3.1. XMP
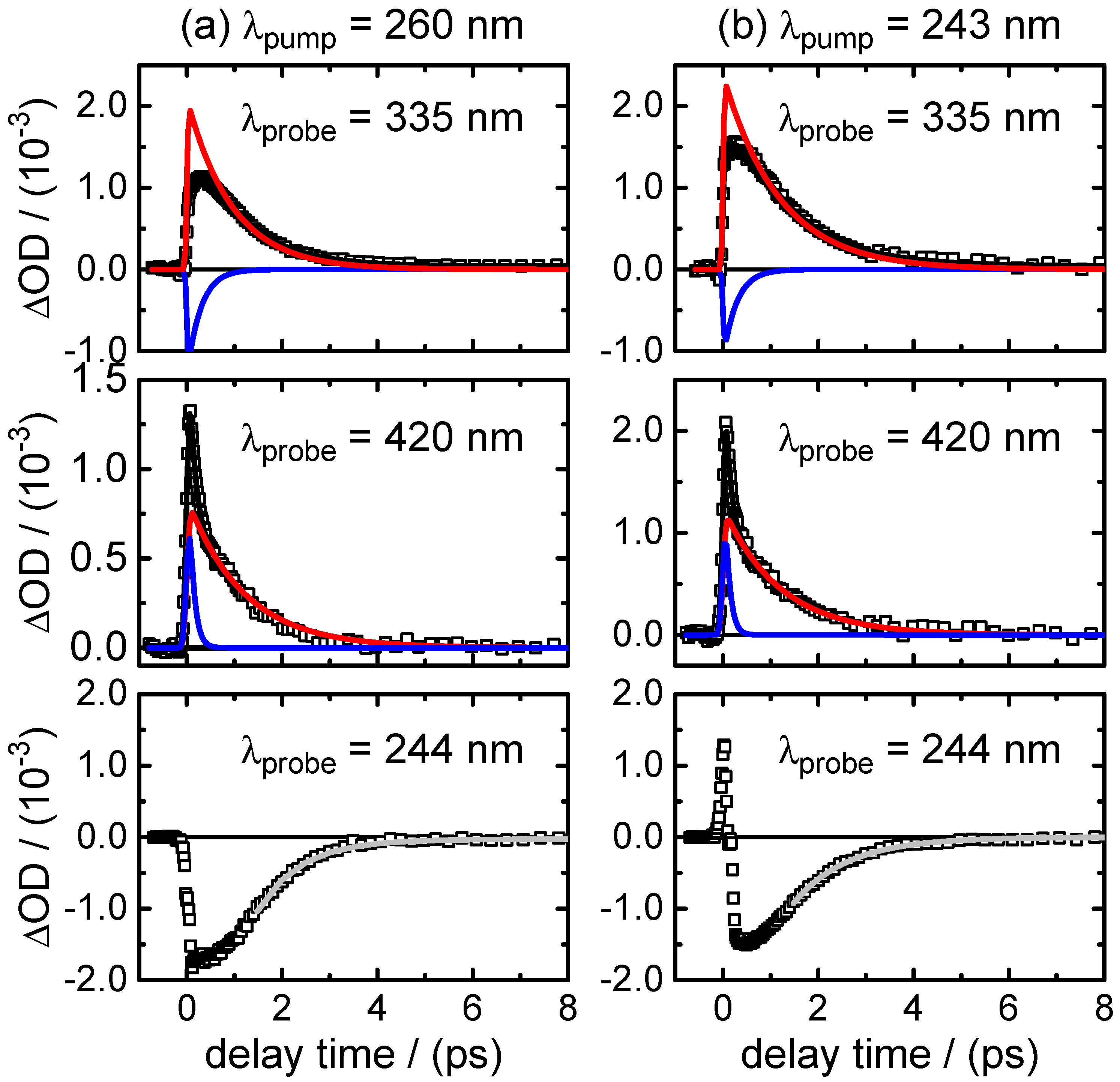
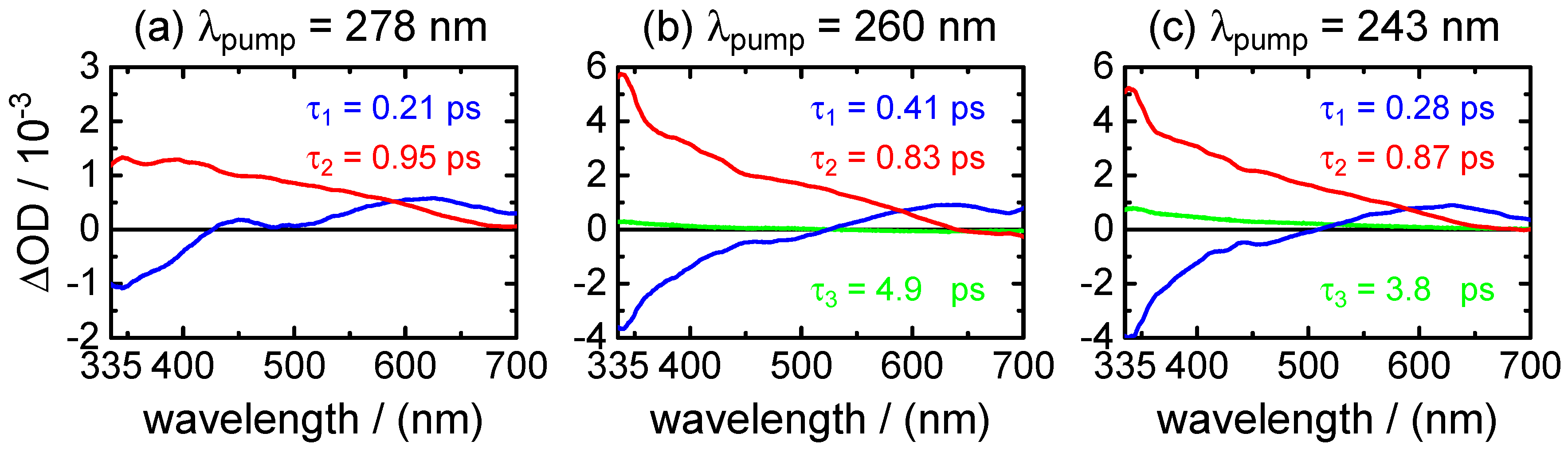
The time-resolved experiments on neutral XMP in aqueous solution under acidic conditions demonstrate an ultrafast radiationless deactivation of the photo-excited electronic state(s) to the ground state that is characterized by two time constants, ps and ps. Both values have to be associated with different processes.
Several arguments let us assign the sub-picosecond lifetime
to the decay dynamics of the initial FC excited state. First,
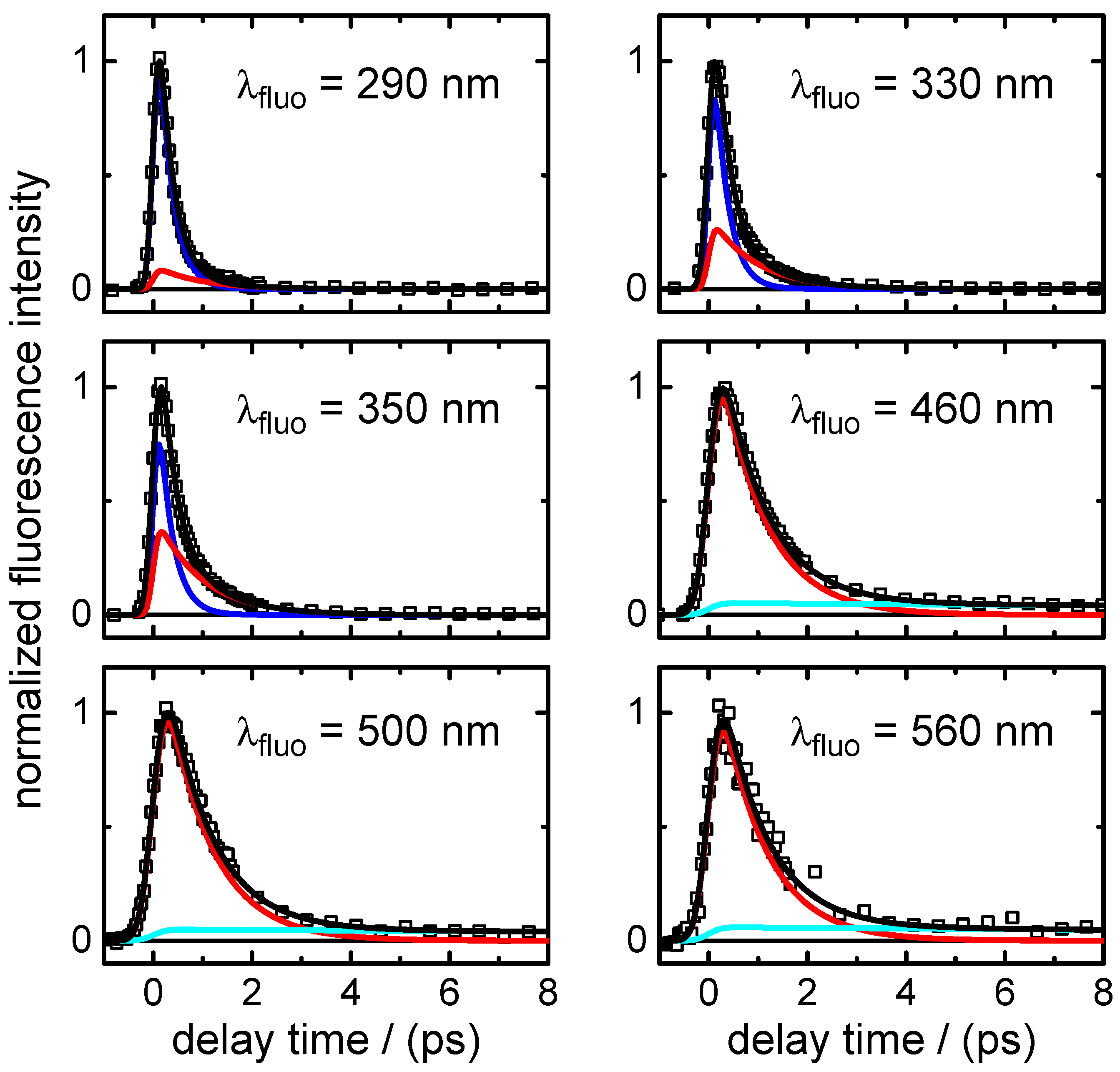
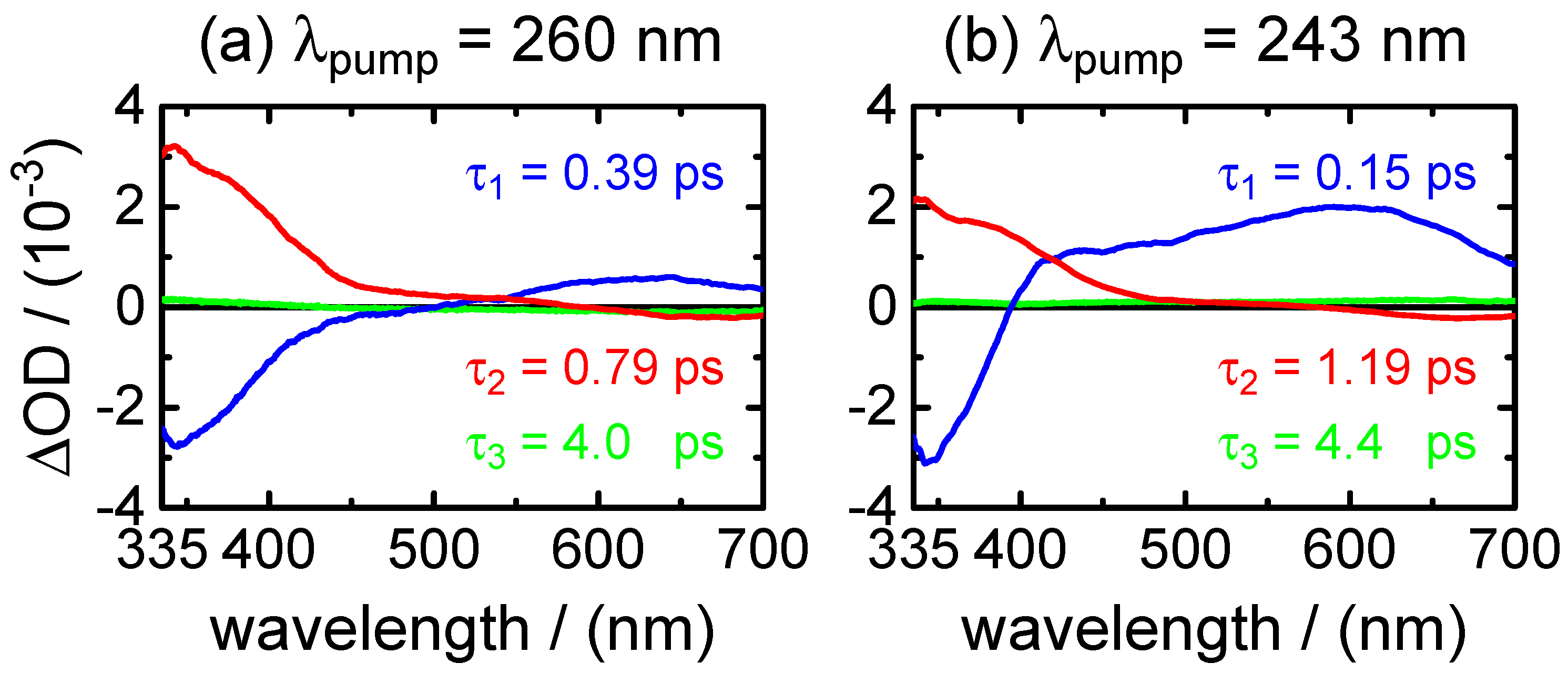
