High Resolution NMR Spectroscopy as a Structural and Analytical Tool for Unsaturated Lipids in Solution



Abstract
:1. Introduction
2. The Pleiotropic Role of Unsaturated Fatty Acids
3. NMR Structural and Analytical Parameters
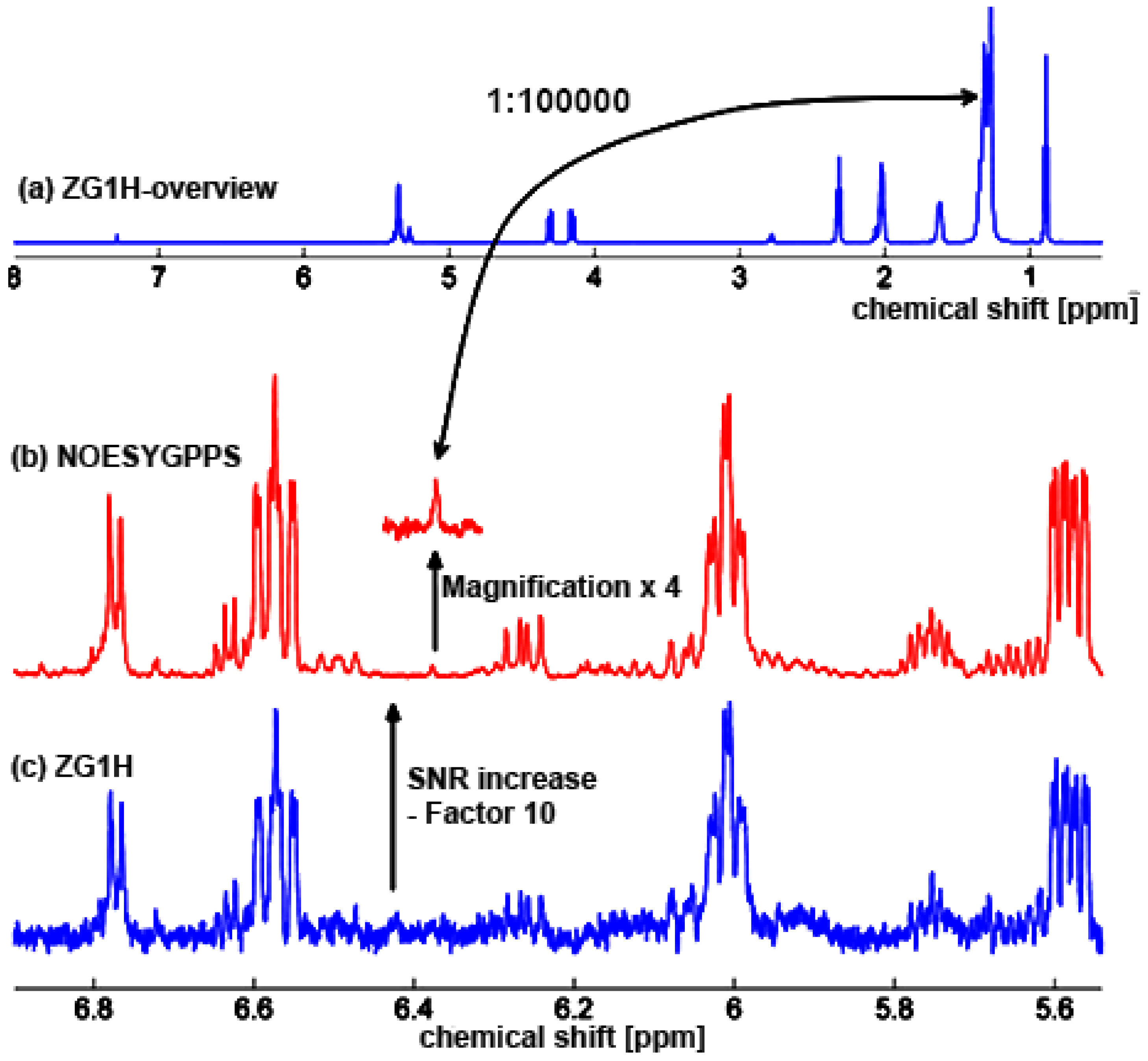
3.1. 1H-NMR Spectroscopy
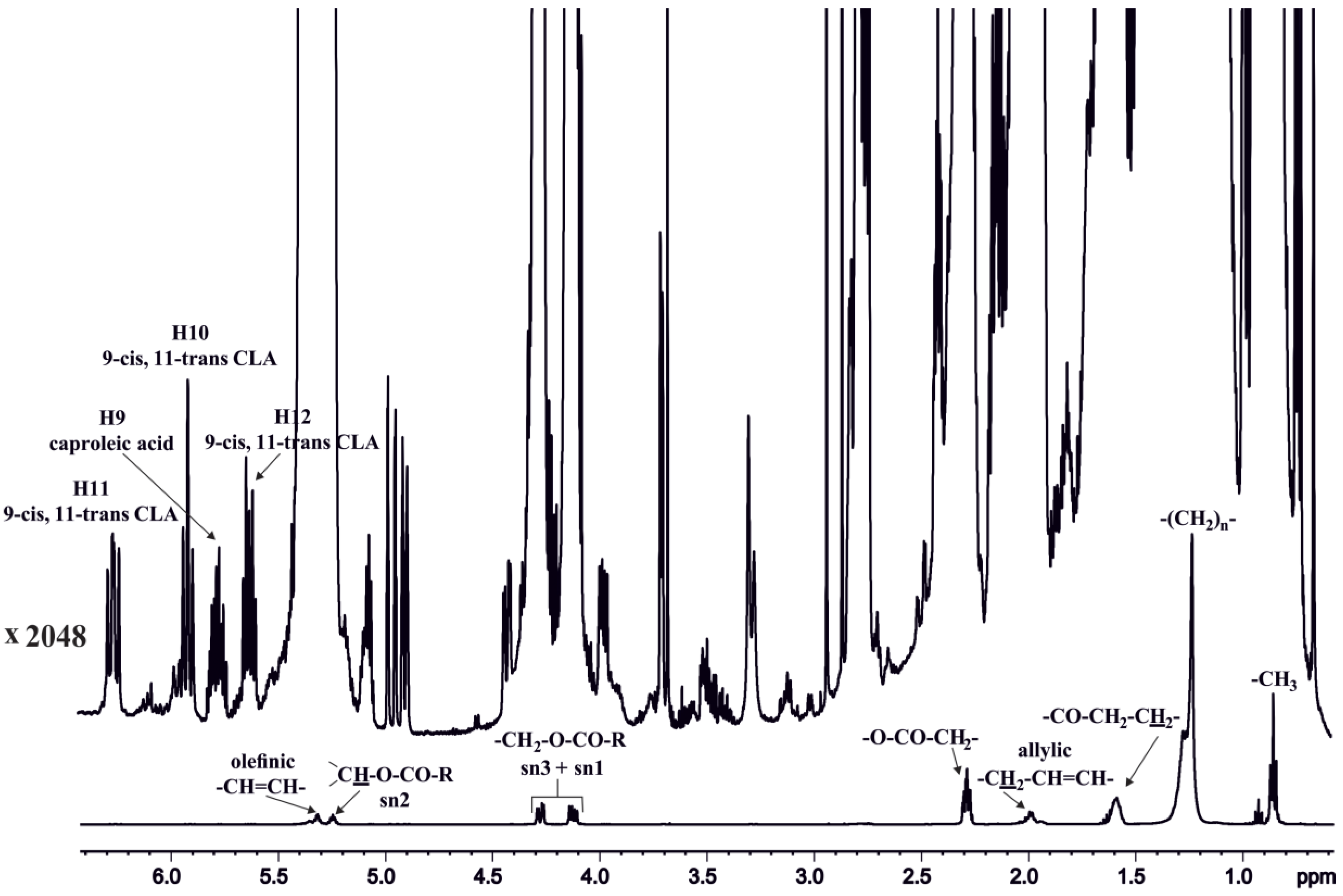
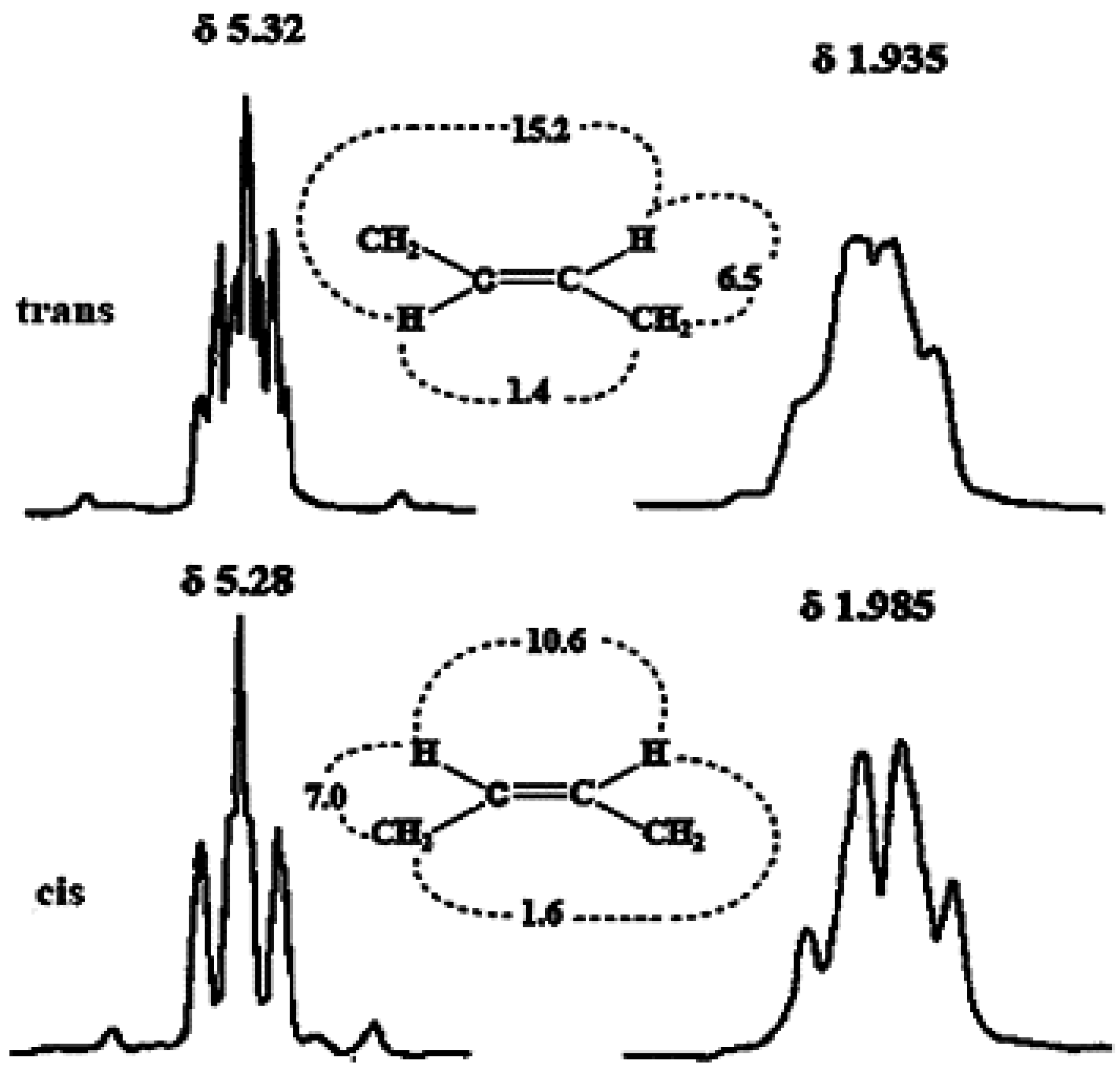
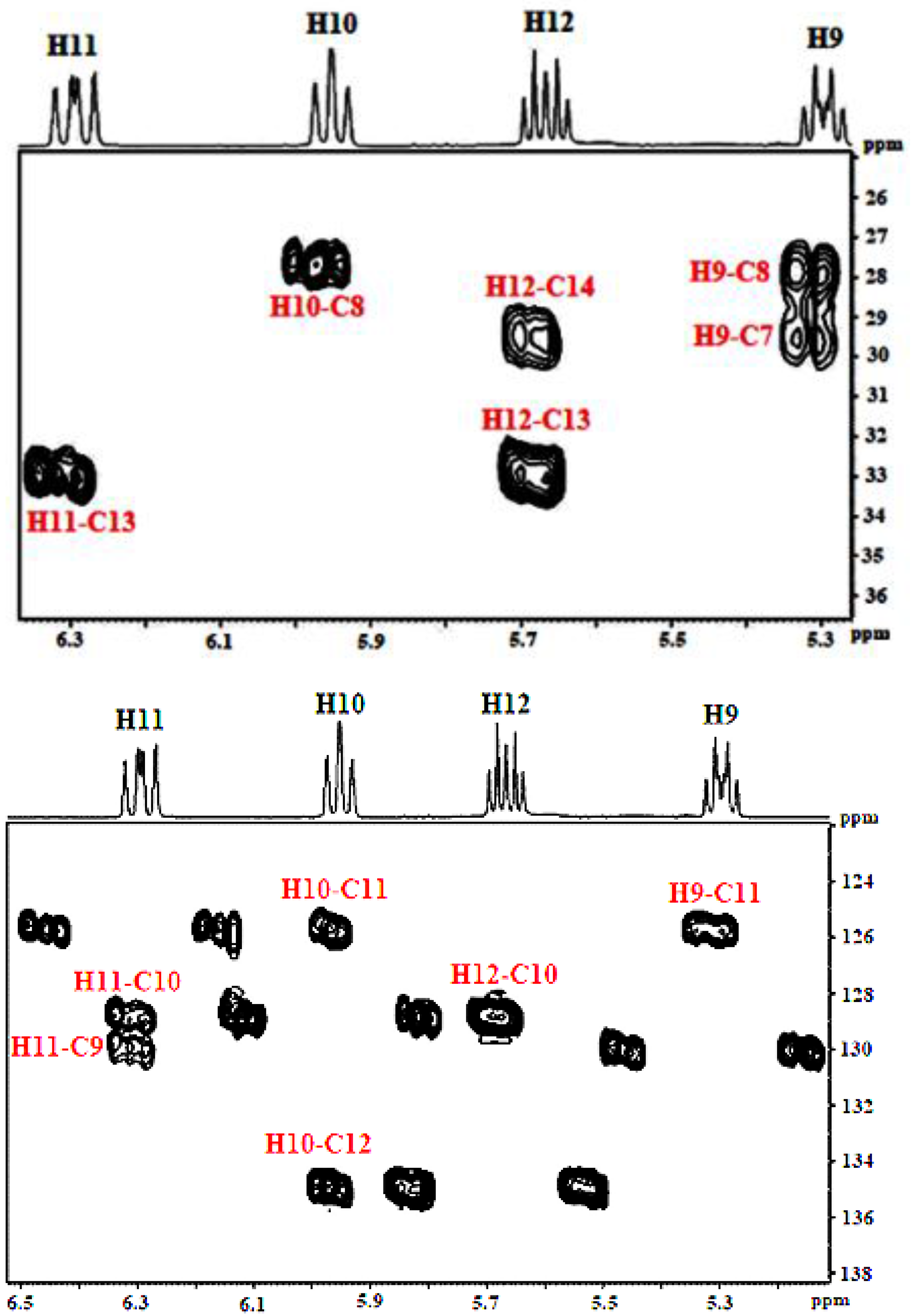
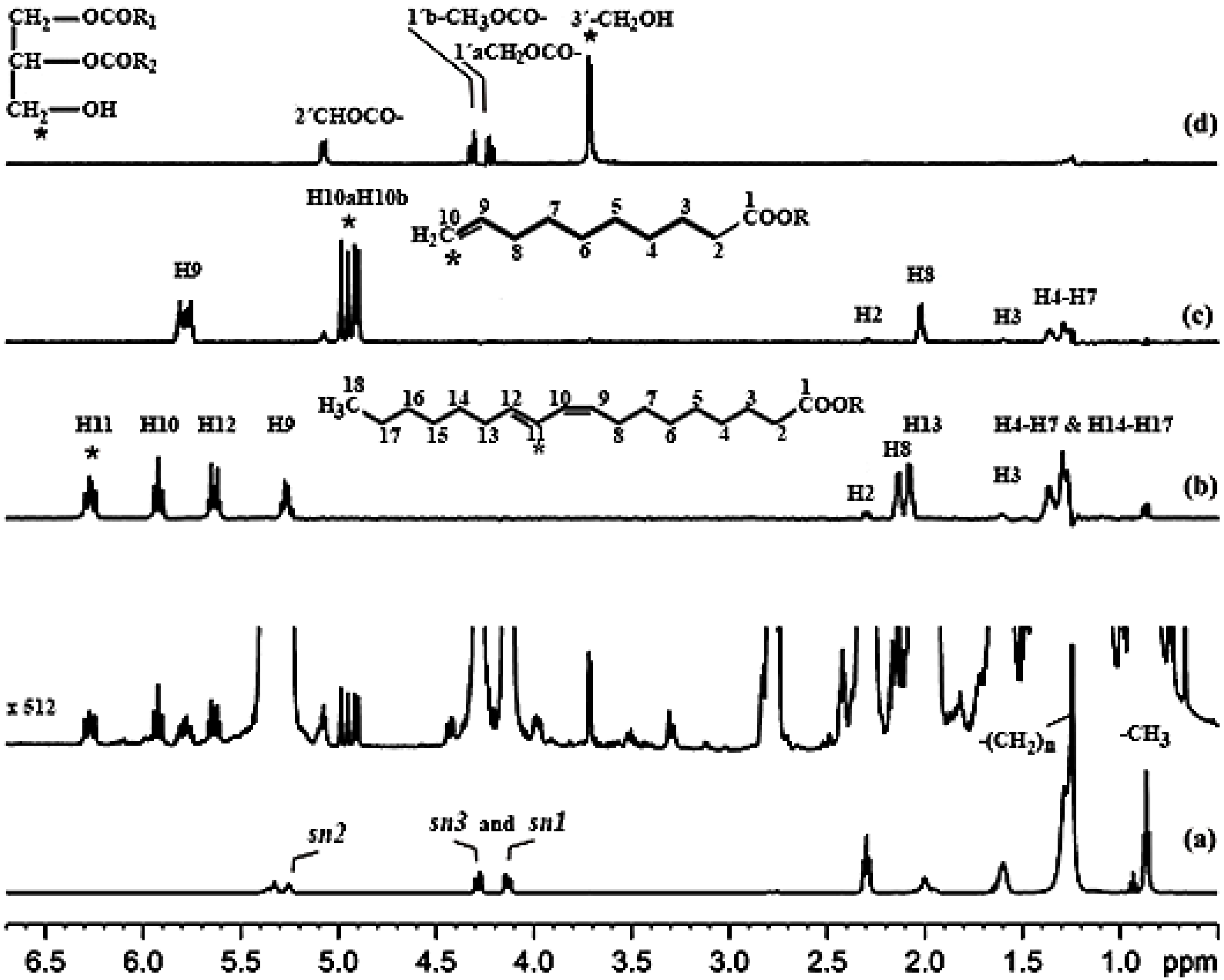
3.1.1. Olefinic Protons
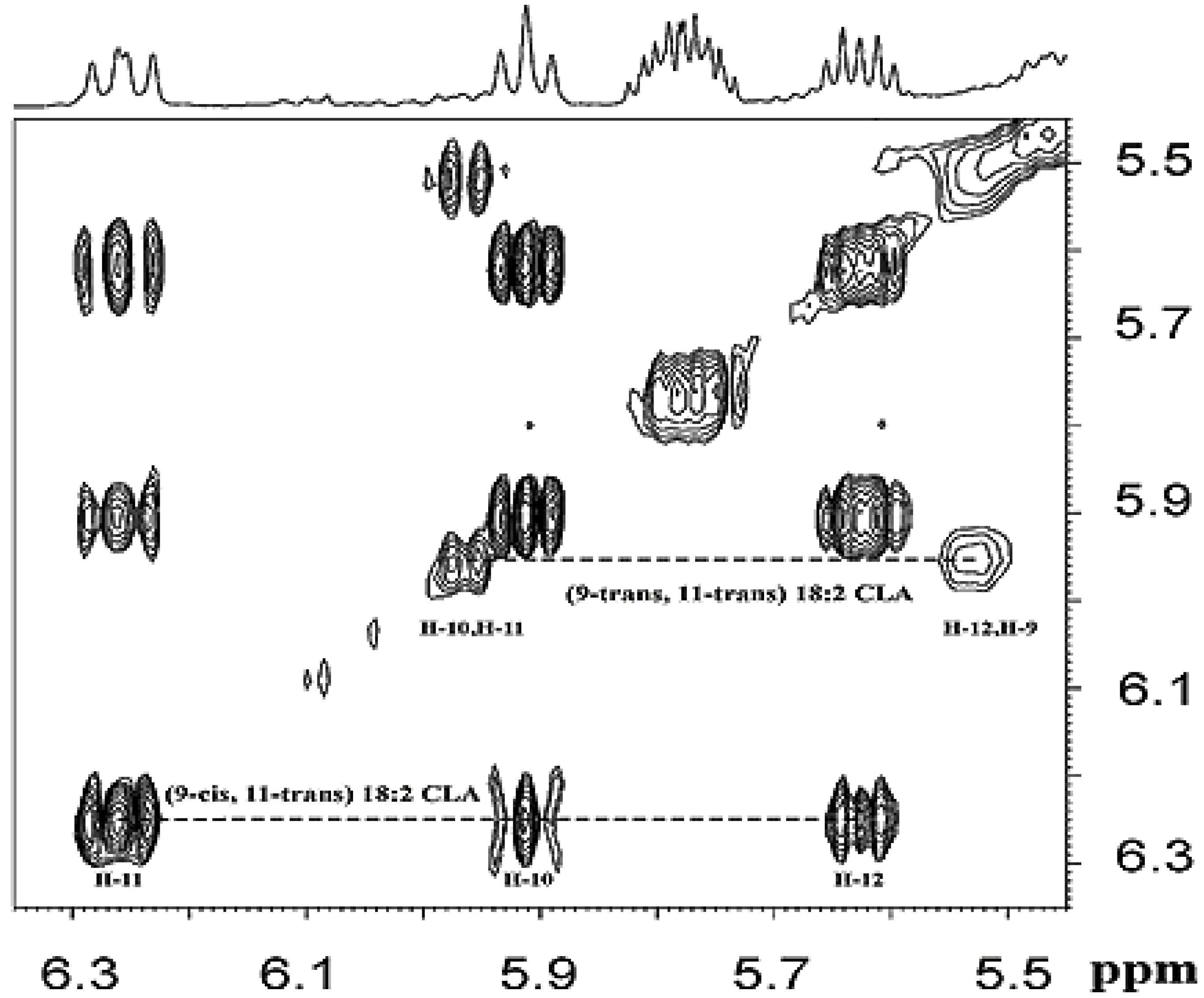
3.1.2. Olefinic Protons in Conjugated Double Bonds
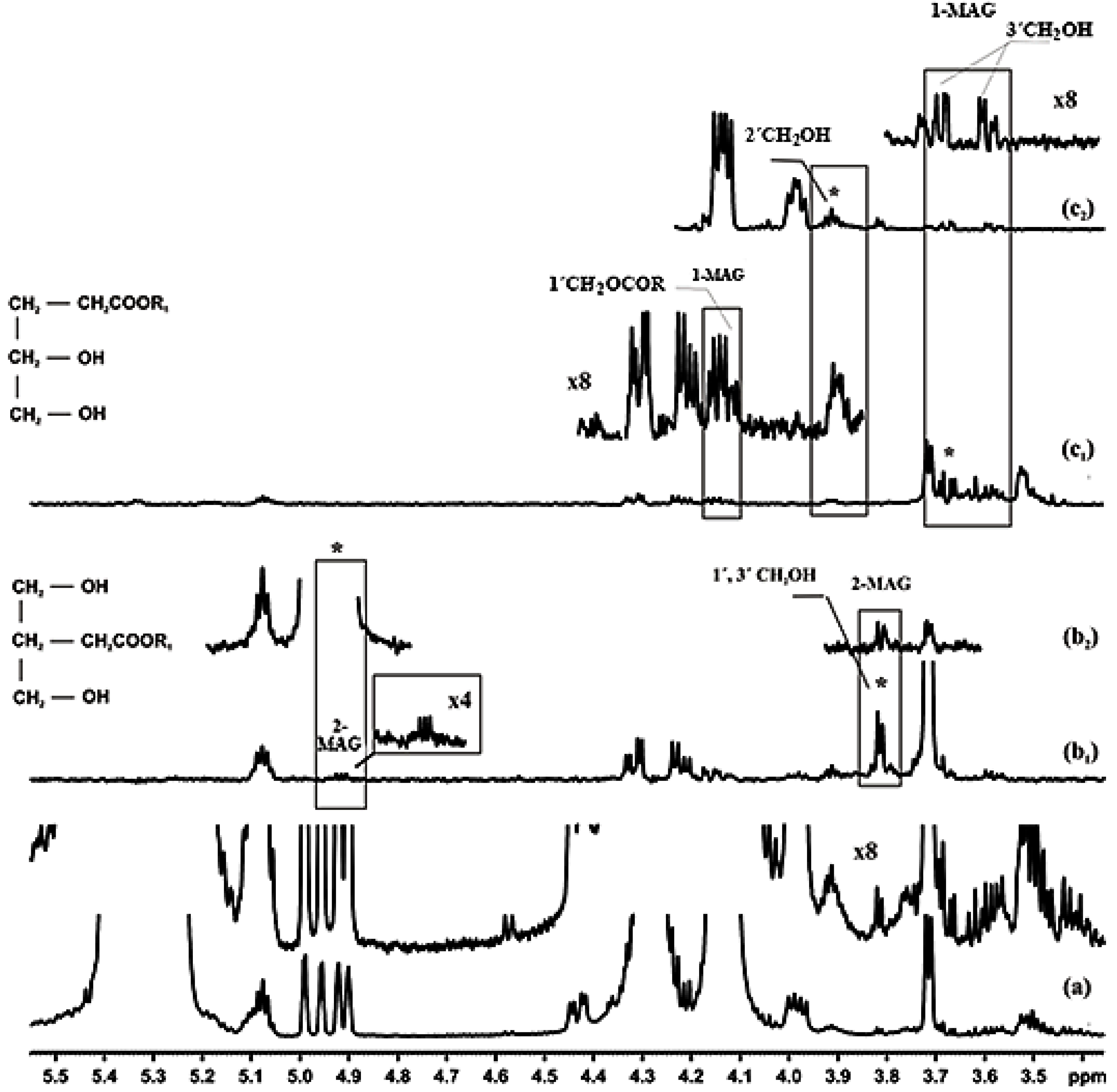
3.1.3. Protons of the Glycerol Moiety
3.1.4 Bis-allylic Protons
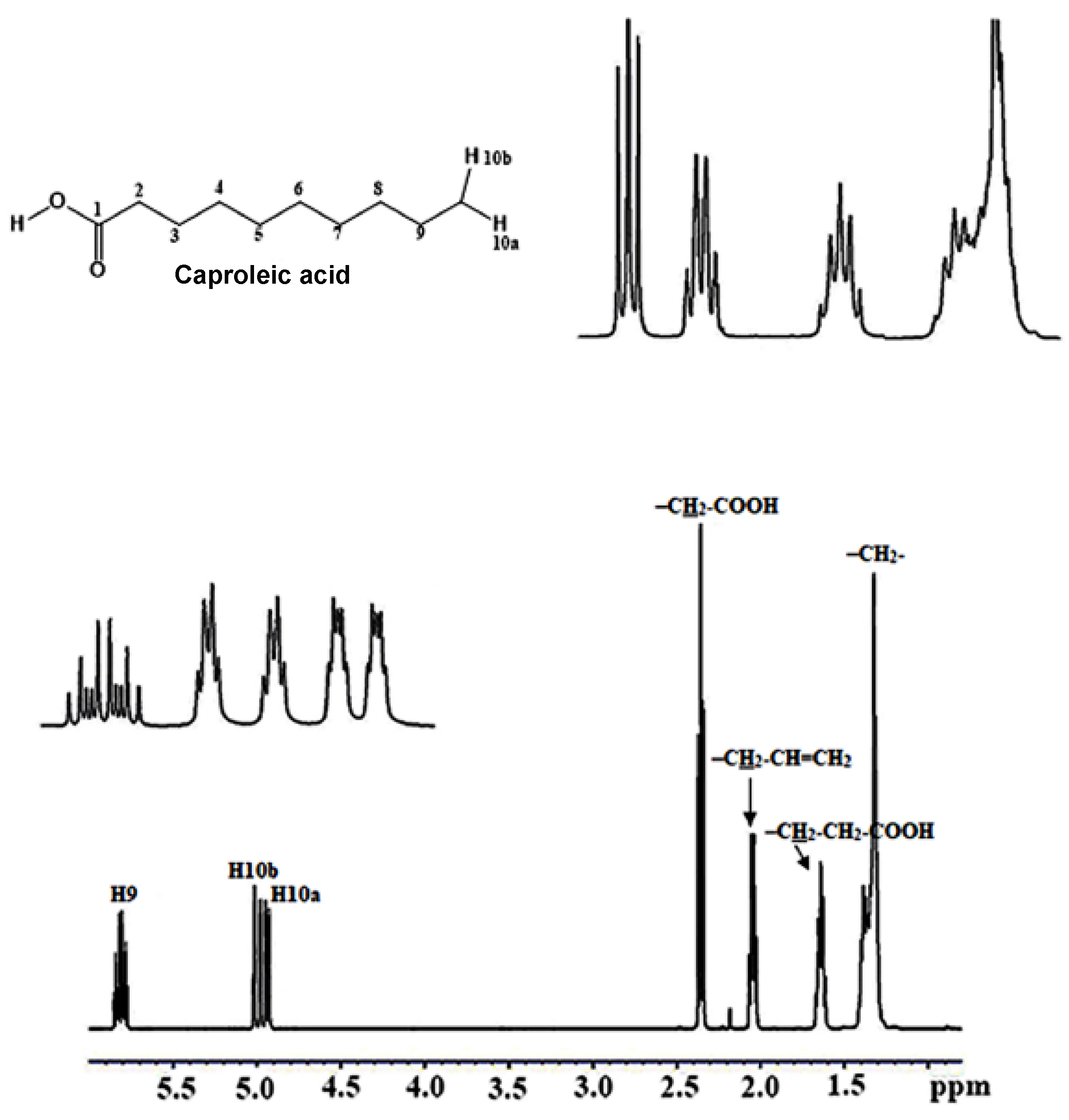
3.1.5. α-CH2, Allylic CH2–CH=CH and (CH2)n Protons
3.1.6. The CH3 Protons
3.1.7. 1H-NMR Relaxation Times
3.2. 13C-NMR Spectroscopy
