33, 315–320. [Google Scholar] [CrossRef]
- Kroon, A.R.; Hoff, W.D.; Fennema, H.P.; Gijzen, J.; Koomen, G.J.; Verhoeven, J.W.; Crielaard, W.; Hellingwerf, K.J. Spectral tuning, fluorescence, and photoactivity in hybrids of photoactive yellow protein, reconstituted with native or modified chromophores. J. Biol. Chem. 1996, 271, 31949–31956. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.; Norinobu, T.; Sato, M.; Arita, K.; Shirakawa, M.; Kikuchi, K. Development of Fluorogenic Probes for Quick No-Wash Live-Cell Imaging of Intracellular Proteins. J. Am. Chem. Soc. 2013, 135, 12360–12365. [Google Scholar] [CrossRef] [PubMed]
- Clouaire, T.; Stancheva, I. Methyl-CpG binding proteins: Specialized transcriptional repressors or structural components of chromatin? Cell Mol. Life Sci. 2008, 65, 1509–1522. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.; Hirayama, S.; Sato, M.; Kikuchi, K. Redesign of a fluorogenic labeling system to improve surface charge, brightness, and binding kinetics for imaging the functional localization of bromodomains. Angew. Chem. Int. Ed. 2015, 54, 14368–14371. [Google Scholar] [CrossRef] [PubMed]
- Kamikawa, Y.; Hori, Y.; Yamashita, K.; Lin, J.; Hirayama, S.; Standley, D.M.; Kikuchi, K. Design of a protein tag and fluorogenic probe with modular structure for live-cell imaging of intracellular proteins. Chem. Sci. 2016, 7, 308–314. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
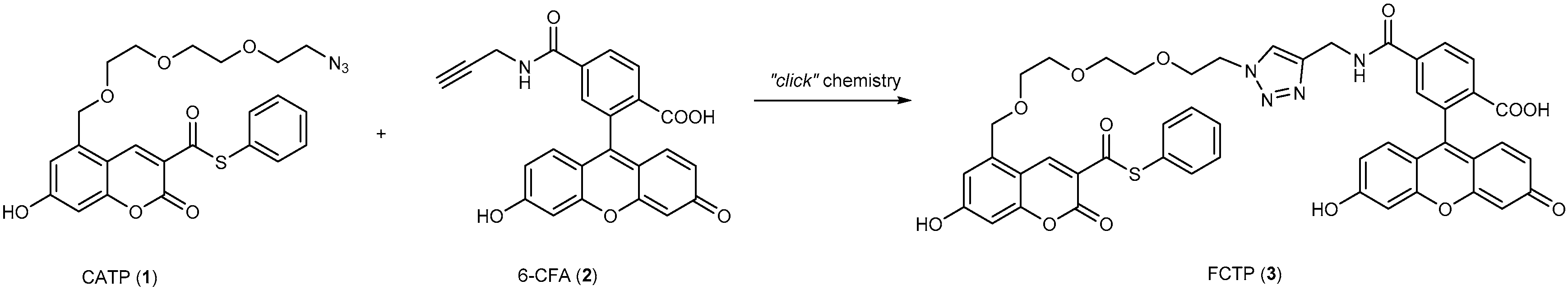{kind=link}
| Species | Function/Pathway |
|---|---|
| Rs. centenum | Polyketide synthase |
| Cyst formation | |
| Phototaxis | |
| Tc. tepidum | |
| Methylomicrobium | Phototaxis |
| Burkholderia | Gas vesicles 4 |
| Haliangium | (Cell buoyancy) |
| Curvibacter | |
| Phodobacter | |
| Rp. palustris | DNA repair (DPL) |
| Rt. Salexigens | Blue light sensor (DGL) |
| Desulfonema | DNA repair (DGL) |
| Idiomarina | Osmotic pressure |
| Halorhodospira | Phototaxis |
| Stigmatella | Stalk formation |
| (cyst formation) | |
| Spirosoma |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, F.; Gao, T.; Zhou, K.; Zeng, W. Small Molecule-Photoactive Yellow Protein Labeling Technology in Live Cell Imaging. Molecules 2016, 21, 1163. https://doi.org/10.3390/molecules21091163
Gao F, Gao T, Zhou K, Zeng W. Small Molecule-Photoactive Yellow Protein Labeling Technology in Live Cell Imaging. Molecules. 2016; 21(9):1163. https://doi.org/10.3390/molecules21091163
Chicago/Turabian StyleGao, Feng, Tang Gao, Kechao Zhou, and Wenbin Zeng. 2016. "Small Molecule-Photoactive Yellow Protein Labeling Technology in Live Cell Imaging" Molecules 21, no. 9: 1163. https://doi.org/10.3390/molecules21091163
APA StyleGao, F., Gao, T., Zhou, K., & Zeng, W. (2016). Small Molecule-Photoactive Yellow Protein Labeling Technology in Live Cell Imaging. Molecules, 21(9), 1163. https://doi.org/10.3390/molecules21091163