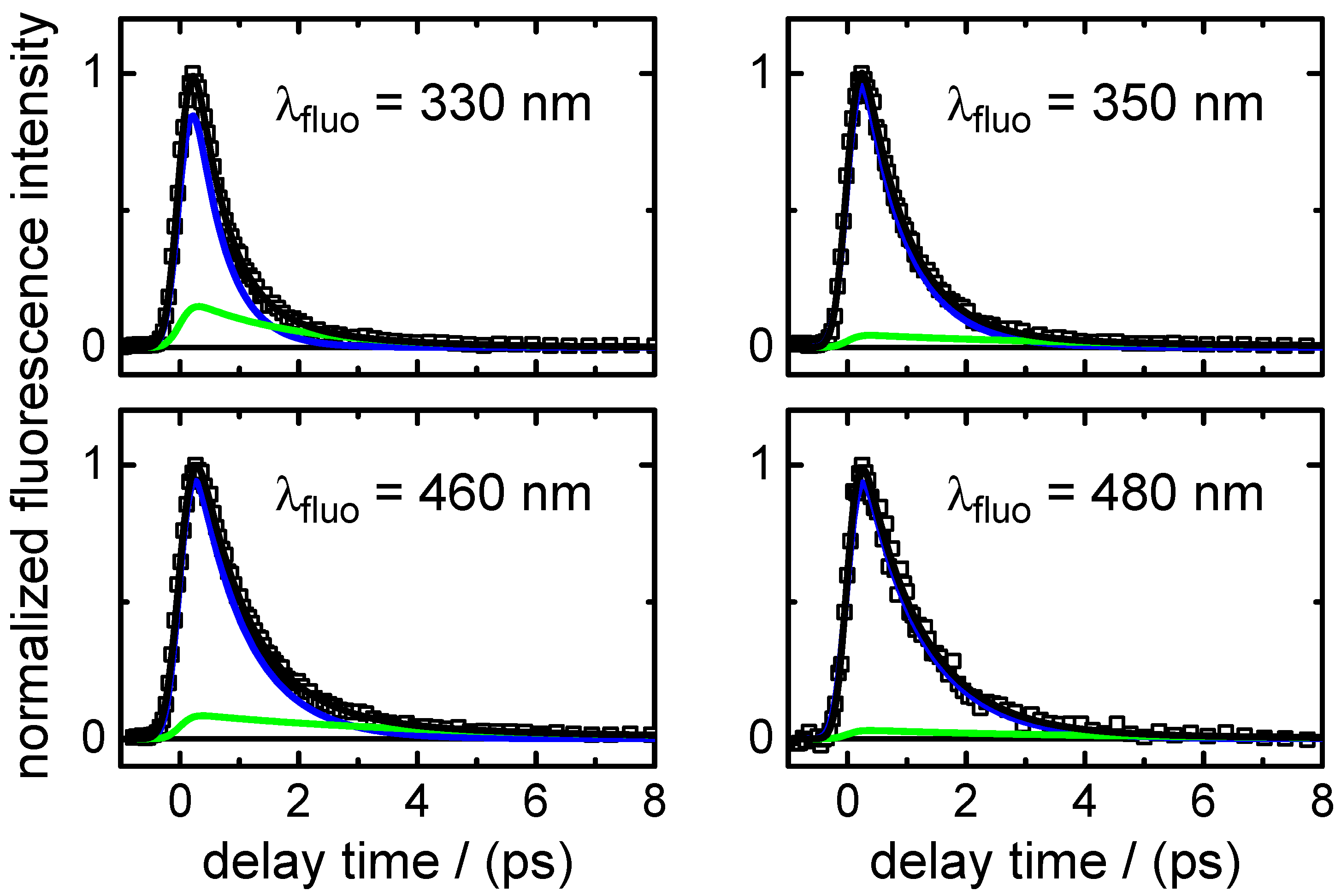
is the predominating decay time of the fluorescence at wavelengths in the UV close to the excitation wavelengths. Second, as shown by the DADS in
Figure 6 and by the selected time profiles in
Figure 5,
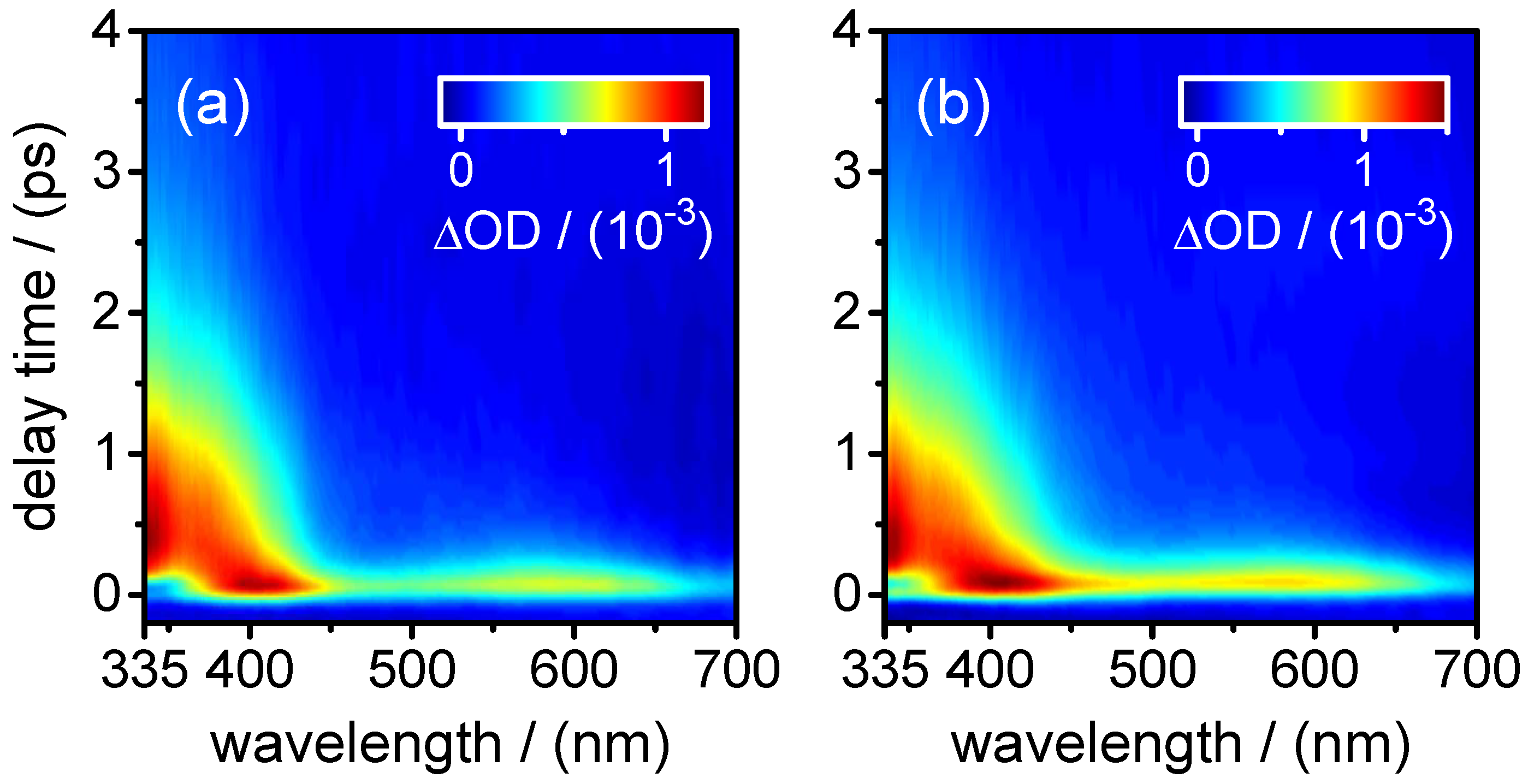
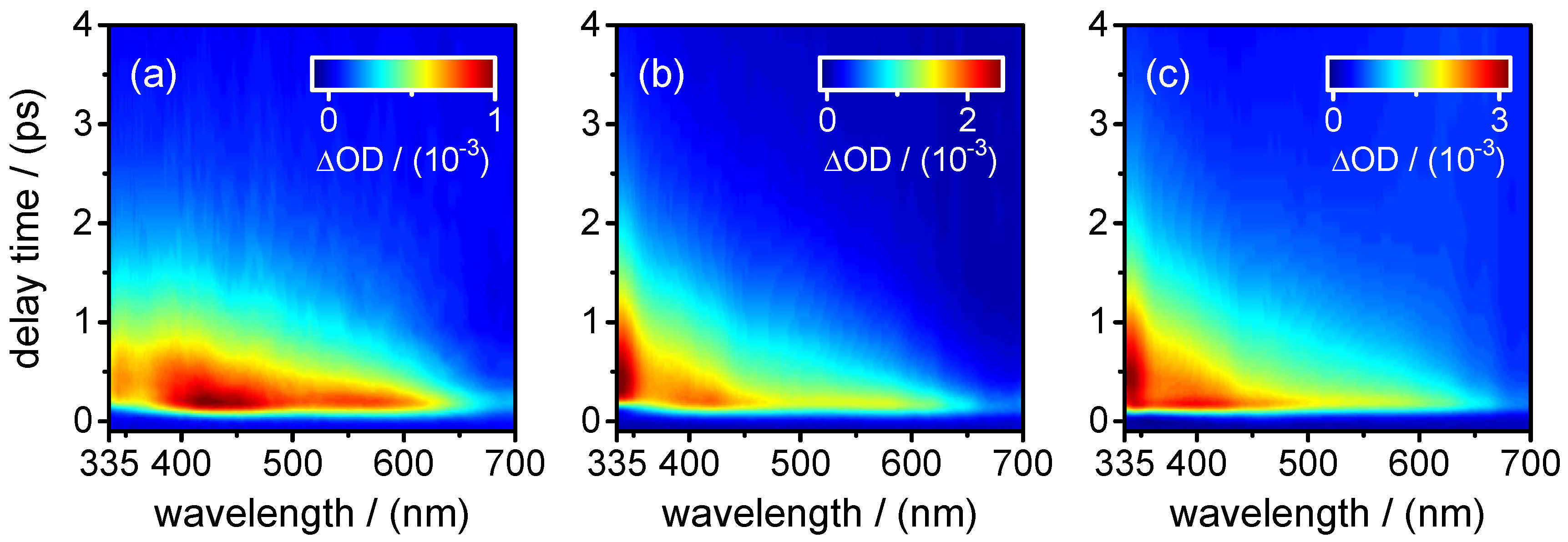
also describes the decay of the SE contribution (negative) in the transient absorption maps observed at
400–450 nm. At the same time, it is associated with the fast decay component of the (positive) fast ESA signal at