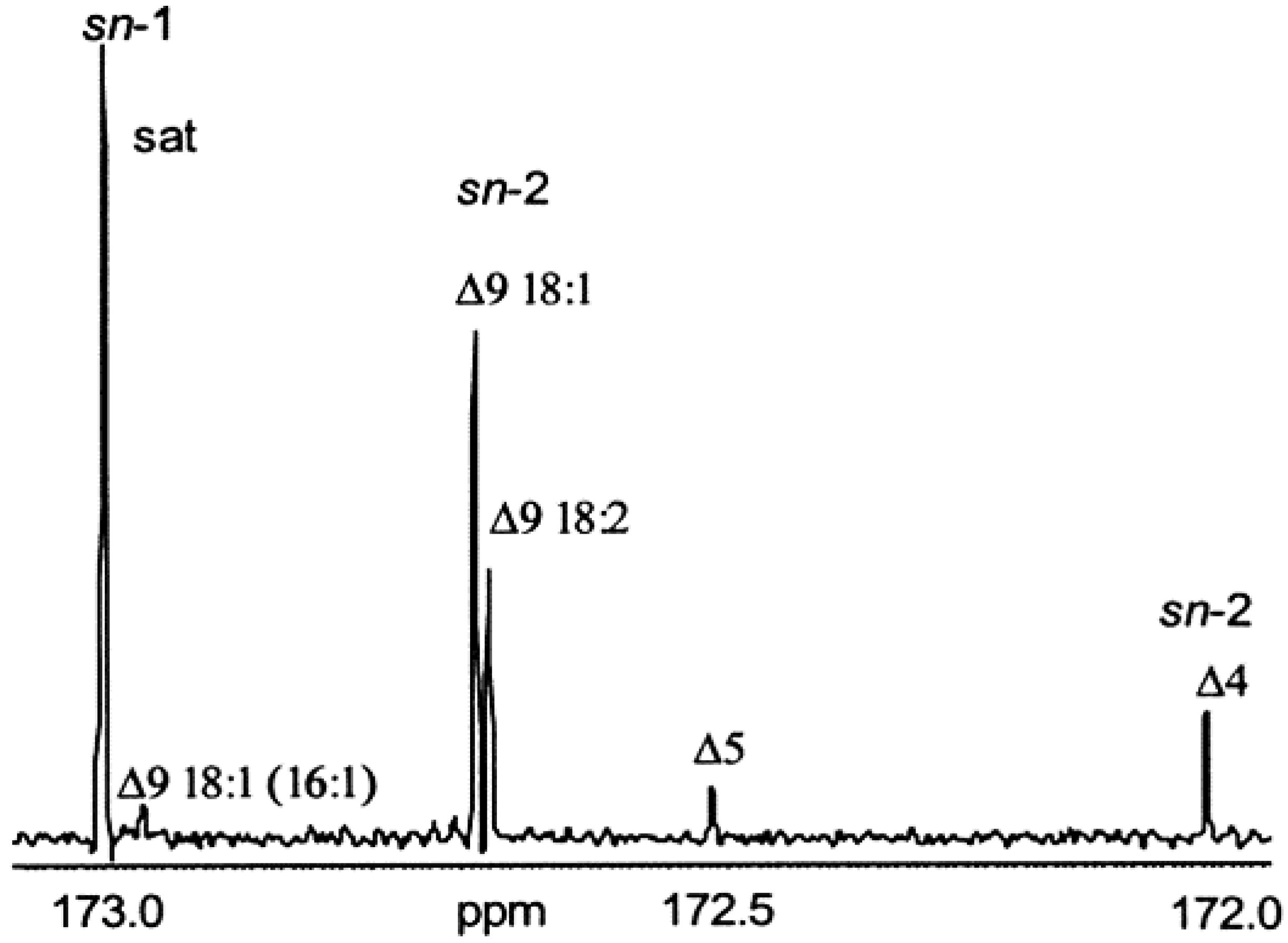
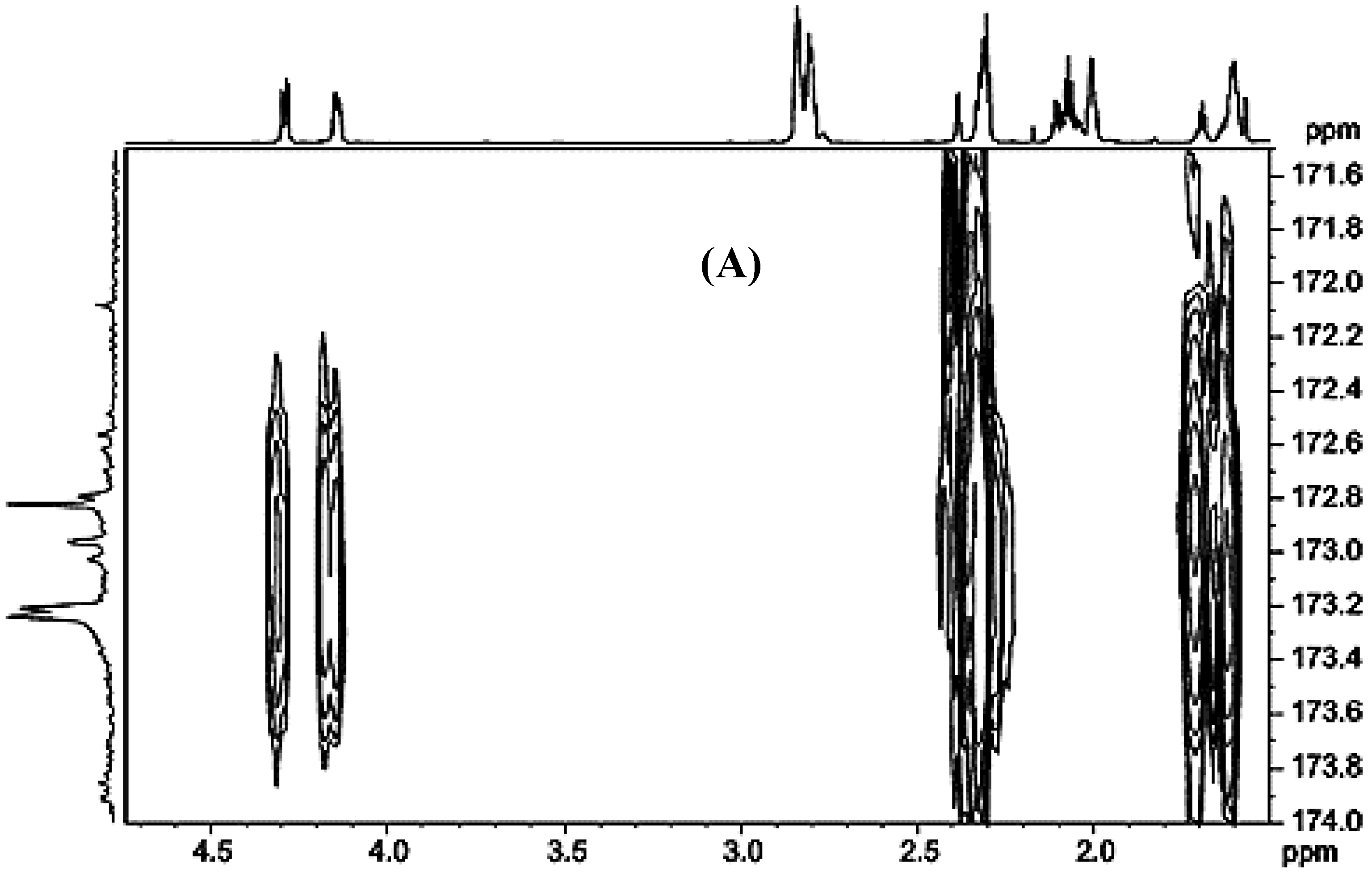
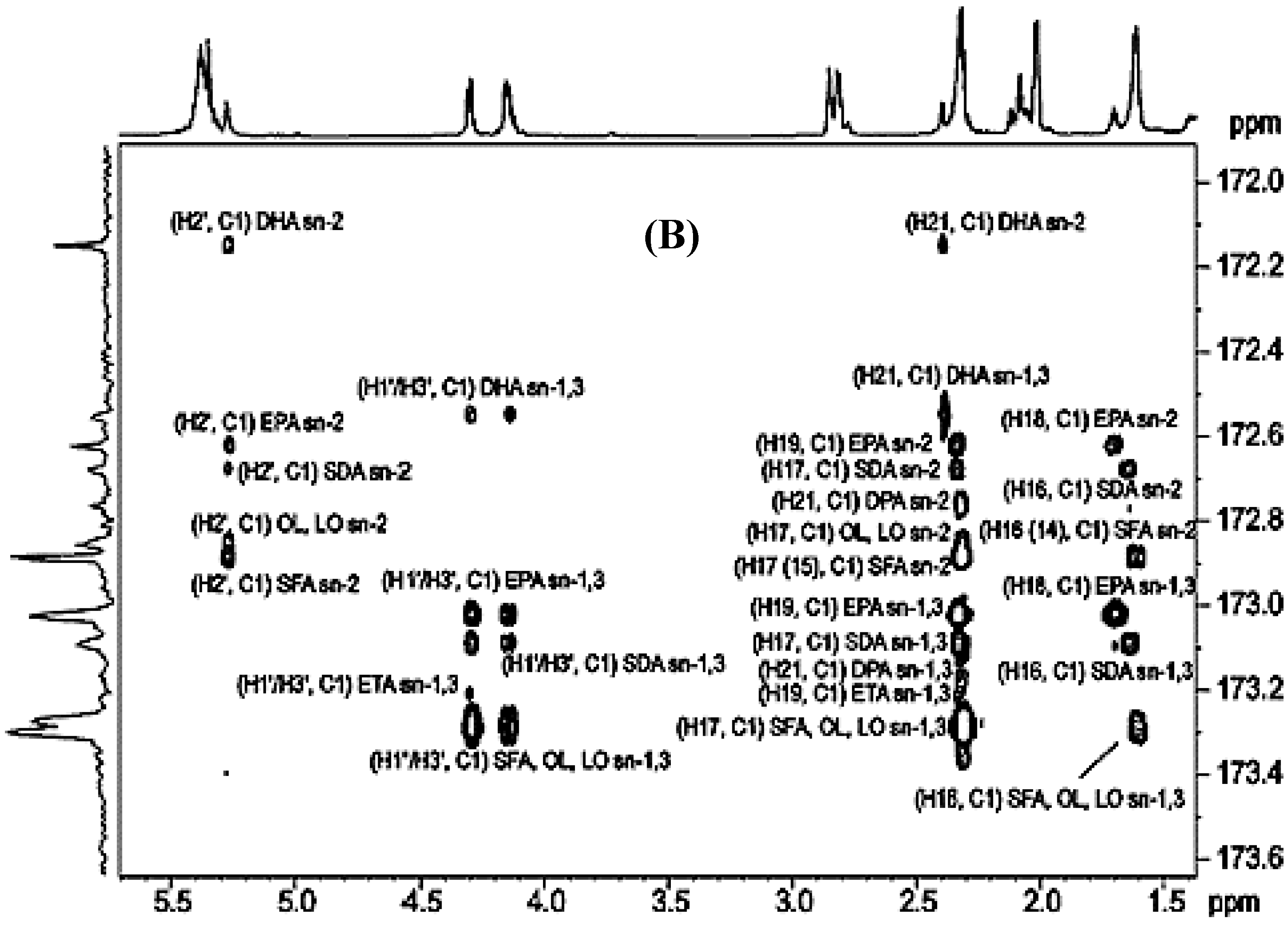
3.2.1. Carbonyl and Carboxyl Carbon Region
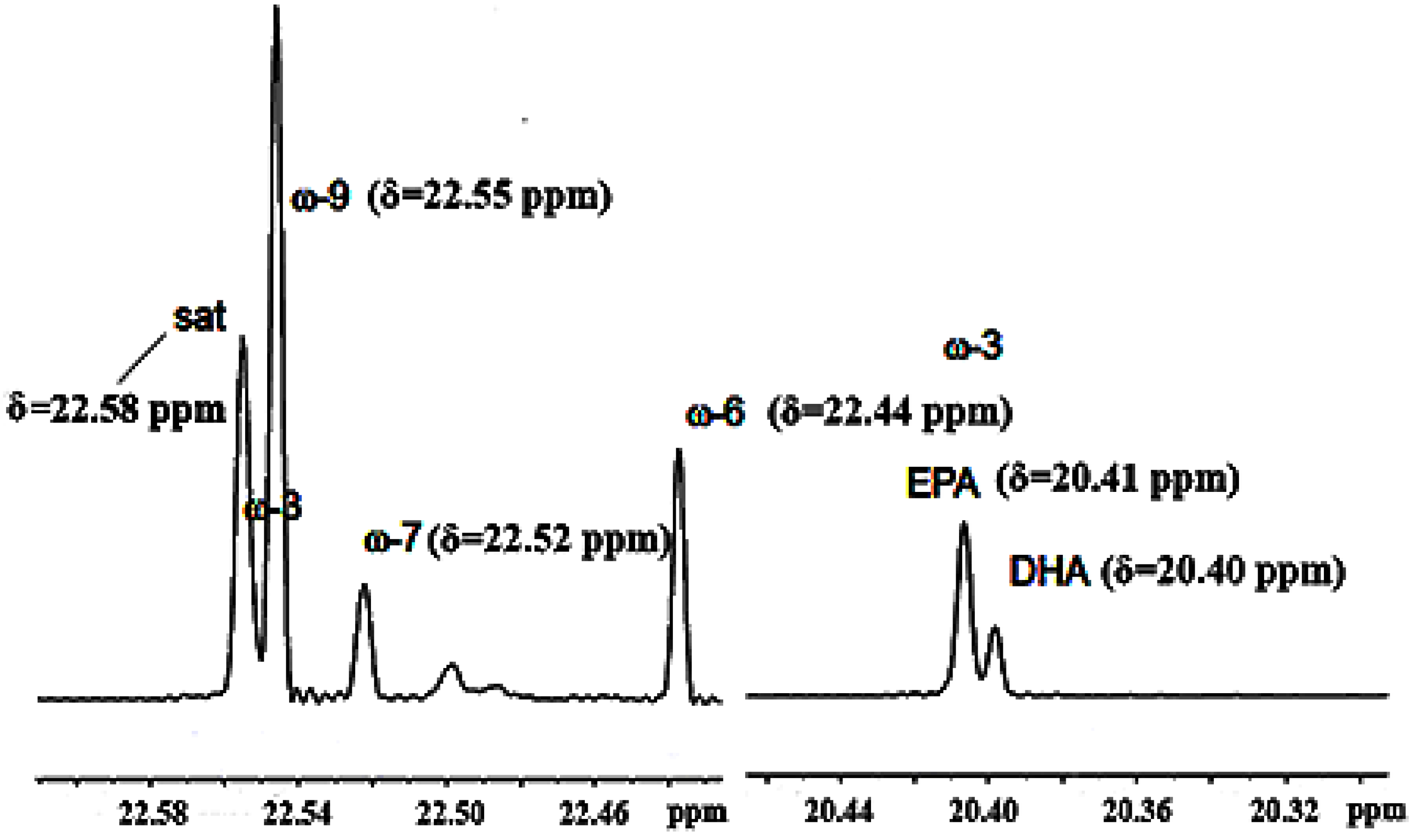
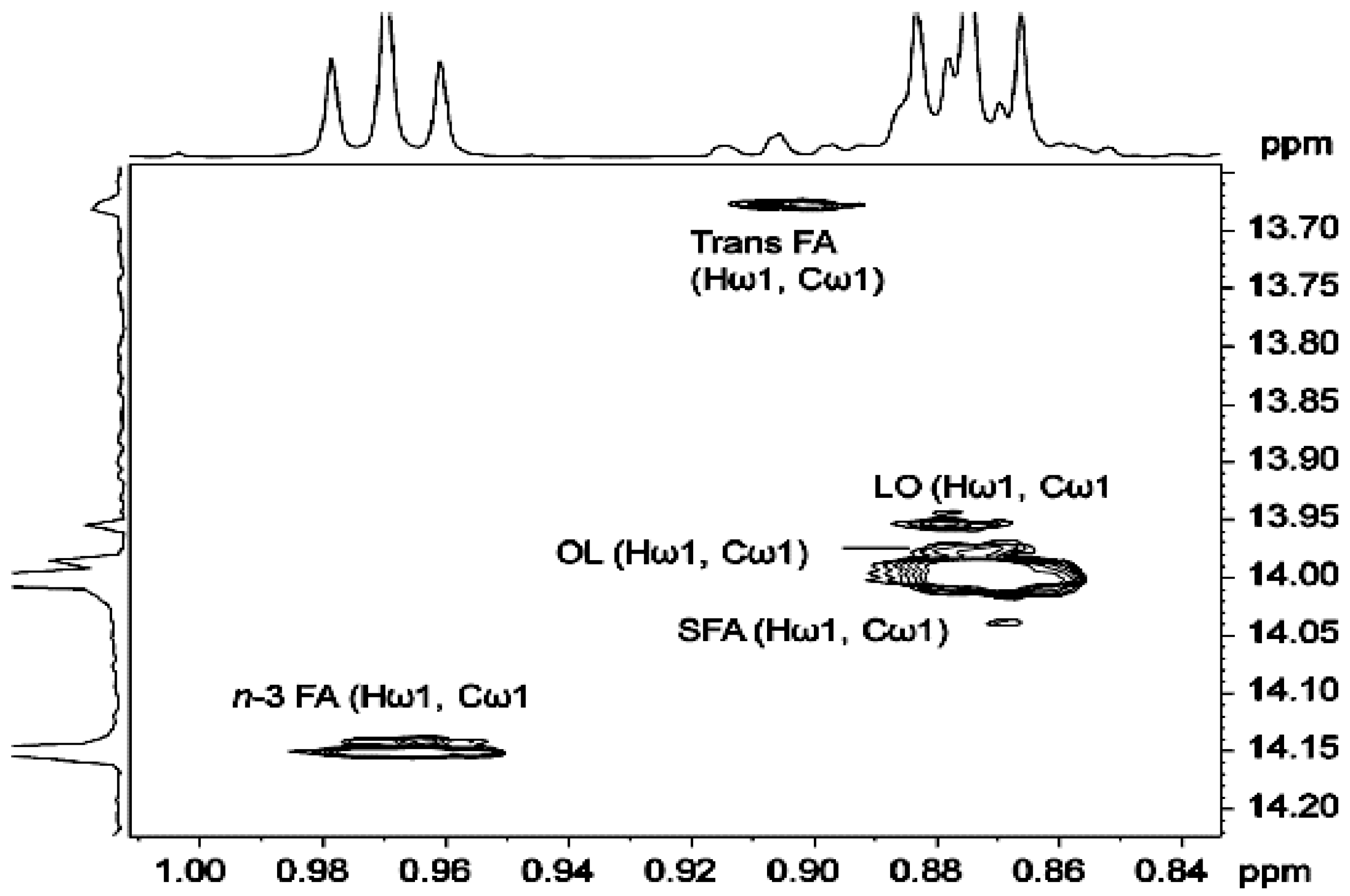
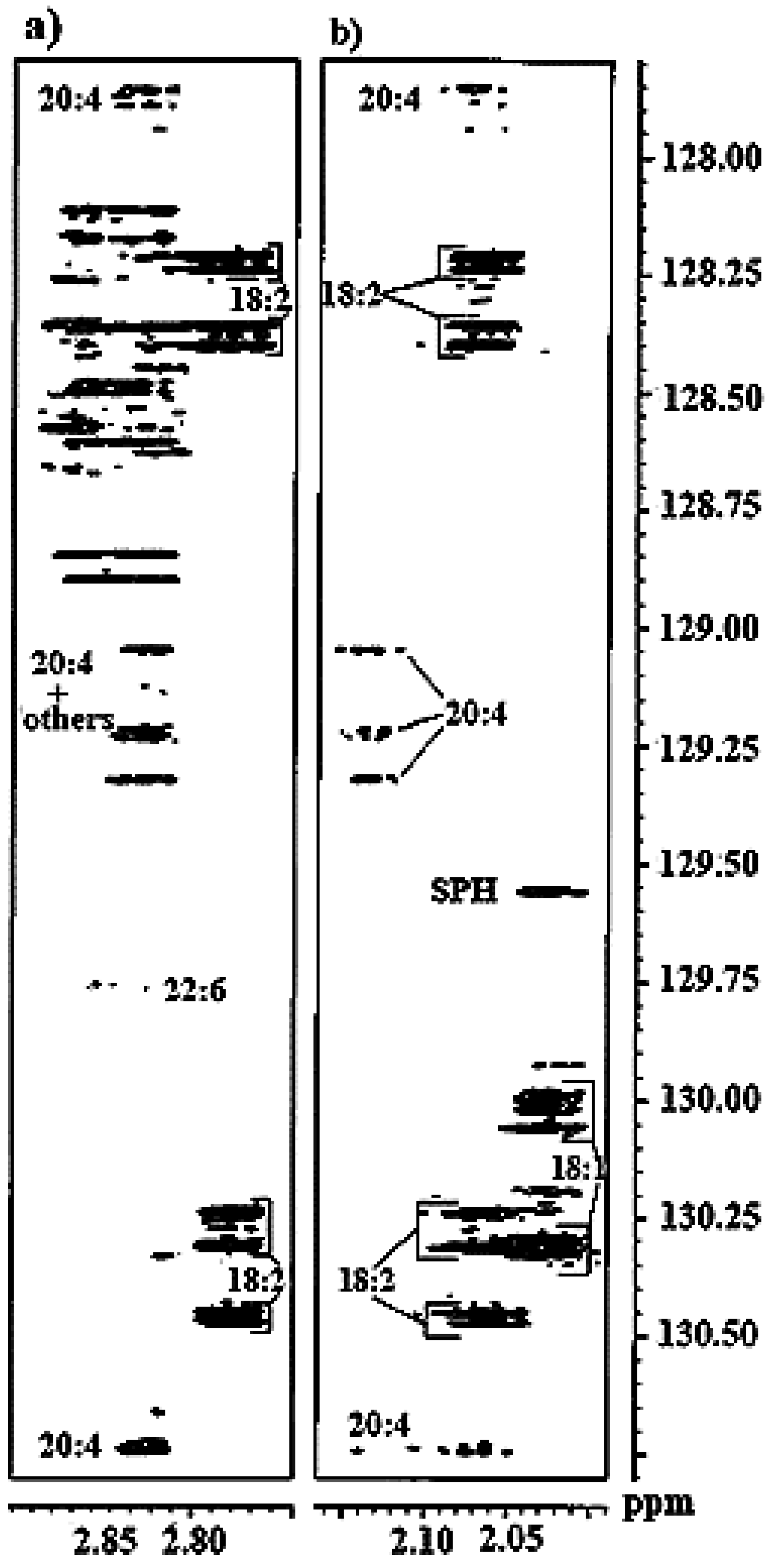
3.2.2. Olefinic Carbon Region
3.2.3. Glycerol Carbons
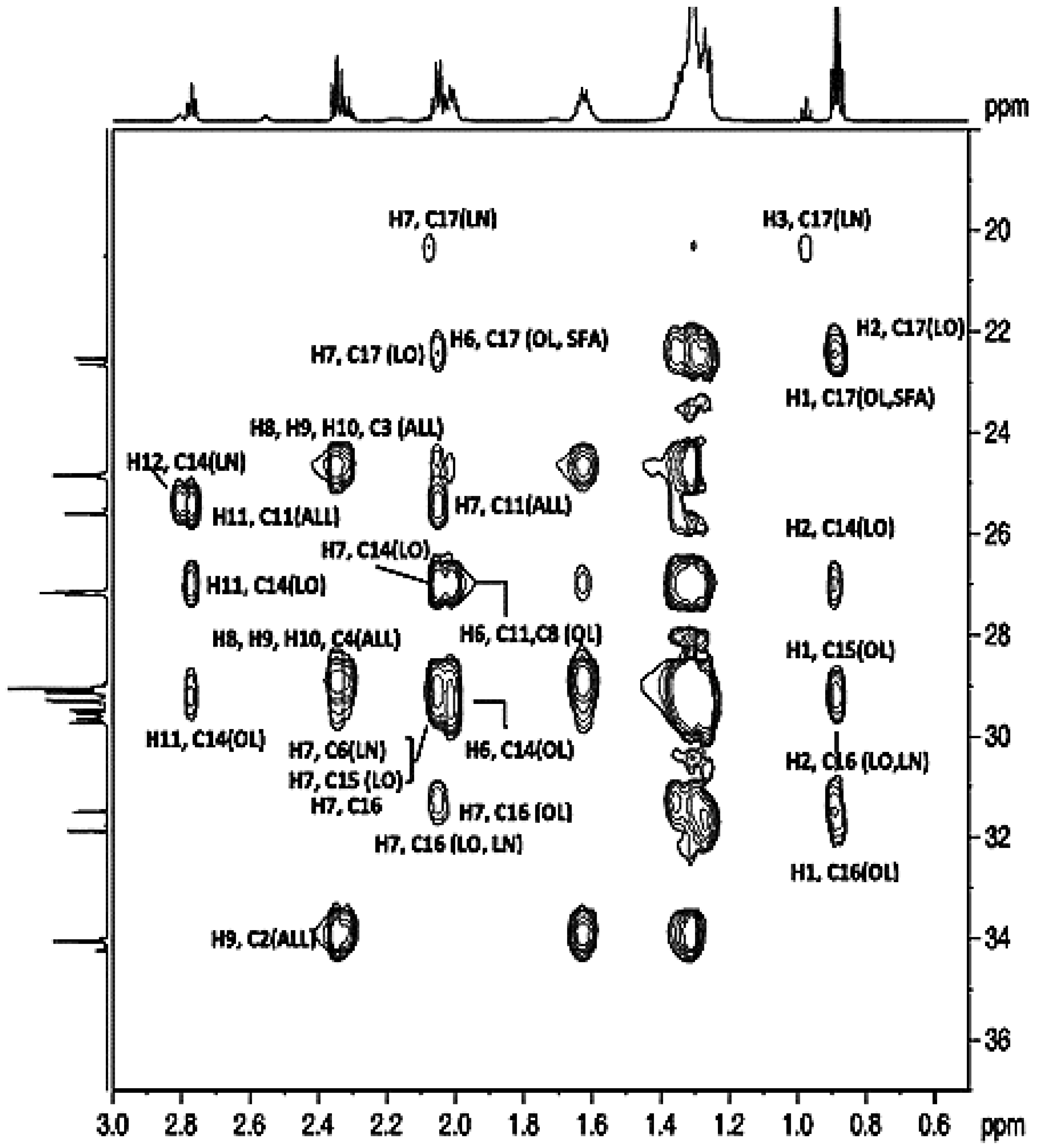
3.2.4. Aliphatic Carbons
3.3. 31P-NMR Spectroscopy
4. NMR Methods for Assignment
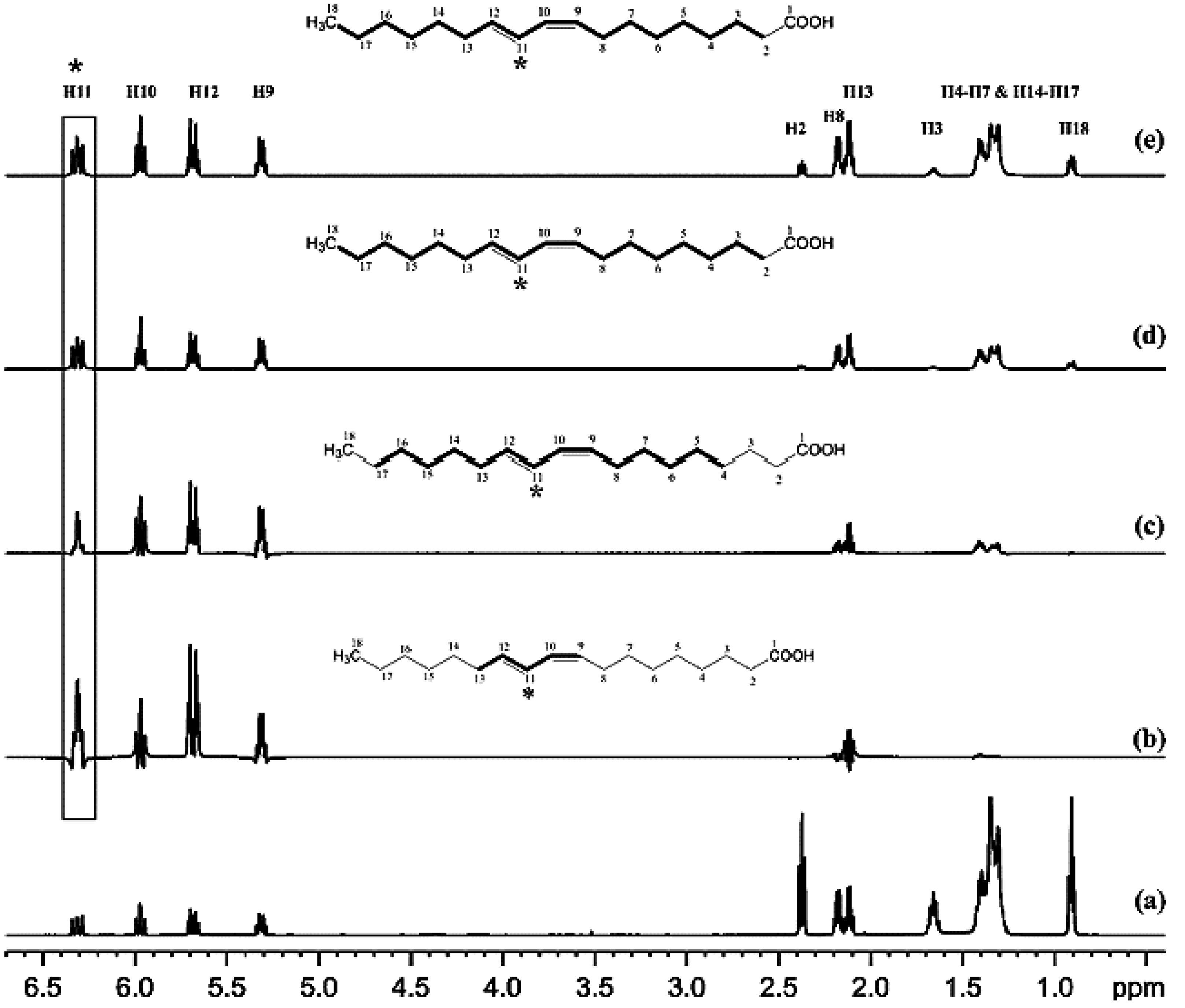
4.1. Selective Suppression of Major Signals
4.2. Selective 1D TOCSY Experiments
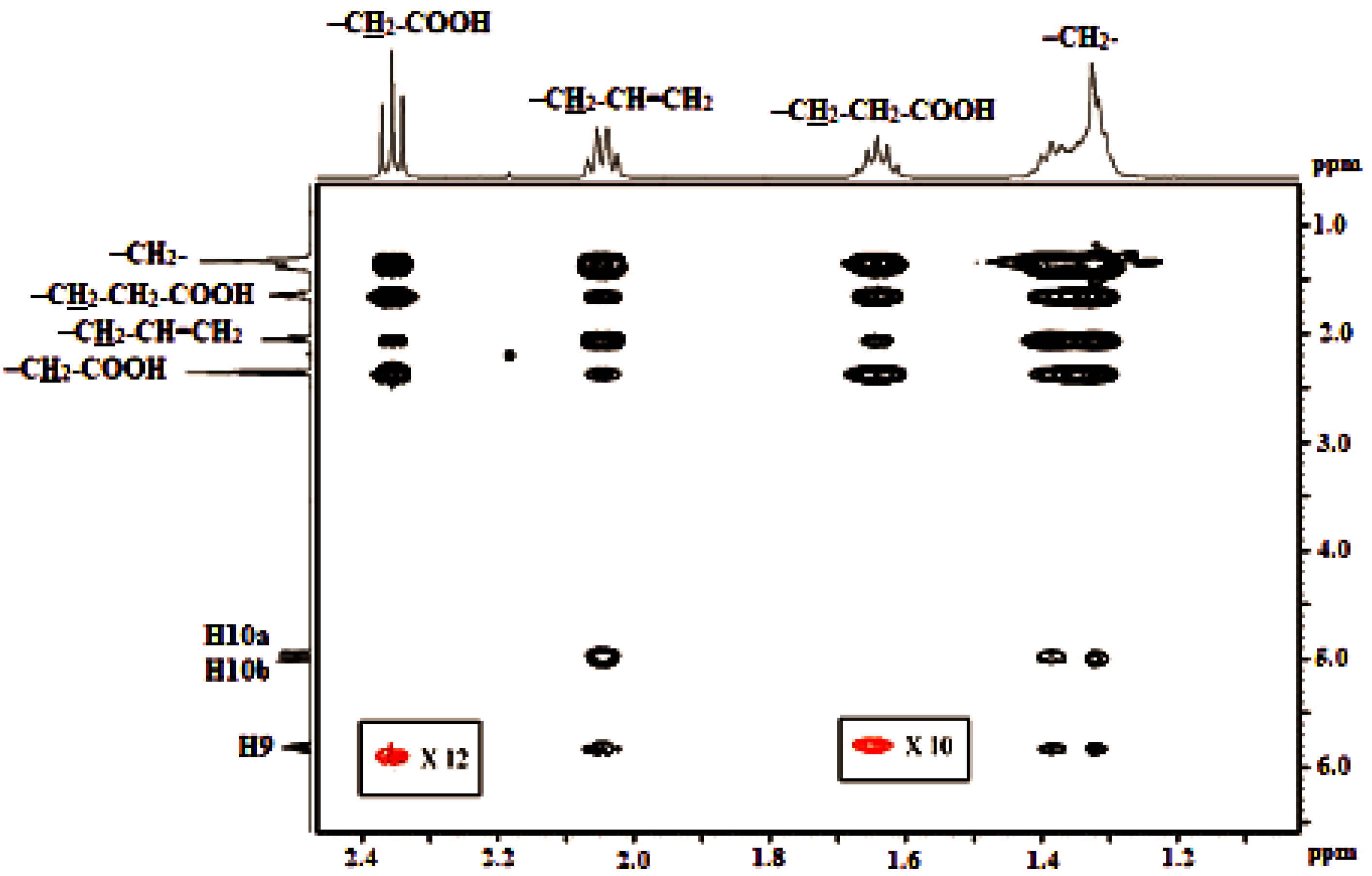
4.3. Homonuclear 2D 1H-1H COSY and 2D 1H-1H TOCSY Experiments [80,81,82]
4.4. 1H-13C Heteronuclear Single-Quantum Correlation Spectroscopy (1H-13C HSQC)-Band Selective 1H-13C HSQC [80,81,82,83]
4.5. Multiplicity Edited 1H-13C HSQC
4.6. 2D 1H-13C HSQC-TOCSY-Band Selective Experiments
4.7. 1H-13C Heteronuclear Multiple-Bond Correlation (1H-13C HMBC) Experiments—Band Selective Constant Time 1H-13C HMBC [80,81,82,83]
4.8. DOSY Experiments
4.9. Chemical Shift Reagents (CSR)
4.10. Database Matching Approach—Resolving NMR Signals Using Multivariate Data Analysis
5. Selected Analytical and Structural Studies
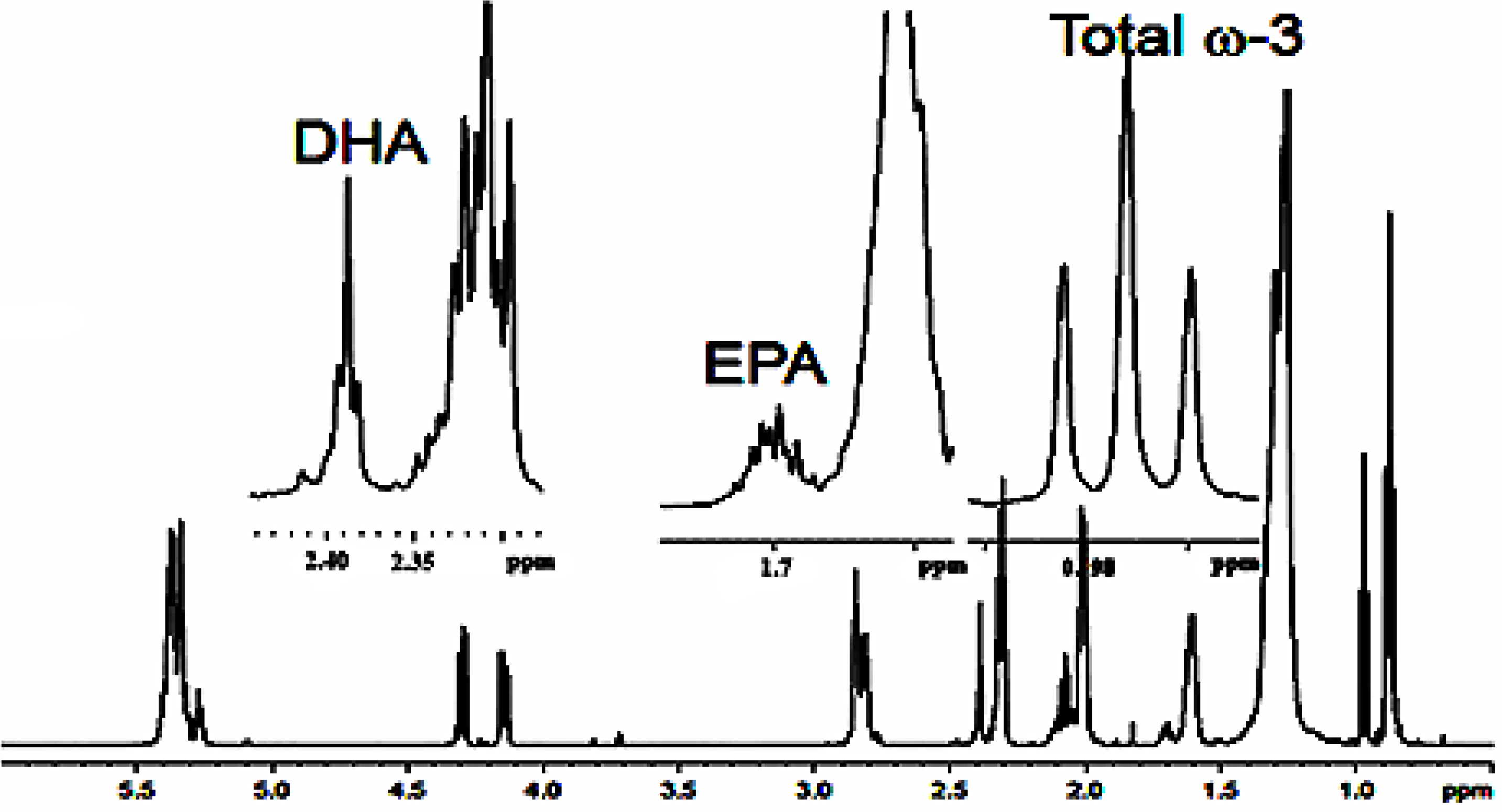
5.1. Identification and Quantification of Unsaturated Fatty Acids in Complex Mixtures
5.2. Identification and Quantification of Minor Lipids in Complex Mixtures
5.3. Identification and Quantification of Free Fatty Acids
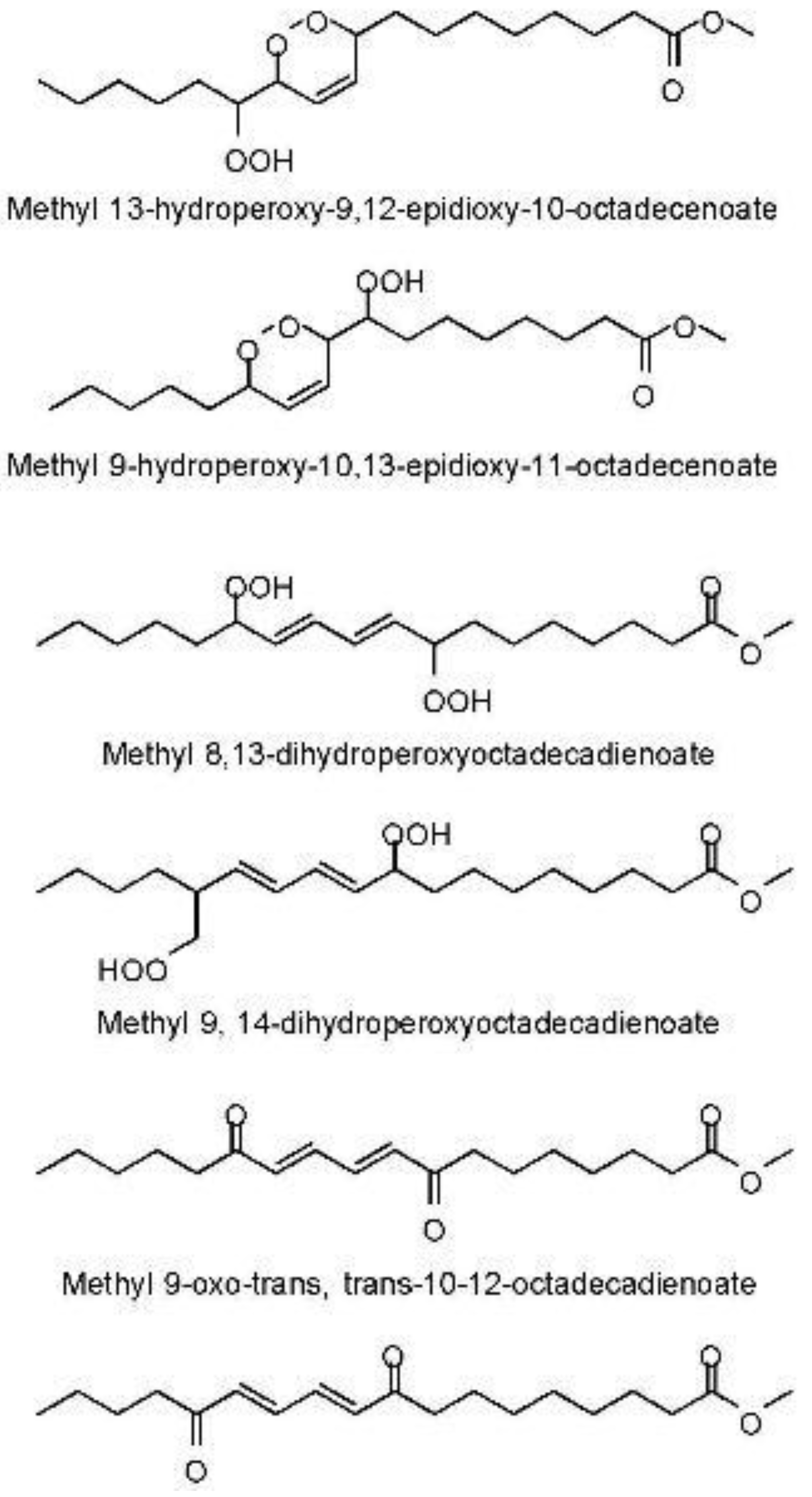
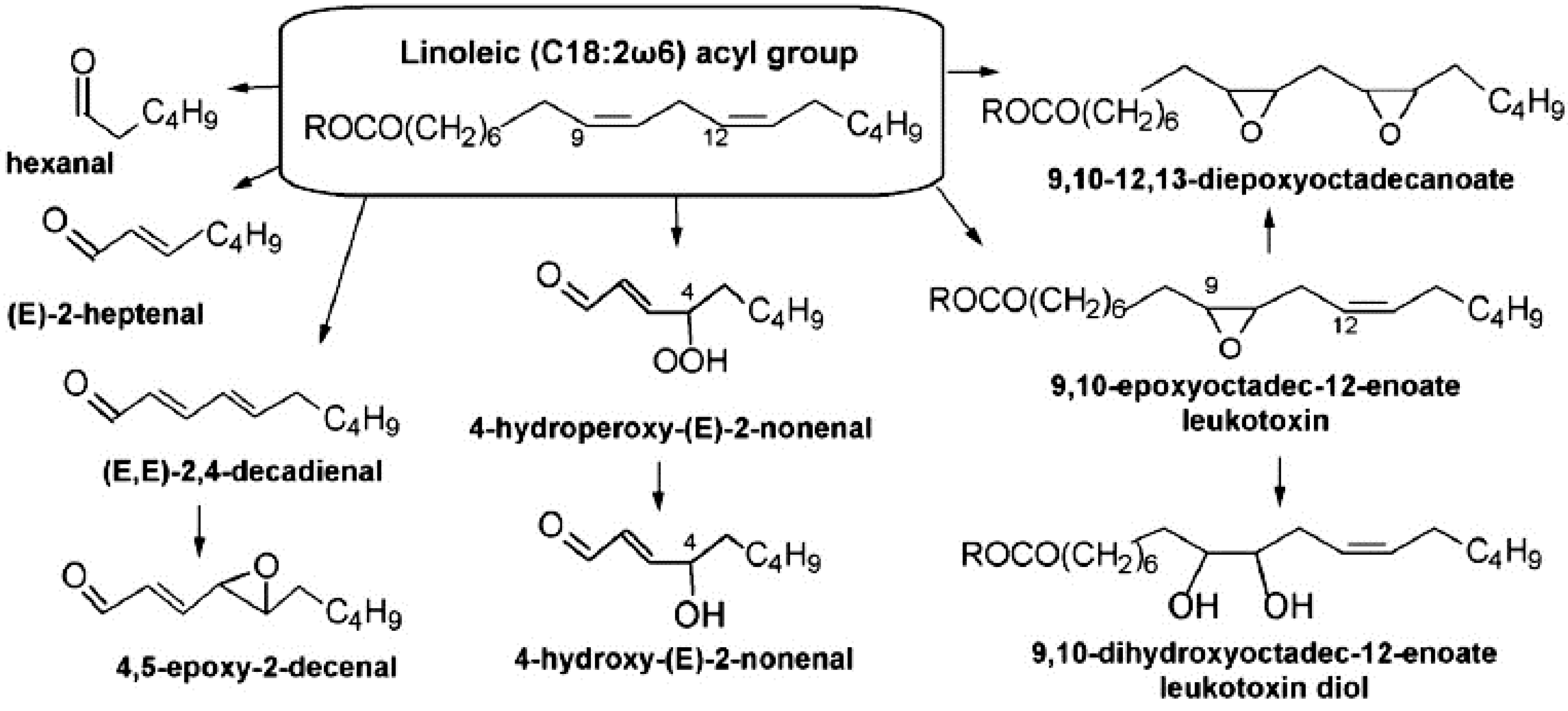
5.4. Investigation of Oxidation Products
5.5. LC-NMR
5.6. Lipidomics
5.6.1. Authentication and Quality Assessment of Oils
5.6.2. Authentication and Quality Assessment of Dairy Products