400–450 nm. The positive (ESA) amplitude increases towards longer probe wavelengths at the expense of the negative (SE) amplitude in the near-UV. The observation that the SE and fast ESA contributions could not be separated by the SVD analysis, but are described by a single DADS component, with a common single time constant (
), strongly supports that they are related to the initial FC excited state.
The DADS component corresponding to time constant
0.8–1.2 ps, on the other hand, remains positive in the entire probe wavelength range. Its observation hints at an ESA contribution from a “relaxed” excited state. The most plausible assignment for
is to the transition through the CI that connects the first
state to the ground state. Its short value indicates a rapid, barrierless wave packet motion in the direction of the CI. A definitive assignment would require QM/MM dynamics simulations of the first three electronically-excited states, as were recently reported for purine and 9-methylpurine [
26]. However, the single-colour experiments at
nm confirmed that the initially excited molecules return to the electronic ground state within
∼3.5 ps. This value has to be slightly longer than
because it includes the vibrational cooling time of the initially very “hot” molecules after their return through the CI to the S
ground state. The vibrational cooling is likely described by the third decay component
∼4.2 ps, which has a DADS with a very low amplitude. Eventually, we note that the absence of sizeable differences after selective photoexcitation to either the first or the second
state hints at an ultrafast internal conversion from S
to S
faster than our experimental time resolution. The longer-lived fluorescence component decaying with
∼36 ps suggested by the up-conversion data was barely above the detection experimental limit, such that it is hard to judge its reliability and origin. A slightly increased scattered light level in the measurement cannot be fully ruled out. A trapping of a small fraction of the excited wave packet in a shallow potential energy well along some different deactivation pathway could give a plausible explanation for the longer-lived component. Another possibility is a transfer of a fraction of the excited-state population to the
n
state [
22]. In this case, the lack of a corresponding excited-state absorption signal could be due to a very weak
n
S
oscillator strength. We note, meanwhile, that no comparable long-lived decay component was needed for modelling the ground state recovery data.
Yamazaki et al. proposed the coordinate connecting the first
excited state with the electronic ground state to be an out-of-plane deformation of the five-membered ring [
15]. This relaxation pathway differs compared to the usual situation in the purine bases, where relaxation is supposed to occur via an ethylenic puckering motion of the six-membered ring. This usual pathway is not available in XMP, as there is no double bond at C2-N3. Given the observed ∼1 ps fluorescence and ESA decay time constants, a slight potential energy barrier, as proposed by the calculations of Yamazaki et al., seems to be highly unlikely at least for XMP in water. This might be a result of the replacement of the H atom at the N(9) position by the ribosyl-phosphate group. Although sugar-phosphate groups do not usually have large impacts on the photodynamics of other purine bases, it may be of larger importance here, where the five-membered ring is involved. A comparative study of xanthine and its nucleoside xanthosine would help to shed light on this question, but is difficult regarding the low solubility of the free nucleic acid base in aqueous solution. Gas phase experiments could be an alternative at this point. In particular, while gas phase studies of xanthine and xanthosine require an elaborate laser desorption setup, XMP
could be brought into a molecular beam using an electrospray source.
The results obtained for the deactivation of XMP in aqueous solution are in good agreement with those obtained for the deactivation of methylated xanthine derivatives, where time scales of several 100 fs, ∼1 ps and
–6.3 ps were found in water and acetonitrile [
22]. Those values were attributed to relaxation dynamics via the five-membered ring, as well. Chen et al. [
22] discussed the existence of long-lived optically dark states, such as
n
, after excitation of methylated xanthine derivatives. Although we cannot strictly rule out the
n
state as a short-lived intermediate, we can exclude a substantial population of a long-lived dark
n
state or triplet states on the grounds of the observed GSR results.
3.2. XMP
The excited-state deactivation of XMP
in phosphate buffered H
O at pH 7 is governed by lifetime constants of
∼0.3 ps and
∼0.9 ps, comparable to the values for XMP. Again,
is ascribed to fast motion away from the FC window and
to the radiationless transition through the CI to the ground state. With
∼4.4–5.9 ps, the ground state recovery time for XMP
appears to be slightly longer than for XMP. As for neutral XMP, excitation to the second
state compared to the first
state seems to cause little changes of the observable dynamics, suggesting a rapid internal conversion from the upper to the lower
state on a time scale faster than our experimental resolution. Somewhat surprisingly, however, as noted in
Section 2.2.2 above, it was not possible to obtain satisfactory fits to the fluorescence decay curves for XMP