5.6.3. Authentication and Quality Assessment of Fish and Meat Lipids
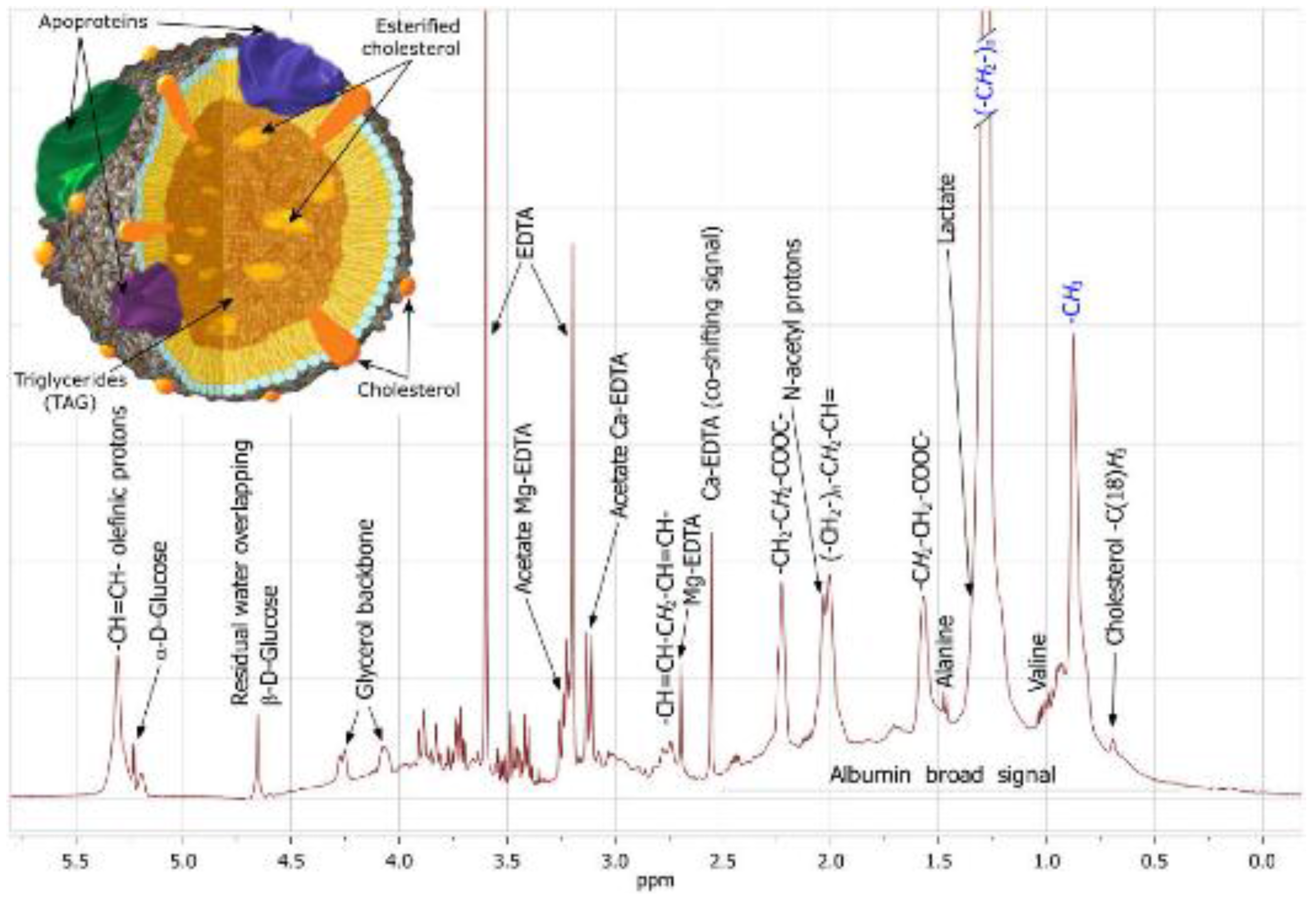
5.6.4. Serum/Plasma Lipoprotein Analysis
5.7. Investigation of Lipid-Derived Molecules
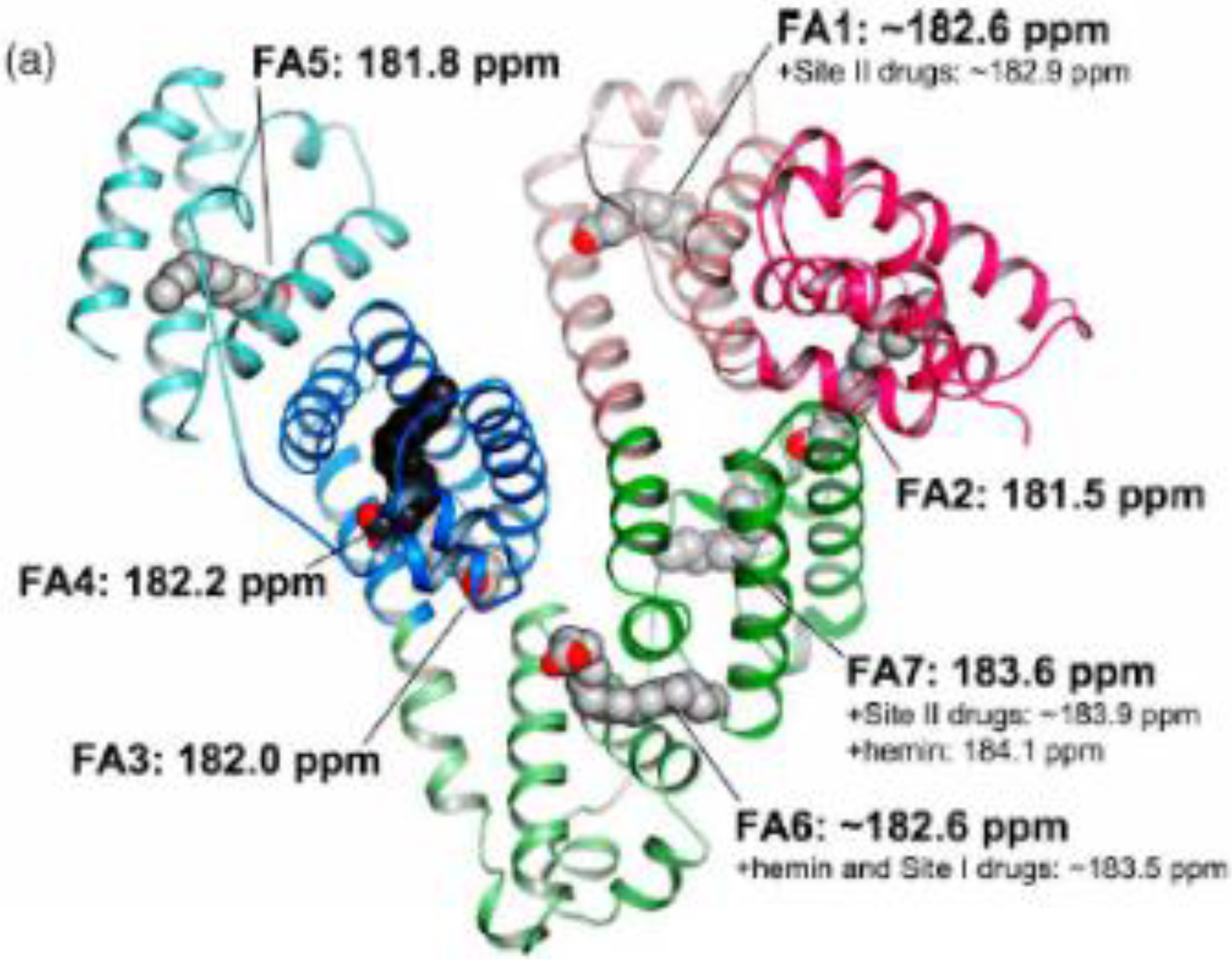
5.8. Protein-Lipid Interactions
6. Practical Considerations
6.1. Preparation of Solid and Liquid Samples
6.2. Lipid Extraction Methods
6.3. NMR Sample Preparation
6.3.1. NMR Solvent, Referenicng and Quantification Methods
6.3.2. Effects of Temperature and Conentration
7. Conclusions and Future Prospects
- (1)
- The excellent resolution and sensitivity advantages of the selective 1D TOCSY and band selective 1H-13C HSQC, 1H-13C HSQC-TOCSY and 1H-13C HMBC experiments show great potential in deciphering complex lipid extracts and oxidation products, even though such methods are still not frequently used in lipid research.
- (2)
- Application of broadband 1H homonuclear decoupled techniques will result in highly resolved 1H-NMR spectra with collapsed singlets, thus minimizing overlap and expediting spectral analysis [248,249]. Several novel 1D and 2D selective experiments and improved slice—selective experiments have been proposed which can provide ultra-highly resolved NMR spectra with great potentialities for accurate determination of very small chemical shift differences, coupling constants, and relaxation times [250,251,252,253,254].
- (3)
- (4)
- Application of hyperpolarizable techniques will greatly improve sensitivity since they are capable of generating spin population levels that are ~5 × 104 times higher than the Boltzman equilibrium at room temperature [257]. Particularly promising results can be obtained in solution state by dissolution dynamic nuclear polarization (DNP), where the sample to be analyzed is mixed with free radicals in a solution frozen to liquid helium temperatures and hyperpolarized by irradiating in the vicinity of the ESR of the unpaired electrons of the radicals [258,259].
- (5)
- Development of automated hyphenated LC-SPE-NMR-MS platforms [260].
- (6)
- More comprehensive and systematic comparisons between NMR and other analytical methods, such as GC-MS, should be performed in order to investigate their limitations, strengths and weaknesses as well as the advantages of their combined use.
- (7)
- Development of officially recognized NMR methodologies for fats and oils through collaboration of various NMR groups.
- (8)
- (9)
- Further developments in DOSY (Diffusion Order Spectroscopy) can make it special powerful tool for the analysis of unsaturated fatty acids and lipids [248].
- (10)
Acknowledgments
Author Contributions
Conflicts of Interest
References and Note
- Gunstone, F.D. Fatty Acid and Lipid Chemistry, 1st ed.; Springer: New York, NY, USA, 1996. [Google Scholar]
- Vance, D.E.; Vance, J.E. (Eds.) Biochemistry of Lipids, Lipoproteins and Membranes, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Akoh, C.C.; Min, D.B. (Eds.) Food Lipids, Chemistry, Nutrition and Biochemistry, 1st ed.; Marcel Dekker Inc.: New York, NY, USA, 2002. [Google Scholar]
- Leray, C. Dietary Lipids for Healthy Brain Function; CRC Press: Boca Raton, FL, USA, 2017. [Google Scholar]
- Kuller, L.H. Nutrition, lipids, and cardiovascular disease. Nutr. Rev. 2006, 64, S15–S26. [Google Scholar] [CrossRef] [PubMed]
- Layden, B.T.; Angueira, A.R.; Brodsky, M.; Durai, V.; Lowe, W.L., Jr. Short chain fatty acids and their receptors: New metabolic targets. Transl. Res. 2013, 161, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Schwager, J.; Richard, N.; Riegger, C.; Salem, N., Jr. ω-3 PUFAs and resveratrol differently modulate acute and chronic inflammatory processes. BioMed. Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Kummerow, F.A. The negative effects of hydrogenated trans fats and what to do about them. Atherosclerosis 2009, 205, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Nishida, C.; Uauy, R. WHO scientific update on health consequences of trans fatty acids: Introduction. Eur. J. Clin. Nutr. 2009, 63, S1–S4. [Google Scholar] [CrossRef] [PubMed]
- US Department of Health and Human Services and US Department of Agriculture. Dietary Guidelines for Americans, 6th ed.; US Government Printing Office: Washington, DC, USA, 2005.
- World Health Organization (WHO). Diet, Nutrition and the Prevention of Chronic Diseases; RWHO/FAO; World Health Organization: Geneva, Switzerland, 2003; pp. 87–89. [Google Scholar]
- Council Directive 90/496/EEC of 24.9.1990 on Nutrition Labelling for Foodstuffs, OJ L 276, 6.10.1990.
- O’Keefe, S.F. Nomenclature and classification of lipids. In Food Lipids, Chemistry, Nutrition and Biochemistry, 1st ed.; Akoh, C.C., Min, D.B., Eds.; Marcel Dekker Inc.: New York, NY, USA, 2002. [Google Scholar]
- Mitchell, T.W.; Brown, S.H.O.; Blanksby, S.J. Structural lipidomics. In Lipidomics: Technologies and Applications; Ekroos, K., Ed.; Wiley VCH: Weinheim, Germany, 2012; pp. 99–128. [Google Scholar]
- Hancock, S.E.; Poad, B.J.; Batarseh, A.; Abbott, S.K.; Mitchell, T.D. Advances and unresolved challenges in the structural characterization of isomeric lipids. Anal. Biochem. 2017, 524, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Medina, I.; Aubourg, S.; Gallardo, J.M.; Pérez-Martín, R. Comparison of six methylation methods for analysis of the fatty acid composition of albacore lipid. Int. J. Food Sci. Technol. 1992, 27, 597–601. [Google Scholar] [CrossRef]
- Igarashi, T.; Aursand, M.; Hirata, Y.; Gribbestad, I.S.; Wada, S.; Nonaka, M. Nondestructive quantitative determination of docosahexaenoic acid and n-3 fatty acids in fish oils by high-resolution 1H nuclear magnetic resonance spectroscopy. J. Am. Oil. Chem. Soc. 2000, 77, 737–748. [Google Scholar] [CrossRef]
- De la Fuente, A.M.; Luna, P.; Juárez, M. Chromatographic techniques to determine conjugated linoleic acid isomers. Trends Anal. Chem. 2006, 25, 917–926. [Google Scholar] [CrossRef]
- Gunstone, F.D. High resolution 13C-NMR spectroscopy of lipids. In Advances in Lipid Methodology—Two; Christie, W.W., Ed.; The Oily Press: Dundee, UK, 1993; pp. 1–68. [Google Scholar]
- Vlahov, G. Application of NMR to the study of olive oils. Prog. Nucl. Magn. Reson. Spectrosc. 1999, 35, 341–357. [Google Scholar] [CrossRef]
- Diehl, B.W.K. High resolution NMR spectroscopy. Eur. J. Lipid Sci. Technol. 2001, 103, 830–834. [Google Scholar] [CrossRef]
- Diehl, B.W.K. Multinuclear high resolution nuclear magnetic resonance spectroscopy. In Lipid Analysis in Oils and Fats; Hamilton, R.J., Ed.; Springer: Boston, MA, USA, 1998; pp. 87–135. [Google Scholar]
- Spyros, A.; Dais, P. 31P NMR spectroscopy in food analysis. Prog. Nucl. Magn. Reson. Spectrosc. 2009, 54, 195–207. [Google Scholar] [CrossRef]
- Mannina, L.; Sobolev, A.P.; Viel, S. Liquid state 1H high field NMR in food analysis. Prog. Nucl. Magn. Reson. Spectrosc. 2012, 66, 1–39. [Google Scholar] [CrossRef] [PubMed]
- Gunstone, F.D.; Knothe, G.H. Nuclear Magnetic Resonance Spectroscopy of Fatty Acids and Their Derivatives. AOCS Lipid Library, Updated 2nd April 2014. Available online: www.aocs.org (accessed on 31 July 2014).
- Μartínez-Yusta, A.; Goicoechea, E.; Guillén, M.D. A review of thermo-oxidative degradation of food lipids studied by 1H NMR spectroscopy: Influence of degradative conditions and food lipid nature. Compr. Rev. Food Sci. Food Saf. 2014, 13, 838–859. [Google Scholar] [CrossRef]
- Pikula, S.; Bandorowicz, J.; Groves, P. NMR of Lipids. In Nuclear Magnetic Resonance: Volume 43; Kamienska-Trela, K., Wojcik, J., Eds.; Royal Society of Chemistry: London, UK, 2014; pp. 378–400. [Google Scholar]
- Hwang, H.-S.; Bakota, E.L. NMR spectroscopy for evaluation of lipid oxidation. In Applications of NMR Spectroscopy; Rahman, A.U., Choudhary, M.I., Eds.; Bentham eBooks: Oak Park, IL, USA; Volume 4, pp. 62–95.
- Hwang, H.-S. Advances in NMR Spectroscopy for Lipid Oxidation Assessment, 1st ed.; Springer Briefs in Food, Health, and Nutrition; Springer International Publishing: Cham, Switzerland, 2017. [Google Scholar]
- Watkins, S.M.; German, J.B. Unsaturated fatty acids. In Food Lipids, Chemistry, Nutrition and Biochemistry, 1st ed.; Akoh, C.C., Min, D.B., Eds.; Marcel Dekker Inc.: New York, NY, USA, 2002. [Google Scholar]
- Lemmon, M.A. Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 2008, 9, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Newton, A.C. Diacylglycerol’s affair with protein kinase C turns 25. Trends Pharmacol. Sci. 2004, 25, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Moolennar, W.H.; van Meeteren, L.A.; Giepmans, B.N.G. The ins and outs of lysophosphatidic acid signaling. Bioassays 2004, 26, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Cook, H.W.; McMaster, C.R. Fatty acid desaturation and chain elongation in eukaryotes. In Biochemistry of Lipids, Lipoproteins and Membranes, 4th ed.; Vance, D.E., Vance, J.E., Eds.; Elsevier Science: Oxford, UK, 2002; pp. 181–204. [Google Scholar]
- Masoodi, M.; Volmer, D.A. Comprehensive quantitative determination of PUFA-related bioactive lipids for functional lipidomics using high-resolution mass spectrometry. Methods Mol. Biol. 2014, 1198, 221–232. [Google Scholar] [PubMed]
- Liu, Z.; Hopkins, M.M.; Zhang, Z.; Quisenberry, C.B.; Fix, L.C.; Galvan, B.M.; Meier, K.E. ω-3 Fatty acids and other FFA4 agonists inhibit growth factor signaling in human prostate cancer cells. J. Pharmacol. Exp. Ther. 2015, 352, 1–15. [Google Scholar]
- Hopkins, M.M.; Zhang, Z.; Liu, Z.; Meier, K.E. Eicosopentaneoic acid and other free ratty acid receptor agonists inhibit lysophosphatidic acid- and epidermal growth factor-induced proliferation of human breast cancer cells. J. Clin. Med. 2016, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Hardman, W.E. Omega-3 fatty acids to augment cancer therapy. J. Nutr. 2002, 132, 3508S–3512S. [Google Scholar] [PubMed]
- Zheng, J.-S.; Hu, X.-J.; Zhao, Y.-M.; Yang, J.; Duo, L. Intake of fish and marine n-3 polyunsaturated fatty acids and risk of breast cancer: Meta-analysis of data from 21 independent prospective cohort studies. Br. Med. J. 2013, 346. [Google Scholar] [CrossRef] [PubMed]
- Simopoulos, A.P. The importance of the ratio of omega-6/omega-3 essential fatty acids. Biomed. Pharmacother. 2002, 56, 365–379. [Google Scholar] [CrossRef]
- Simopoulos, A.P. The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp. Biol. Med. 2008, 233, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Lakshmy, R.; Abraham, R.A.; Reddy, K.S.; Jeemon, P.; Prabhakaran, D. Serum omega-6/omega-3 ratio and risk markers for cardiovascular disease in an industrial population of Delhi. Food Nutr. Sci. 2013, 4, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Blasbalg, T.L.; Hibbeln, J.R.; Ramsden, C.E.; Majchrzak, S.F.; Rawlings, R.R. Changes in consumption of omega-3 and omega-6 fatty acids in the United States during the 20th century. Am. J. Clin. Nutr. 2011, 93, 950–962. [Google Scholar] [CrossRef] [PubMed]
- Sebedio, J.L.; Christie, W.W.; Adolf, R.O. (Eds.) Advances in Conjugated Linoleic Acid Research; American Oil Chemists’ Society: Champaign, IL, USA, 2003; Volume 2. [Google Scholar]
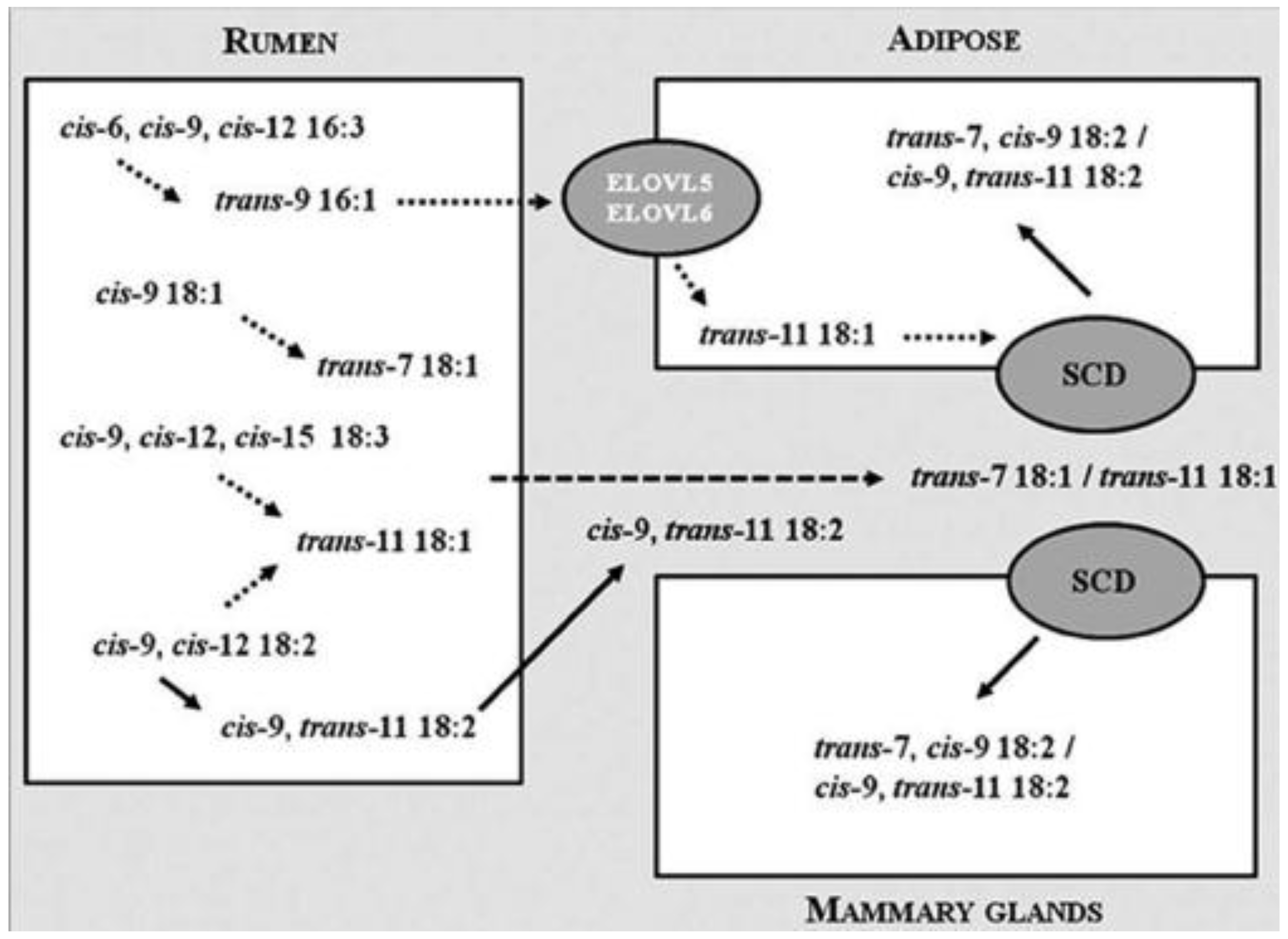
- Shingfield, K.J.; Wallace, R.J. Synthesis of conjugated linoleic acid in ruminants and humans. In Conjugated Linoleic Acids and Conjugated Vegetable Oils; Sels, B., Philippaerts, A., Eds.; Royal Society of Chemistry: Cambridge, UK, 2014; pp. 1–65. [Google Scholar]
- Dilzer, A.; Park, Y. Implication of conjugated linoleic acid (CLA) in human health. Crit. Rev. Food Sci. Nutr. 2012, 52, 488–513. [Google Scholar] [CrossRef] [PubMed]
- Wahle, K.W.; Heys, S.D.; Rotondo, D. Conjugated linoleic acids: Are they beneficial or detrimental to health? Prog. Lipid Res. 2004, 43, 553–587. [Google Scholar] [CrossRef] [PubMed]
- Tricon, S.; Burdge, G.C.; Williams, C.M.; Calder, P.C.; Yaqoob, P. The effects of conjugated linoleic acid on human health-related outcomes. Proc. Nutr. Soc. 2005, 64, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.K.; Mann, B.E. (Eds.) NMR and the Periodic Table, 5th ed.; Academic Press: London, UK, 1978; pp. 5–7. [Google Scholar]
- Gerothanassis, I.P. Oxygen-17 NMR spectroscopy: Basic principles and applications (Part I). Prog. Nucl. Magn. Reson. Spectrosc. 2010, 56, 95–197. [Google Scholar] [CrossRef] [PubMed]
- Gerothanassis, I.P. Oxygen-17 NMR spectroscopy: Basic principles and applications (Part II). Prog. Nucl. Magn. Reson. Spectrosc. 2010, 57, 1–110. [Google Scholar] [CrossRef] [PubMed]
- Gerothanassis, I.P.; Lauterwein, J. An evaluation of various pulse sequences for the suppression of acoustic ringing in oxygen-17 NMR. J. Magn. Reson. 1986, 66, 32–42. [Google Scholar] [CrossRef]
- Andersson, H.; Carlsson, A.-C.C.; Nekoueishahraki, B.; Brath, U.; Erdélyi, M. Solvent effects on nitrogen chemical shifts. Annu. Rep. NMR Spectrosc. 2015, 86, 73–210. [Google Scholar]
- Frost, D.J.; Gunstone, F.D. The PMR analysis of non-conjugated alkenoic and alkynoic acids and esters. Chem. Phys. Lipids 1975, 15, 53–85. [Google Scholar] [CrossRef]
- Kellersmann, C.; Steinhart, Η.; Francke, W. Syntheses of conjugated octadecadienoic acids. Lipids 2006, 41, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Tsiafoulis, C.G.; Skarlas, T.; Tzamaloukas, O.; Miltiadou, D.; Gerothanassis, I.P. Direct nuclear magnetic resonance identification and quantification of geometric isomers of conjugated linoleic acid in milk lipid fraction without derivatization steps: Overcoming sensitivity and resolution barriers. Anal. Chim. Acta 2014, 821, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Tulloch, A.P.; Bergter, L. Analysis of the conjugated trienoic acid containing oil from Fevillea trilobata by 13C nuclear magnetic resonance spectroscopy. Lipids 1979, 14, 996–1002. [Google Scholar] [CrossRef]
- Cao, Y.H.-L.; Chen, J.-N.; Chen, Z.-Y.; Yang, L. Identification and characterization of conjugated linolenic acid isomers by Ag+-HPLC and NMR. J. Agric. Food Chem. 2006, 54, 9004–9009. [Google Scholar] [CrossRef] [PubMed]
- Hatzakis, E.; Agiomyrgianaki, A.; Kostidis, S.; Dais, P. High-resolution NMR spectroscopy: An alternative fast tool for qualitative and quantitative analysis of diacylglycerol (DAG) oil. J. Am. Oil. Chem. Soc. 2011, 88, 1695–1708. [Google Scholar] [CrossRef]
- Dais, P.; Misiaka, M.; Hatzakis, E. Analysis of marine dietary supplements using NMR spectroscopy. Anal. Methods 2015, 7, 5226–5238. [Google Scholar] [CrossRef]
- Shoolery, J.N. Some quantitative applications of 13C NMR spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc. 1977, 11, 79–93. [Google Scholar] [CrossRef]