using two exponentials of varying amplitudes, but common lifetimes at the different emission wavelengths. Instead, a gradual increase of the fluorescence lifetime with increasing emission wavelength was evident. This observation, made after excitation mainly to the second
excited state, may hint at a sizeable gradient of the PEHS of the emitting state of XMP
. The motion of the excited wave packet following the potential energy gradient should lead to a rapid temporal red-shift of the fluorescence. Unfortunately, the amplitudes of our up-converted fluorescence signals cannot be quantitatively compared for experimental reasons; time-resolved measurements of the entire fluorescence spectra as possible by Kerr-gating would be needed here. We note, however, that the static fluorescence spectrum of XMP
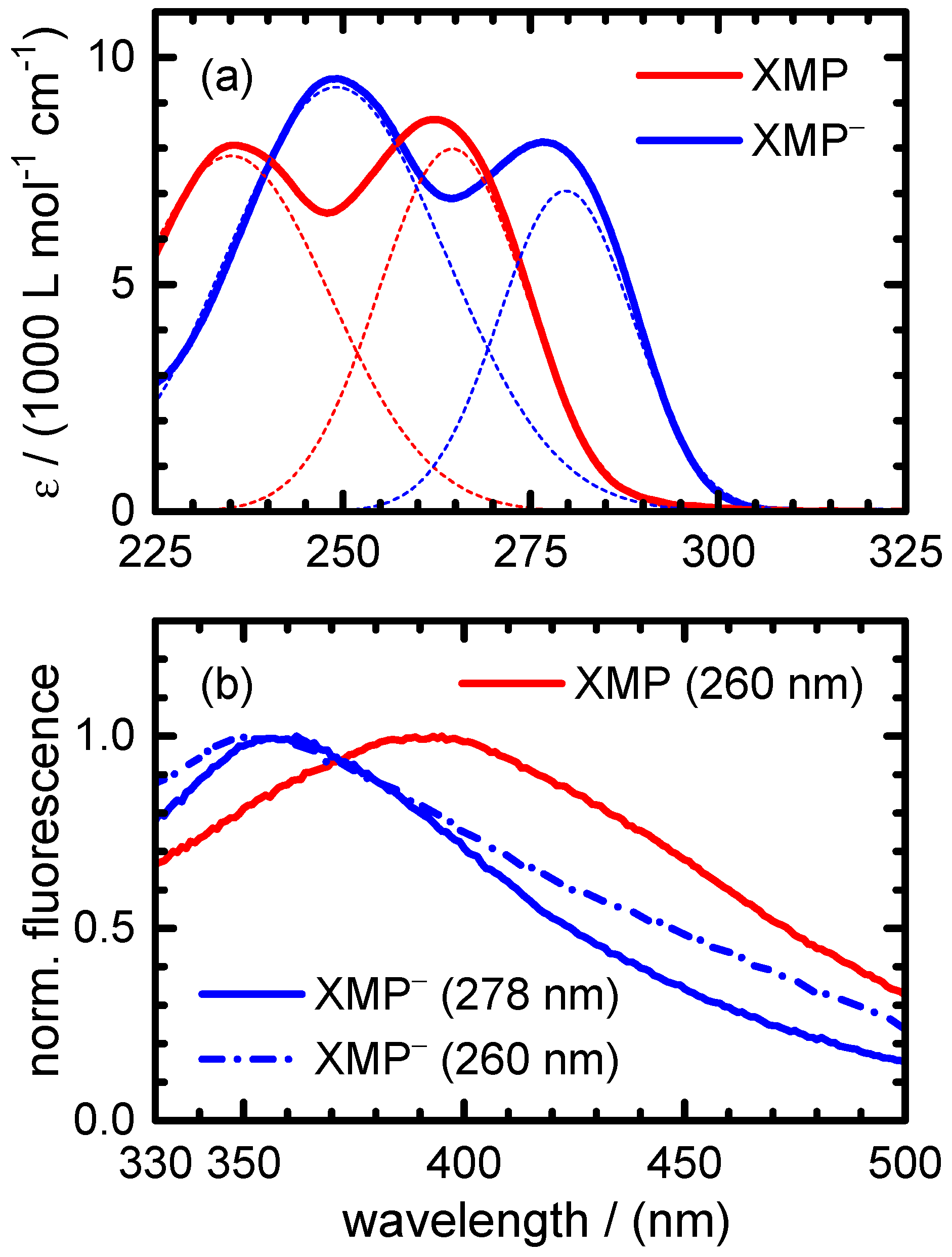
is blue-shifted despite its red-shifted absorption spectrum in comparison to XMP (
Figure 2). In some not yet well understood way, this hints at significant differences between the excited potential energy hypersurfaces of XMP
and XMP.
Since the results of the time-resolved experiments for XMP
are similar to those for XMP, the relaxation mechanism can be expected to be similar, as well. This is not unlikely because deprotonation takes place at the N(3) position in the six-membered ring, where, as already noted in the Introduction, it might not have a large impact on the electronic structure of the xanthine moiety [
9,
13]. Interestingly, drastically different behaviours upon deprotonation were observed for the electronic deactivation dynamics of hypoxanthine and its nucleoside inosine [
22,
27,
28,
29]. Both molecules are deactivated via involvement of the out-of-plane deformation of the six-membered ring. However, deprotonation of inosine, which takes place at the six-membered ring, causes little difference in the excited-state dynamics [
29]. Hypoxanthine, on the other hand, is deprotonated at the five-membered ring and shows an almost 20-fold increase of the fluorescence lifetime. The striking change has been explained by different electronic structures [
29].
Besides these effects arising from deprotonation, a large impact on the electronic deactivation dynamics can also arise from protonation. For monoprotonated hypoxanthine and guanosine, for example, an increase of the excited-state lifetimes was found and attributed to the change of the potential energy topography compared to the neutral molecule [
29,
30]. Neutral hypoxanthine and its nucleoside inosine have been found to exhibit the fastest electronic deactivation rates of all purine bases studied to date. The observed fluorescence and ESA lifetimes of
and
ps, respectively, were assigned to the out-of-plane puckering deactivation channel involving the six-membered ring similar to guanine, but different from xanthine [
28].



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