- Gunstone, F.D. 13C-NMR studies of mono-, di-and tri-acylglycerols leading to qualitative and semiquantitative information about mixtures of these glycerol esters. Chem Phys. Lipids 1991, 58, 219–224. [Google Scholar]
- Wollenberg, K.F. Quantitative high resolution 13C nuclear magnetic resonance of the olefinic and carbonyl carbons of edible vegetable oils. J. Am. Oil Chem. Soc. 1990, 67, 487–494. [Google Scholar] [CrossRef]
- Howarth, O.W.; Samuel, C.J.; Vlahov, G. The σ-inductive effects of C=C double bond, and C≡C bonds: Predictability of NMR shifts at sp2 carbon in non-conjugated polyenoic acids, esters and glycerides. J. Chem. Soc. Perkin Trans. 2 1995, 12, 2307–2310. [Google Scholar]
- Bianchi, G.; Howarth, C.W.; Samuel, C.J.; Vlahov, G. σ-Inductive interaction through up to fourteen saturated C–C bonds. J. Chem. Soc. Chem. Commun. 1994, 5, 627–628. [Google Scholar] [CrossRef]
- Bianchi, G.; Howarth, C.W.; Samuel, C.J.; Vlahov, G. Long-range σ-inductive interactions through saturated C–C bonds in polymethylene chains. J. Chem. Soc. Perkin Trans. 2 1995, 97, 1427–1432. [Google Scholar] [CrossRef]
- Batchelor, J.G.; Prestegard, J.H.; Cushley, R.J.; Lipsky, S.R. Electric field effects in the carbon-13 nuclear magnetic resonance spectra of unsaturated fatty acids. Potential tool for conformational analysis. J. Am. Chem. Soc. 1973, 95, 6358–6364. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.L.; Mc Neill, G.P.; Caswell, D.C. Analysis of conjugated linoleic acid isomers by 13C NMR spectroscopy. Chem. Phys. Lipids 1999, 97, 155–156. [Google Scholar] [CrossRef]
- Meneses, P.; Glonek, T. High resolution 31P NMR of extracted phospholipids. J. Lipid Res. 1988, 29, 679–689. [Google Scholar] [PubMed]
- Vigli, G.; Philippidis, A.; Spyros, A.; Dais, P. Classification of edible oils by employing 31P and 1H NMR spectroscopy in combination with multivariate statistical analysis. A proposal for the detection of seed oil adulteration in virgin olive oils. J. Agric. Food Chem. 2003, 51, 5715–5722. [Google Scholar] [CrossRef] [PubMed]
- Dais, P.; Spyros, A. 31P NMR spectroscopy in the quality control and authentication of extra-virgin olive oil: A review of recent progress. Magn. Reson. Chem. 2007, 45, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Hatzakis, E.; Dagounakis, G.; Agiomyrgianaki, A.; Dais, P. A facile NMR method for the quantification of total, free and esterified sterols in virgin olive oil. Food Chem. 2010, 122, 346–352. [Google Scholar] [CrossRef]
- Longobardi, F.; Ventrella, A.; Napoli, C.; Humpfer, E.; Schütz, B.; Schäfer, H.; Kontominas, M.G.; Sacco, A. Classification of olive oils according to geographical origin by using 1H NMR fingerprinting combined with multivariate analysis. Food Chem. 2012, 130, 177–183. [Google Scholar] [CrossRef]
- Parella, T. High-quality 1D spectra by implementing pulsed-field gradients as the coherence pathway selection procedure. Magn. Reson. Chem. 1996, 34, 329–347. [Google Scholar] [CrossRef]
- Sharman, G.J. Development of a selective TOCSY experiment and its use in analysis of a mixture of related compounds. Chem. Commun. 1999, 14, 1319–1320. [Google Scholar] [CrossRef]
- Sandusky, P.; Raftery, D. Use of selective TOCSY NMR experiments for quantifying minor components in complex mixtures: Application to the metabonomics of amino acids in honey. Anal. Chem. 2005, 77, 2455–2463. [Google Scholar] [CrossRef] [PubMed]
- Koda, M.; Furihata, K.; Wei, F.; Miyakawa, T.; Tanokura, M. Metabolic discrimination of mango juice from various cultivars by band-selective NMR spectroscopy. J. Agric. Food. Chem. 2012, 60, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanouil, C.; Tsiafoulis, C.G.; Alivertis, D.; Tzamaloukas, O.; Miltiadou, D.; Tzakos, A.; Gerothanassis, I.P. Selective 1D TOCSY NMR experiments for a rapid identification of minor components in the lipid fraction of milk and dairy products: Towards spin-chromatography? J. Agric. Food. Chem. 2015, 63, 5381–5387. [Google Scholar] [CrossRef] [PubMed]
- Kontogianni, V.G.; Tsiafoulis, C.G.; Roussis, I.G.; Gerothanassis, I.P. Selective 1D TOCSY NMR method for the determination of glutathione in white wine. Anal. Methods 2017, 9, 4464–4470. [Google Scholar] [CrossRef]
- Günther, H. NMR Spectroscopy: Basic Principles, Concepts and Applications in Chemistry, 3rd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Rahman, A.-U.; Choudhary, M.I.; Wahab, A.-T. Solving Problems with NMR Spectroscopy, 2nd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Claridge, T. High-Resolution NMR Techniques in Organic Chemistry, 3rd ed.; Elsevier: Oxford, UK, 2016. [Google Scholar]
- Berger, S.; Braun, S. (Eds.) 200 and More NMR Experiments: A Practical Course; Wiley VCH: Weinheim, Germany, 2004. [Google Scholar]
- Exarchou, V.; Troganis, A.; Gerothanassis, I.P.; Tsimidou, M.; Boskou, D. Analysis of phenolic acids in complex phenolic mixtures by the use of variable temperature two dimensional 1H-1H COSY, 1H-13C HMQC and 1H-13C HMBC gradient enhanced NMR spectroscopy: Application to methanolic extracts of several oregano species. J. Agric. Food Chem. 2001, 49, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Kontogianni, V.G.; Exarchou, V.; Troganis, A.; Gerothanassis, I.P. Rapid and novel discrimination and quantification of oleanolic and ursolic acids in complex plant extracts using two-dimensional nuclear magnetic resonance spectroscopy-comparison with HPLC methods. Anal. Chim. Acta 2009, 635, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Willker, W.; Flögel, U.; Leibfritz, D. Ultra-high-resolved HSQC spectra of multiple-13C-labeled biofluids. J. Magn. Reson. 1997, 125, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Fiori, L.; Solana, M.; Manfrini, M.; Guella, G. Lipid profiles of oil from trout (Oncorhynchus mykiss) heads, spines and viscera: Trout by-products as a possible source of omega-3 lipids? Food Chem. 2012, 134, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Parella, T.; Sanchez-Ferrando, F.; Virgili, A. Improved sensitivity in gradient-based 1D and 2D multiplicity-edited HSQC experiments. J. Magn. Reson. 1997, 126, 274–277. [Google Scholar] [CrossRef]
- Willker, W.; Leibfritz, D. Assignments of mono- and polyunsaturated fatty acids in lipids of tissue and body fluids. Magn. Reson. Chem. 1998, 36, S79–S84. [Google Scholar] [CrossRef]
- Vatèle, J.-M.; Fenet, B.; Eynard, T. Complete 13C assignments and structural elucidation of n-3 polyunsaturated fatty acids by the use of a new 2D NMR technique: SAPHIR-HSQC. Chem. Phys. Lipids 1998, 94, 239–250. [Google Scholar] [CrossRef]
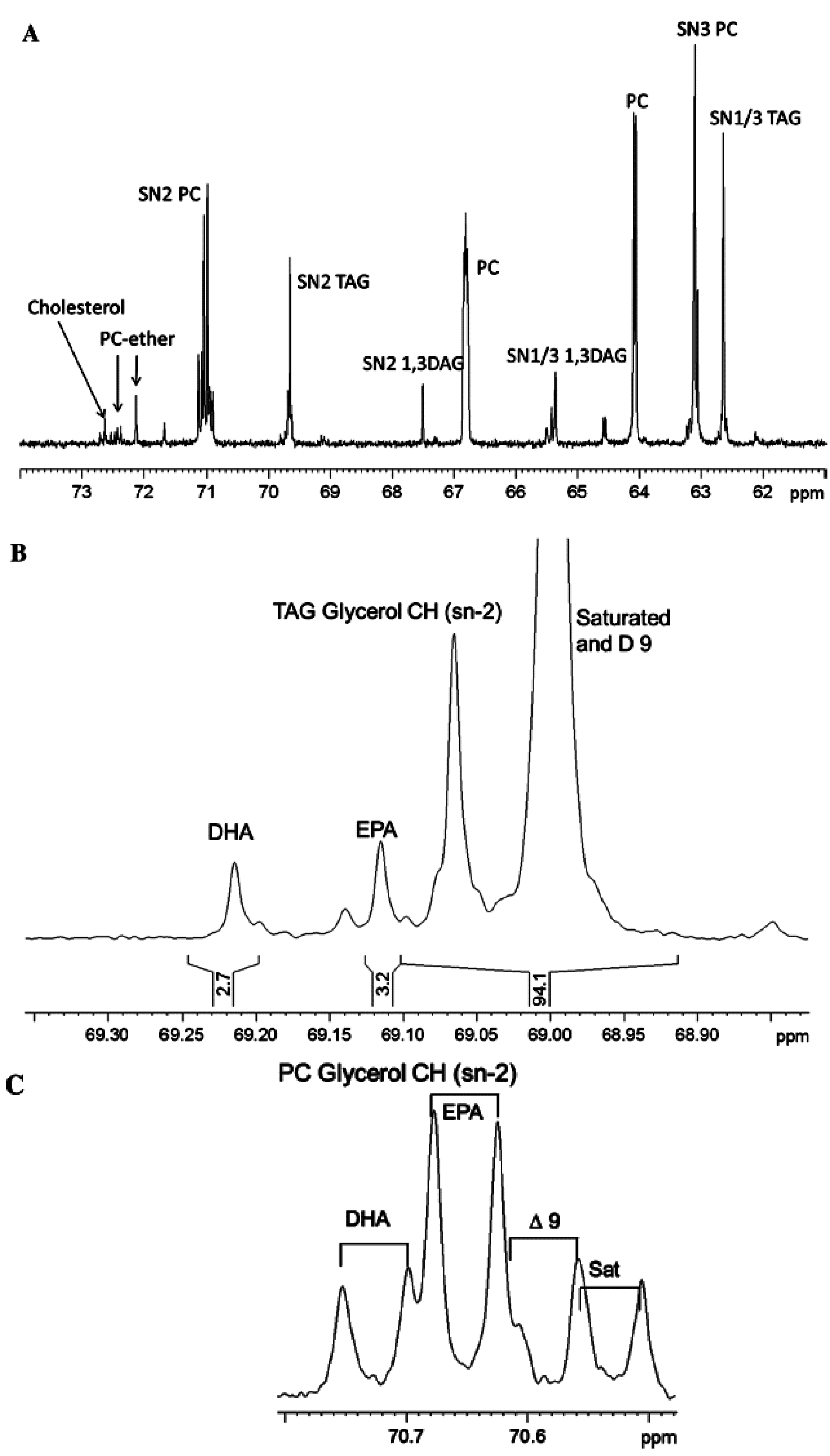
- Simova, S.; Ivanova, G.; Spassov, S.L. Alternative NMR method for quantitative determination of acyl positional distribution in triacylglycerols and related compounds. Chem. Phys. Lipids 2003, 126, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Vlahov, G.; Giuliani, A.A.; Del Re, P. 13C NMR spectroscopy for determining the acylglycerol positional composition of lampante olive oils. Chemical shift assignments and their dependence on sample concentration. Anal. Methods 2010, 2, 916–923. [Google Scholar] [CrossRef]
- Gaillet, C.; Lequart, C.; Debeire, P.; Nuzillard, J.-M. Band-selective HSQC and HMBC experiments using excitation sculpting and PFGSE. J. Magn. Reson. 1999, 139, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Claridge, T.D.W.; Pérez-Victoria, I.P. Enhanced 13C resolution in semi-selective HMBC: A band-selective, constant-time HMBC for complex organic structure elucidation by NMR. Org. Biomol. Chem. 2003, 1, 3632–3634. [Google Scholar] [CrossRef] [PubMed]
- Cohen, Y.; Avram, L.; Frish, L. Diffusion NMR spectroscopy in supramolecular and combinatorial chemistry: An old parameter-new insights. Angew. Chem. Int. Ed. 2005, 44, 520–554. [Google Scholar] [CrossRef] [PubMed]
- Caldareli, S. Chromatographic NMR: A tool for the analysis of mixtures of small molecules. Magn. Reson. Chem. 2007, 45, S48–S55. [Google Scholar] [CrossRef] [PubMed]
- Gil, A.M.; Duarte, I.; Cabrita, E.; Goodfellow, B.J.; Spraul, M.; Kerssebaum, R. Exploratory applications of diffusion ordered spectroscopy to liquid foods: An aid towards spectral assignment. Anal. Chim. Acta 2004, 506, 215–223. [Google Scholar] [CrossRef]
- Rodrigues, E.D.; Silva, D.B.; Oliveira, D.C.R.; Silva, G.V.J. DOSY NMR applied to analysis of flavonoid glycosides from Bidens sulphurea. Magn. Reson. Chem. 2009, 47, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Primikyri, A.; Kyriakou, E.; Charisiadis, P.; Tsiafoulis, C.; Stamatis, C.; Tzakos, A.G.; Gerothanassis, I.P. A fine-tuning of the diffusion dimension of –OH groups for high resolution DOSY NMR applications in crude enzymatic trans formations and mixtures of organic compounds. Tetrahedron 2012, 68, 6887–6891. [Google Scholar] [CrossRef]
- Vieira, M.G.S.; Gramosa, N.V.; Ricardo, N.M.P.S.; Morris, G.A.; Adams, R.W.; Nilsson, M. Natural product mixture analysis by matrix-assisted DOSY using Brij surfactants in mixed solvents. RSC Adv. 2014, 4, 42029–42034. [Google Scholar] [CrossRef]
- Swern, D.; Wineburg, J.P. NMR chemical shift reagents. Application to structural determination of lipid derivatives. J. Am. Oil. Chem. Soc. 1971, 48, 371–372. [Google Scholar] [CrossRef]
- Cockerill, A.F.; Davies, G.L.O.; Harden, R.C.; Rackham, D.M. Lanthanide shift reagents for nuclear magnetic resonance spectroscopy. Chem. Rev. 1973, 73, 553–588. [Google Scholar] [CrossRef]
- Wineburg, J.P.; Swern, D. NMR chemical shift reagents in structural determination of lipid derivatives: II. Methyl petroselinate and methyl oleate. J. Am. Oil. Chem. Soc. 1972, 49, 267–273. [Google Scholar] [CrossRef]
- Wineburg, J.P.; Swern, D. NMR chemical shift reagents in structural determination of lipid derivatives: III. Methyl ricinoleate and methyl 12-hydroxystearate. J. Am. Oil. Chem. Soc. 1973, 50, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Iida, T.; Tamura, T.; Matsumoto, T. Proton nuclear magnetic resonance identification and discrimination of side chain isomers of phytosterols using a lanthanide shift reagent. J. Lipid Res. 1980, 21, 326–338. [Google Scholar] [PubMed]
- Agiomyrgianaki, A.; Sedman, J.; Van de Voort, F.R.; Dais, P. Cis and trans components of lipids: Analysis by 1H NMR and silver shift reagents. Eur. J. Lipid Sci. Technol. 2012, 114, 504–509. [Google Scholar] [CrossRef]
- Kim, H.K.; Choi, Y.H.; Verpoorte, R. NMR-based metabolomic analysis of plants. Nat. Protoc. 2010, 5, 536–549. [Google Scholar] [CrossRef] [PubMed]
- López-Perez, J.L.; Therón, R.; del Olmo, E.; Díaz, D. NAPROC-13: A database for the dereplication of natural product mixtures in bioassay-guided protocols. Bioinformatics 2007, 23, 3256–3257. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, S.; Egert, B.; Neumann, S.; Steinbeck, C. Building blocks for automated elucidation of metabolites: Machine learning methods for NMR prediction. BMC Bioinform. 2008, 9, 400. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.C.; Jarak, I.; Carvalho, R.A. Resolving NMR signals of short chain fatty acid mixtures using unsupervised component analysis. Magn. Reson. Chem. 2017, 55, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Asif, M. Chemical characteristics and nutritional potentials of unsaturated fatty acids. Chem. Internsh. 2015, 1, 118–133. [Google Scholar]
- Aursand, M.; Rainuzzo, R.J.; Grasdalen, H. Quantitative high-resolution 13C and 1H nuclear magnetic resonance of ω3 fatty acids from white muscle of atlantic salmon (Salmo salar). J. Am. Oil. Chem. Soc. 1993, 70, 971–981. [Google Scholar] [CrossRef]
- Sacchi, R.; Medina, I.; Aubourg, S.P.; Addeo, F.; Paolillo, L. Proton nuclear magnetic resonance rapid and structure-specific determination of ω-3 polyunsaturated fatty acids in fish lipids. J. Am. Oil. Chem. Soc. 1993, 70, 225–228. [Google Scholar] [CrossRef]
- Sacchi, R.; Addeo, F.; Paolillo, L. 1H and 13C-NMR of virgin olive oil. An overview. Magn. Reson. Chem. 1997, 35, S133–S145. [Google Scholar] [CrossRef]
- Miyake, Y.; Yokomizo, K.; Matsuzaki, N. Determination of unsaturated fatty acid composition by high-resolution nuclear magnetic resonance spectroscopy. J. Am. Oil. Chem. Soc. 1998, 75, 1091–1094. [Google Scholar] [CrossRef]
- Miyake, Y.; Yokomizo, K.; Matsuzaki, N. Rapid determination of iodine value by 1H nuclear magnetic resonance spectroscopy. J. Am. Oil. Chem. Soc. 1998, 75, 15–19. [Google Scholar] [CrossRef]
- Fauhl, C.; Reniero, F.; Guillou, C. 1H NMR as a tool for the analysis of mixtures of virgin olive oil with oils of different botanical origin. Magn. Reson. Chem. 2000, 38, 436–443. [Google Scholar] [CrossRef]
- Sacco, A.; Brescia, M.A.; Liuzzi, V.; Reniero, F.; Guillou, G.; Ghelli, S.; van der Meer, P. Characterization of italian olive oils based on analytical and nuclear magnetic resonance determinations. J. Am. Oil. Chem. Soc. 2000, 77, 619–625. [Google Scholar] [CrossRef]
- Knothe, G. Monitoring a progressing trans esterification reaction by fiber-optic near-infrared spectroscopy with correlation to 1H nuclear magnetic resonance spectroscopy. J. Am. Oil. Chem. Soc. 2001, 77, 489–493. [Google Scholar] [CrossRef]
- Knothe, G.; Kenar, J.A. Determination of the fatty acid profile by 1H-NMR spectroscopy. Eur. J. Lipid Sci. Technol. 2004, 106, 88–96. [Google Scholar] [CrossRef]
- Mannina, L.; Segre, A. High resolution nuclear magnetic resonance: From chemical structure to food authenticity. Grasas y Aceites 2002, 53, 22–33. [Google Scholar] [CrossRef]
- Williamson, Κ.; Hatzakis, E. NMR spectroscopy as a robust tool for the rapid evaluation of the lipid profile of fish oil supplements. J. Vis. Exp. 2017, 123, e55547. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanouil, C.; Tsiafoulis, C.G.; Alivertis, D.; Tzamaloukas, O.; Miltiadou, D.; Balayssac, S.; Malt-Martino, M.; Gerothanassis, I.P. Unpublished work. 2017.
- Sedman, J.; Gao, L.; García-González, D.; Ehsan, S.; van de Voort, F.R. Determining nutritional labeling data for fats and oils by 1H NMR. Eur. J. Lipid Sci. Technol. 2010, 112, 439–451. [Google Scholar] [CrossRef]
- Guillén, M.D.; Ruiz, A. High resolution 1H nuclear magnetic resonance in the study of edible oils and fats. Trends Food Sci. Technol. 2001, 12, 328–338. [Google Scholar] [CrossRef]
- Lewis, I.A.; Schommer, S.C.; Hodis, B.; Robb, K.A.; Tonelli, M.; Westler, W.M.; Sussman, M.R.; Markley, J.L. Method for determining molar concentrations of metabolites in complex solutions from two-dimensional 1H-13C NMR spectra. Anal. Chem. 2007, 79, 9385–9390. [Google Scholar] [CrossRef] [PubMed]
- Sandusky, P.; Appiah-Amponsah, E.; Raftery, D. Use of optimized 1D TOCSY NMR for improved quantitation and metabolomic analysis of biofluids. J. Biomol. NMR 2011, 49, 281–290. [Google Scholar] [CrossRef] [PubMed]
- International Organization of Standardization (ISO). Milk Fat—Preparation of Fatty Acid Methyl Esters; ISO 15884:2002 (IDF 182:2002); ISO: Geneva, Switzerland, 2002; p. 6. [Google Scholar]
- Skiera, C.; Steliopoulos, P.; Kuballa, T.; Diehl, B.; Holzgrabe, U. Determination of free fatty acids in pharmaceutical lipids by 1H NMR and comparison with the classical acid value. J. Pharm. Biomed. Anal. 2014, 93, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Pokorny, J. Volumetric analysis of oxidized lipids. In Analysis of Lipid Oxidation; Kamal-Eldin, A., Pocorny, J., Eds.; AOCS Press: Champaign, IL, USA, 2005; pp. 8–16. [Google Scholar]
- Satyarthi, J.K.; Srinivas, D.; Ratnasamy, P. Estimation of free fatty acid content in oils, fats, and biodiesel by 1H NMR Spectroscopy. Energy Fuels 2009, 23, 2273–2277. [Google Scholar] [CrossRef]
- Skiera, C.; Steliopoulos, P.; Kuballa, T.; Holzgrabe, U.; Diehl, B. Determination of free fatty acids in edible oils by 1H NMR spectroscopy. Lipid Technol. 2012, 24, 279–281. [Google Scholar] [CrossRef]
- Charisiadis, P.; Exarchou, V.; Troganis, A.N.; Gerothanassis, I.P. Exploring the “forgotten” –OH-1H-NMR spectral region in natural products. Chem. Commun. 2010, 46, 3589–3591. [Google Scholar] [CrossRef] [PubMed]
- Kontogianni, V.; Primikyri, A.; Exarchou, V.; Charisiadis, P.; Tzakos, A.; Gerothanassis, I.P. Ηydrogen bonding probes of phenol –OH groups: Shielding ranges, solvent effects and temperature coefficients of 1H-NMR shieldings and –OH diffusion coefficients. Org. Biomol. Chem. 2013, 11, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Neratzaki, A.A.; Tsiafoulis, C.G.; Charisiadis, P.; Kontogianni, V.G.; Gerothanassis, I.P. Novel determination of the total phenolic content in crude plant extracts by the use of 1H-NMR of the –OH spectral region. Anal. Chim. Acta 2011, 688, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Charisiadis, P.; Kontogianni, V.G.; Tsiafoulis, C.G.; Tzakos, A.G.; Siskos, M.; Gerothanassis, I.P. 1H-NMR as a structural and analytical tool of intra- and intermolecular hydrogen bonds of phenol-containing natural products and model compounds. Molecules 2014, 19, 13643–13682. [Google Scholar] [CrossRef] [PubMed]
- Dayrit, F.M.; Buenafe, O.E.; Chainani, E.T.; De Vera, I.M. Analysis of monoglycerides, diglycerides, sterols, and free fatty acids in coconut (Cocos nucifera L.) oil by 31P NMR spectroscopy. J. Agric. Food Chem. 2008, 56, 5765–5769. [Google Scholar] [CrossRef] [PubMed]
- Spyros, A.; Dais, P. Application of 31P NMR spectroscopy in food analysis. 1. Quantitative determination of the mono- and diglyceride composition of olive oils. J. Agric. Food Chem. 2000, 48, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Dais, P.; Spyros, A.; Cristophoridou, S.; Hatzakis, E.; Fragaki, G.; Agiomyrgianaki, A.; Salivaras, E.; Siragakis, G.; Daaskalaki, D.; Tasioula-Margari, M.; et al. Comparison of analytical methodologies based on 1H and 31P NMR spectroscopy with conventional methods of analysis for the determination of some olive oil constituents. J. Agric. Food Chem. 2007, 55, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Sacchi, R.; Medina, I.; Aubourg, S.P.; Giudicianni, I.; Paolillo, L.; Addeo, F. Quantitative high resolution 13C NMR analysis of lipids extracted from the wine muscle of Atlantic Tuna (Thunnus alalunga). J. Agric. Food Chem. 1993, 41, 1247–1253. [Google Scholar] [CrossRef]
- Ng, S. Quantitative analysis of partial acylglycerols and free fatty acids in palm oil by 13C Nuclear Magnetic Resonance Spectroscopy. J. Am. Oil Chem. Soc. 2000, 77, 749–755. [Google Scholar] [CrossRef]
- Chan, H.W.; Levett, G. Autoxidation of methyl linoleate. Separation and analysis of isomeric mixtures of methyl linoleate hydroperoxides and methyl hydroxylinoleates. Lipids 1977, 12, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Neff, W.E.; Frankel, E.N.; Selke, E.; Weisleder, D. Photosensitized oxidation of methyl linoleate monohydroperoxides: Hydroperoxy cyclic peroxides, dihydroperoxides, keto esters and volatile thermal decomposition products. Lipids 1983, 18, 868–876. [Google Scholar] [CrossRef]
- Saito, H. Estimation of the oxidative deterioration of fish oils by measurements of Nuclear Magnetic Resonance. Agric. Biol. Chem. 1987, 51, 3433–3435. [Google Scholar]
- Saito, H.; Udagawa, M. Application of NMR to evaluate the oxidative deterioration of brown fish meal. J. Sci. Food Agric. 1992, 58, 135–137. [Google Scholar] [CrossRef]
- Silwood, C.J.L.; Grootveld, M. Application of high-resolution, two-dimensional 1H and 13C nuclear magnetic resonance techniques to the characterization of lipid oxidation products in autoxidized linoleoyl/linolenoylglycerol. Lipids 1999, 34, 741–756. [Google Scholar] [CrossRef] [PubMed]
- Claxson, A.W.D.; Hawkes, G.E.; Richardson, D.P.; Naughton, D.P.; Haywooday, R.M.; Chander, C.L.; Atherton, M.; Lynch, E.J.; Grootveld, M.C. Generation of lipid peroxidation products in culinary oils and fats during episodes of thermal stressing: A high field 1H NMR study. FEBS Lett. 1994, 355, 81–90. [Google Scholar] [CrossRef]
- Haywood, R.M.; Claxson, A.W.D.; Hawkes, G.E.; Richardson, D.P.; Naughton, D.P.; Coumbarides, G.; Hawkes, J.; Lynch, E.J.; Grootveld, M.C. Detection of aldehydes and their conjugated hydroperoxydiene precursors in thermally-stressed culinary oils and fats: Investigations using high resolution proton NMR spectroscopy. Free Radic. Res. 1995, 22, 441–482. [Google Scholar] [CrossRef] [PubMed]
- Guillen, M.D.; Goicoechea, E. Oxidation of corn oil at room temperature: Primary and secondary oxidation products and determination of their concentration in the oil liquid matrix from 1H nuclear magnetic resonance data. Food Chem. 2009, 116, 183–192. [Google Scholar] [CrossRef]
- Porter, N.A.; Mills, K.A.; Carter, R.L. A mechanistic study of oleate autoxidation: Competing peroxyl H-atom abstraction and rearrangement. J. Am. Chem. Soc. 1994, 116, 6690–6696. [Google Scholar] [CrossRef]
- Kuklev, D.V.; Christie, W.W.; Durand, T.; Rossi, J.C.; Vidal, J.P.; Kasyanov, S.P.; Akulin, V.N.; Bezuglov, V.V. Synthesis of keto- and hydroxydienoic compounds from linoleic acid. Chem. Phys. Lipids 1997, 85, 125–134. [Google Scholar] [CrossRef]
- Jie, M.S.F.L.K.; Lam, C.N.W. Reaction of mono-epoxidized conjugated linoleic acid ester with boron trifluoride etherate complex. Lipids 2004, 39, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Guillen, M.D.; Ruiz, A. Study of the oxidative stability of salted and unsalted salmon fillets by 1H nuclear magnetic resonance. Food Chem. 2004, 86, 297–304. [Google Scholar] [CrossRef]
- Guillen, M.D.; Ruiz, A. Formation of hydroperoxy- and hydroxyalkenals during thermal oxidative degradation of sesame oil monitored by proton NMR. Eur. J. Lipid Sci. Technol. 2004, 106, 680–687. [Google Scholar] [CrossRef]
- Guillen, M.D.; Ruiz, A. Monitoring the oxidation of unsaturated oils and formation of oxygenated aldehydes by proton NMR. Eur. J. Lipid Sci. Technol. 2005, 107, 36–47. [Google Scholar] [CrossRef]
- Guillen, M.D.; Goicoechea, E. Detection of primary and secondary oxidation products by Fourier transform infrared spectroscopy (FTIR) and 1H nuclear magnetic resonance (NMR) in sunflower oil during storage. J. Agric. Food Chem. 2007, 55, 10729–10736. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Zhang, J.; Sayre, L.M. Synthesis of six epoxyketooctadecenoic acid (EKODE) isomers, their generation from nonenzymatic oxidation of linoleic acid, and their reactivity with imidazole nucleophiles. J. Org. Chem. 2007, 72, 9471–9480. [Google Scholar] [CrossRef] [PubMed]
- Guillen, M.D.; Uriarte, P.S. Contribution to further understanding of the evolution of sunflower oil submitted to frying temperature in a domestic fryer: Study by 1H nuclear magnetic resonance. J. Agric. Food Chem. 2009, 57, 7790–7799. [Google Scholar] [CrossRef] [PubMed]
- Guillen, M.D.; Uriarte, P.S. Study by 1H NMR spectroscopy of the evolution of extra virgin olive oil composition submitted to frying temperature in an industrial fryer for a prolonged period of time. Food Chem. 2012, 134, 162–172. [Google Scholar] [CrossRef]
- Guillen, M.D.; Uriarte, P.S. Monitoring by 1H nuclear magnetic resonance of the changes in the composition of virgin linseed oil heated at frying temperature. Comparison with the evolution of other edible oils. Food Control 2012, 28, 59–68. [Google Scholar] [CrossRef]
- Guillen, M.D.; Uriarte, P.S. Simultaneous control of the evolution of the percentage in weight of polar compounds, iodine value, acyl groups proportions and aldehydes concentrations in sunflower oil submitted to frying temperature in an industrial fryer. Food Control 2012, 24, 50–56. [Google Scholar] [CrossRef]
- Goicoechea, E.; Guillen, M.D. Analysis of hydroperoxides, aldehydes and epoxides by 1H nuclear magnetic resonance in sunflower oil oxidized at 70 and 100 °C. J. Agric. Food Chem. 2010, 58, 6234–6245. [Google Scholar] [CrossRef] [PubMed]
- Martınez-Yusta, A.; Guillen, M.D. A study by 1H nuclear magnetic resonance of the influence on the frying medium composition of some soybean oil-food combinations in deep-frying. Food Res. 2014, 55, 347–355. [Google Scholar] [CrossRef]
- Skiera, C.; Steliopoulos, P.; Kuballa, T.; Holzgrabe, U.; Diehl, B. 1H-NMR spectroscopy as a new tool in the assessment of the oxidative state in edible oils. J. Am. Oil Chem. Soc. 2012, 89, 1383–1391. [Google Scholar] [CrossRef]
- Charisiadis, P.; Primikyri, A.; Exarchou, V.; Tzakos, A.; Gerothanassis, I.P. Unprecedented ultra-high-resolution hydroxy group 1H-NMR spectroscopic analysis of plant extracts. J. Nat. Prod. 2011, 74, 2462–2466. [Google Scholar] [CrossRef] [PubMed]
- Charisiadis, P.; Tsiafoulis, C.G.; Exarchou, V.; Tzakos, A.G.; Gerothanassis, I.P. Rapid and direct low micromolar NMR method for the simultaneous detection of hydrogen peroxide and phenolics in plant extracts. J. Agric. Food Chem. 2012, 60, 4508–4513. [Google Scholar] [CrossRef] [PubMed]
- Tsiafoulis, C.; Gerothanassis, I.P. A novel NMR method for the determination and monitoring of evolution of hydrogen peroxide in aqueous solution. Anal. Bioanal. Chem. 2014, 406, 3371–3375. [Google Scholar] [CrossRef] [PubMed]
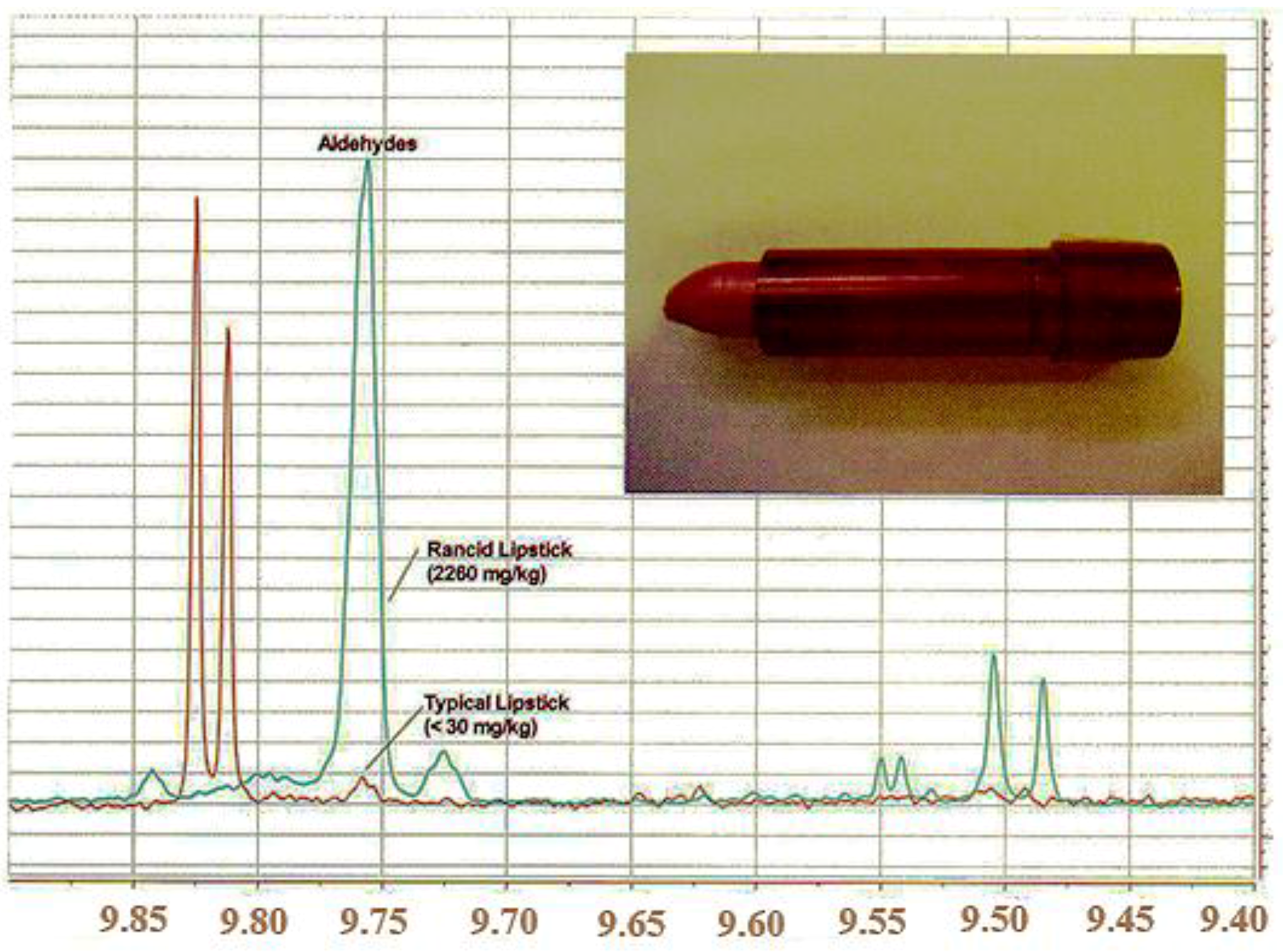
- Lachenmeier, D.W.; Gary, M.; Monakhova, Y.B.; Kuballa, T.; Mildau, G. Rapid NMR screening of total aldehydes to detect oxidative rancidity of vegetable oils and decorative cosmetics. Spectrosc. Eur. 2010, 22, 11–14. [Google Scholar]
- Albert, K. On Line LC-NMR and Related Techniques; John Wiley & Sons: Hoboken, NJ, USA, 2002. [Google Scholar]
- Exarchou, V.; Godejohann, M.; van Beek, T.A.; Gerothanassis, I.P.; Vervoort, J. LC-UV-solid phase extraction-NMR-MS combined with a cryogenic flow probe and its application to the identification of compounds present in Greek oregano. Anal. Chem. 2003, 75, 6288–6294. [Google Scholar] [CrossRef] [PubMed]
- Exarchou, V.; Krucker, M.; van Beek, T.A.; Vervoort, J.; Gerothanassis, I.P.; Albert, K. LC-NMR coupling technology: Recent advancements and applications in natural products analysis. Magn. Reson. Chem. 2005, 43, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Tatsis, E.C.; Boeren, S.; Exarchou, V.; Troganis, A.N.; Vervoort, J.; Gerothanassis, I.P. Identification of the major constituents of hypericum perforatum by LC/SPE/NMR and/or LC/MS. Phytochemistry 2007, 68, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Charisiadis, P.; Kontogianni, V.G.; Tsiafoulis, C.G.; Tzakos, A.G.; Gerothanassis, I.P. Determination of polyphenolic phytochemicals using highly deshielded –OH 1H-NMR signals. Phytochem. Anal. 2017, 28, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Sýkora, J.; Bernášek, P.; Zarevúcká, M.; Kurfürst, M.; Sovová, H.; Schraml, J. High-performance liquid chromatography with nuclear magnetic resonance detection—A method for quantification of α- and γ-linolenic acids in their mixtures with free fatty acids. J. Chromatogr. A 2007, 1139, 152–155. [Google Scholar] [CrossRef] [PubMed]
- Gross, R.W.; Han, Χ. Lipidomics at the interface of structure and function in systems biology. Chem. Biol. 2011, 18, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes, A. Special Issue: Advanced separation methods in food analysis. J. Chromatogr. A 2009, 1216, 7109–7358. [Google Scholar] [CrossRef] [PubMed]
- Laghi, L.; Ricone, G.; Capozzi, F. Nuclear magnetic resonance for foodomics beyond food analysis. Trends Anal. Chem. 2014, 59, 93–102. [Google Scholar] [CrossRef]
- Esslinger, S.; Riedl, J.; Fauhl-Hassek, C. Potential and limitations of non-targeted fingerprinting for authentication of food in official control. Food Res. Int. 2014, 60, 189–204. [Google Scholar] [CrossRef]
- Hidalgo, F.J.; Zamora, R. Edible oil analysis by high-resolution nuclear magnetic resonance spectroscopy: Recent advances and future perspectives. Trends Food Sci. Technol. 2003, 14, 199–506. [Google Scholar] [CrossRef]
- Mannina, L.; Sobolev, A.P. High resolution NMR characterization of olive oils in terms of quality, authenticity and geographical origin. Magn. Reson. Chem. 2011, 49, S3–S11. [Google Scholar] [CrossRef] [PubMed]
- Dais, P.; Hatzakis, E. Analysis of bioactive microconstituents in olives, olive oil and olive leaves by NMR spectroscopy: An overview of the last decade. In Olives and Olive Oil Bioactive Constituents; Boskou, D., Ed.; AOCS Press: Urbana, IL, USA, 2015; pp. 321–324. [Google Scholar]
- Dais, P.; Hatzakis, E. Quality assessment and authentication of virgin olive oil by NMR spectroscopy: A critical review. Anal. Chim. Acta. 2013, 765, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Salces, R.M.; Héberger, K.; Holland, M.V.; Moreno-Rojas, J.M.; Mariani, C.; Bellan, G.; Reniero, F.; Guillou, C. Multivariate analysis of NMR fingerprint of the unsaponifiable fraction of virgin olive oils for authentication purposes. Food Chem. 2010, 118, 956–965. [Google Scholar] [CrossRef]
- Alonso-Salces, R.M.; Moreno-Rojas, J.M.; Holland, M.V.; Reniero, F.; Guillou, C.; Héberger, K. Virgin olive oil authentication by multivariate of 1H NMR fingerprints and δ13C and δ2H data. J. Agric. Food Chem. 2010, 58, 5586–5596. [Google Scholar] [CrossRef] [PubMed]
- Mannina, L.; Marini, F.; Gobbino, M.; Sobolev, A.P.; Capitani, D. NMR and chemometrics in tracing European olive oils: The case study of Ligurian samples. Talanta 2010, 80, 2141–2148. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Salces, R.M.; Segebarth, N.; Garmón-Lobato, S.; Holland, M.V.; Moreno-Rojas, J.M.; Fernández-Pierna, J.A.; Baeten, V.; Fuselli, S.R.; Gallo, B.; Berrueta, L.A.; et al. 1H-NMR and isotopic fingerprinting of olive oil and its unsaponifiable fraction: Geographical origin of virgin olive oils by pattern recognition. Eur. J. Lipid Sci. Technol. 2015, 117, 1991–2006. [Google Scholar] [CrossRef]
- Mannina, L.; D'Imperio, M.; Capitani, D.; Rezzi, S.; Guillou, C.; Mavromoustakos, T.; Vilchez, M.D.; Fernández, A.H.; Thomas, F.; Aparicio, R. 1H NMR-based protocol for the detection of adulterations of refined olive oil with refined hazelnut oil. J. Agric. Food Chem. 2009, 57, 11550–11556. [Google Scholar] [CrossRef] [PubMed]
- Brescia, M.A.; Mazzilli, V.; Sgaramella, A.; Ghelli, S.; Fanizzi, F.P.; Sacco, A. 1H NMR characterization of milk lipids: A comparison between cow and buffalo milk. J. Am. Oil Chem. Soc. 2004, 81, 431–436. [Google Scholar] [CrossRef]
- Schievano, E.; Pasini, G.; Cozzi, G.; Mammi, S. Identification of the production chain of Asiago d’Allevo cheese by nuclear magnetic resonance spectroscopy and principal component analysis. J. Agric. Food Chem. 2008, 56, 7208–7214. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Furihata, K.; Ito-Ishida, M.; Kaminogawa, S.; Tanokura, M. Nondestructive observation of bovine milk by NMR spectroscopy: Analysis of existing states of compounds and detection of new compounds. J. Agric. Food Chem. 2004, 52, 4969–4974. [Google Scholar] [CrossRef] [PubMed]
- Erich, S.; Schill, S.; Annweiler, E.; Waiblinger, H.-U.; Kuballa, T.; Lachenmeier, D.W.; Monakhova, Y.B. Combined chemometric analysis of 1H NMR, 13C NMR and stable isotope data to differentiate organic and conventional milk. Food Chem. 2015, 188, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zheng, N.; Zhao, X.; Zhang, Y.; Han, R.; Yang, J.; Zhao, S.; Li, S.; Guo, T.; Zhang, C.; et al. Metabolomic biomarkers identify differences in milk produced by Holstein cows and other minor dairy animals. J. Proteom. 2016, 136, 174–182. [Google Scholar] [CrossRef] [PubMed]
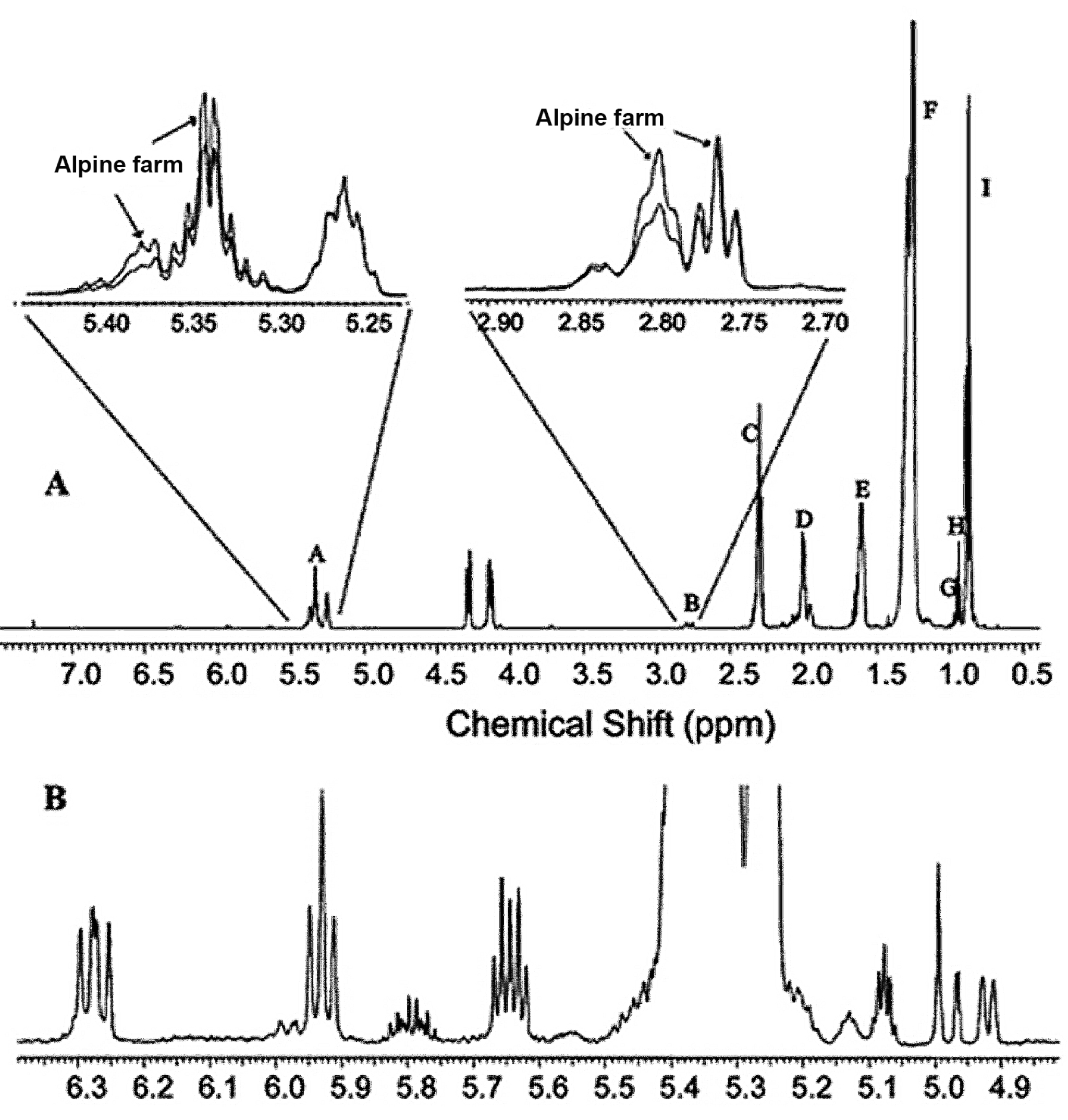
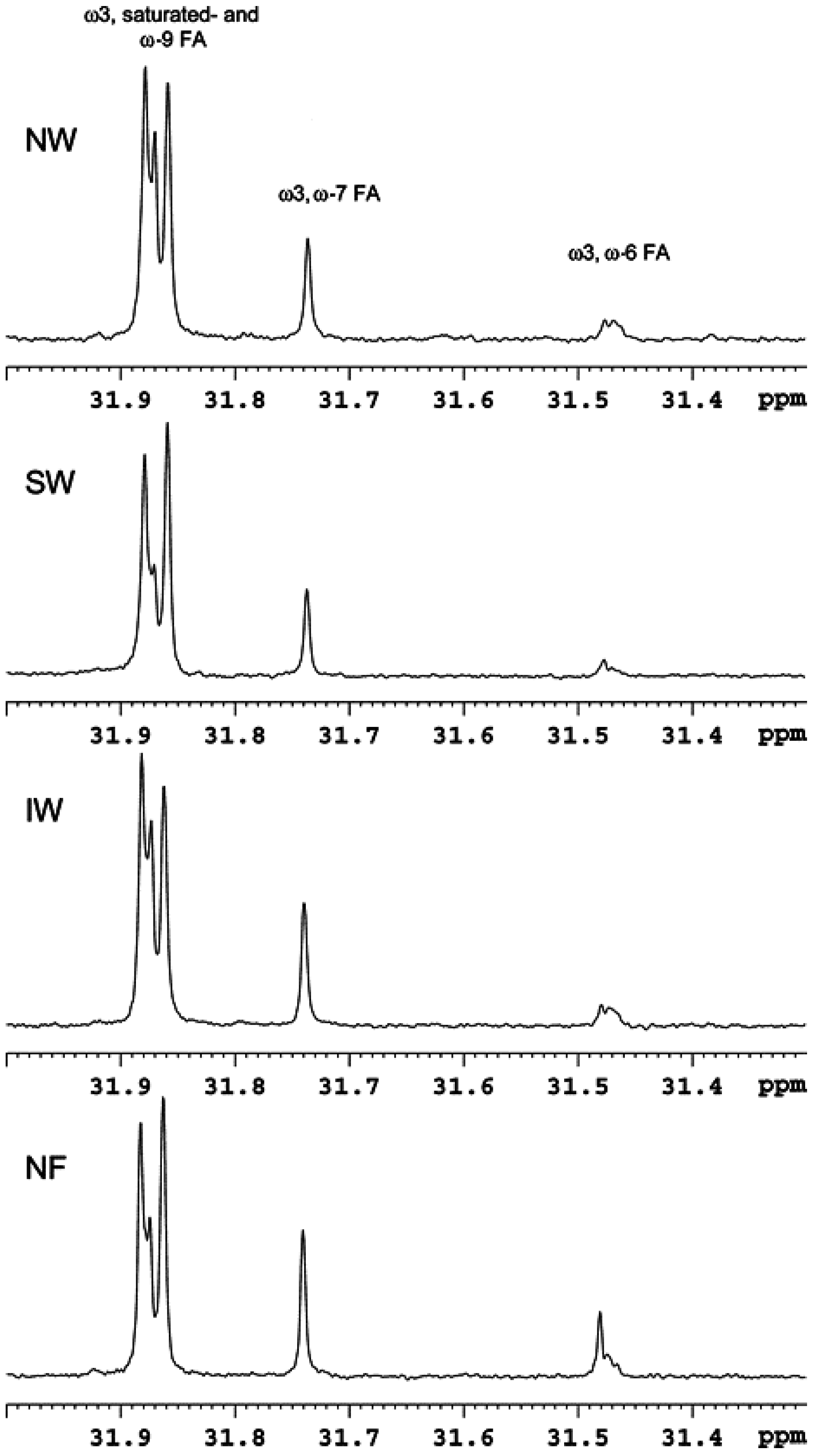
- Aursand, M.; Standal, I.B.; Praël, A.; McEvoy, L.; Irvine, J.; Axelson, D.E. 13C NMR pattern recognition techniques for the classification of atlantic salmon (Salmo salar L.) According to their wild, farmed, and geographical origin. J. Agric. Food Chem. 2009, 57, 3444–3451. [Google Scholar] [CrossRef] [PubMed]
- Standal, I.B.; Axelson, D.E.; Aursand, M. Differentiation of fish oils according to species by 13C-NMR regiospecific analyses of triacyglycerols. J. Am. Oil Chem. Soc. 2009, 86, 401–407. [Google Scholar] [CrossRef]
- Aursand, M.; Standal, I.B.; Axelson, D.E. High-resolution 13C nuclear magnetic resonance spectroscopy pattern recognition of fish oil capsules. J. Agric. Food Chem. 2007, 55, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Rezzi, S.; Giani, I.; Héberger, K.; Axelson, D.E.; Moretti, V.M.; Reniero, F.; Guillou, C. Classification of gilthead sea bream (Sparus aurata) from 1H NMR lipid profiling combined with principal component and linear discriminant analysis. J. Agric. Food Chem. 2007, 55, 9963–9968. [Google Scholar] [CrossRef] [PubMed]
- Burri, L.; Hoem, N.; Monakhova, Y.B.; Diehl, B.W.K. Fingerprinting krill oil by 31P, 1H and 13C NMR spectroscopies. J. Am. Oil Chem. Soc. 2016, 93, 1037–1049. [Google Scholar] [CrossRef]
- Al-Jowder, O.; Casuscelli, F.; Defernez, M.; Kemsley, E.K.; Wilson, R.H.; Colquhoun, I.J. High resolution NMR studies of meat composition and authenticity. In Magnetic Resonance in Food Science: A View to the Future; Webb, G.A., Belton, P.S., Gil, A.M., Delgadillo, I., Eds.; Royal Society of Chemistry: Cambridge, UK, 2001; pp. 232–238. [Google Scholar]
- Jakes, W.; Gerdova, A.; Defernez, M.; Watson, A.; McCallum, C.; Limer, W.; Colquhoun, I.J.; Williamson, D.; Kemsley, E.K. Authentication of beef versus horse meat using 60 MHz 1H NMR spectroscopy. Food Chem. 2015, 175, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zanardi, E.; Caligiani, A.; Padovani, E.; Mariani, M.; Ghidini, S.; Palla, G.; Ianier, A. Detection of irradiated beef by nuclear magnetic resonance lipid profiling combined with chemometric techniques. Meat Sci. 2013, 93, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Renou, J.-P.; Bielicki, G.; Deponge, C.; Gachon, P.; Micol, D.; Ritz, P. Characterization of animal products according to its geographic origin and feeding diet using Nuclear Magnetic Resonance and Isotope Ratio Mass Spectrometry Part II: Beef meat. Food Chem. 2004, 86, 251–256. [Google Scholar] [CrossRef]
- Savorani, F.; Kristensen, M.; Larsen, F.H.; Astrup, A.; Engelsen, S.B. High throughput prediction of chylomicron triglycerides in human plasma by nuclear magnetic resonance and chemometrics. Nutr. Metab. 2010, 7, 43. [Google Scholar] [CrossRef] [PubMed]
- NMR Lipo Profile Test, LipoScience Inc., 2011. Available online: https://www.liposcience.com (accessed on 9 June 2011).
- Mallol, R.; Rodriguez, M.A.; Brezmes, J.; Masana, L.; Correig, X. Human serum/plasma lipoprotein analysis by NMR: Application to the study of diabetic dyslipidemia. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 70, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Sears, B.; Deckelbaum, R.J.; Janiak, M.J.; Shipley, G.G.; Small, D.M. Temperature-dependent carbon-13 nuclear magnetic resonance studies of human serum low density lipoproteins. Biochemhistry 1976, 15, 4151–4157. [Google Scholar] [CrossRef]
- Seip, R.L.; Otvos, J.; Bilbie, C.; Tsongalis, G.J.; Miles, M.; Zoeller, R.; Visich, P.; Gordon, P.; Angelopoulos, T.J.; Pescatello, L.; et al. The effect of apolipoprotein E genotype on serum lipoprotein particle response to exercise. Atherosclerosis 2006, 188, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Burdge, G.C.; Powell, J.; Dadd, T.; Talbot, D.; Civil, J.; Calder, P.C. Acute consumption of fish oil improves postprandial VLDL profiles in healthy men aged 50–65 years. Br. J. Nutr. 2009, 102, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Makinen, V.-P.; Soininen, P.; Forsblom, C.; Parkkonen, M.; Ingman, P.; Kaski, K.; Groop, P.-H.; Korpela, M.A. 1H NMR metabonomics approach to the disease continuum of diabetic complications and premature death. Mol. Syst. Biol. 2008, 4, 167. [Google Scholar] [CrossRef] [PubMed]
- Kostara, C.E.; Papathanasiou, A.; Psychogios, N.; Cung, M.T.; Elisaf, M.S.; Goudevenos, J.; Bairaktari, E.T. NMR-based lipidomic analysis of blood lipoproteins differentiates the progression of coronary heart disease. J. Proteome Res. 2014, 13, 2585–2598. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Tang, H.; Nicholson, J.K.; Lindon, J.C. Use of 1H NMR-determined diffusion coefficients to characterize lipoprotein fractions in human blood plasma. Magn. Reson. Chem. 2002, 40, S83–S88. [Google Scholar] [CrossRef]
- Ala-Korpela, M.; Korhonen, A.; Keisala, J.; Horkko, S.; Korpi, P.; Ingman, L.P.; Jokisaari, J.; Savolainen, M.J.; Kesaniemi, Y.A. 1H NMR-based absolute quantitation of human lipoproteins and their lipid contents directly from plasma. J. Lipid Res. 1994, 35, 2292–2304. [Google Scholar] [PubMed]
- Petersen, M.; Dyrby, M.; Toubro, S.; Engelsen, S.B.; Nørgaard, L.; Pedersen, H.T.; Dyerberg, J. Quantification of lipoprotein subclasses by proton nuclear magnetic resonance–based partial least-squares regression models. Clin. Chem. 2005, 51, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Mallol, R.; Rodríguez, M.A.; Heras, M.; Vinaixa, M.; Cañellas, N.; Brezmes, J.; Plana, N.; Masana, L.; Correig, X. Surface fitting of 2D diffusion-edited 1H NMR spectroscopy data for the characterisation of human plasma lipoproteins. Metabolomics 2011, 7, 572–582. [Google Scholar] [CrossRef]
- Pikula, S.; Bandorowicz, J.; Groves, P. NMR of Lipids. In Nuclear Magnetic Resonance: Volume 44 (Specialist Periodical Reports); Kamienska-Trela, K., Ed.; Royal Society of Chemistry: London, UK, 2015; pp. 385–406. [Google Scholar]
- Brash, A.R.; Boeglin, W.E.; Stec, D.F.; Voehler, M.; Schneider, C.; Cha, J.K. Isolation and characterization of two geometric allene oxide isomers synthesized from 9S-hydroperoxylinoleic acid by cytochrome P450 CYP74C3. J. Biol. Chem. 2013, 288, 20797–20806. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Chen, P.; Véricel, E.; Lelli, M.; Béguin, L.; Lagarde, M.; Guichardant, M. Characterization and biological effects of di-hydroxylated compounds deriving from the lipoxygenation of ALA. J. Lipid Res. 2013, 54, 2083–2094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobson, E.P.; Barrow, C.J.; Kralovec, J.A.; Adcock, J.L. Controlled formation of mono- and dihydroxy-resolvins from EPA and DHA using soybean 15-lipoxygenase. J. Lipid Res. 2013, 54, 1439–1447. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.-H.; Makino, A.; Sudesh, K.; Greimel, P.; Kobayashi, T. Spectroscopic evidence for the unusual stereochemical configuration of an endosome-specific lipid. Angew. Chem. Int. Ed. 2012, 51, 533–535. [Google Scholar] [CrossRef] [PubMed]
- Quehenberger, O.; Dennis, E.A. The human plasma lipidome. N. Engl. J. Med. 2011, 365, 1812–1823. [Google Scholar] [CrossRef] [PubMed]
- Yli-Jama, P.; Meyer, H.E.; Ringstad, J.; Pedersen, J.I. Serum free fatty acid pattern and risk of myocardial infarction: A case-control study. J. Intern. Med. 2002, 251, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.H.; Kleinfeld, A.M. Unbound free fatty acid profiles in human plasma and the unexpected absence of unbound palmitoleate. J. Lipid Res. 2017, 58, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Xe, X.M.; Carter, D.C. Atomic structure and chemistry of human serum albumin. Nature 1992, 358, 209–215. [Google Scholar]
- Sugio, S.; Kashima, A.; Mochizuki, S.; Noda, M.; Kobayashi, K. Crystal structure of human serum albumin at 2.5 Å resolution. Protein Eng. 1999, 12, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Curry, S.; Mandelkow, H.; Brick, P.; Franks, N. Crystal structure of human serum albumin complexed with fatty acid reveals an asymmetric distribution of binding sites. Nat. Struct. Biol. 1998, 5, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Curry, S.; Brick, P.; Franks, N.P. Fatty acid binding to human serum albumin: New insights from crystallographic studies. Biochim. Biophys. Acta. 1999, 1441, 131–140. [Google Scholar] [CrossRef]
- Hamilton, J.A.; Era, S.; Bhamidipati, S.P.; Reed, R.G. Locations of the three primary binding sites for long-chain fatty acids on bovine serum albumin. Proc. Natl. Acad. Sci. USA 1991, 88, 2051–2054. [Google Scholar] [CrossRef] [PubMed]
- Reed, R.G. Location of long chain fatty acid-binding sites of bovine serum albumin by affinity labeling. J. Biol. Chem. 1986, 261, 15619–15624. [Google Scholar] [PubMed]
- Sklar, L.A.; Hudson, B.S.; Simoni, R.D. Conjugated polyene fatty acids as fluorescent probes: Binding to bovine serum albumin. Biochemistry 1977, 16, 5100–5108. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.A.; Cistola, D.P.; Morrisett, J.D.; Sparrow, J.T.; Small, D.M. Interactions of myristic acid with bovine serum albumin: A 13C NMR study. Proc. Natl. Acad. Sci. USA 1984, 81, 3718–3722. [Google Scholar] [CrossRef] [PubMed]
- Parks, J.S.; Cistola, D.P.; Small, D.M.; Hamilton, J.A. Interactions of the carboxyl group of oleic acid with bovine serum albumin: A 13C NMR study. J. Biol. Chem. 1983, 258, 9262–9269. [Google Scholar] [PubMed]
- Cistola, D.P.; Small, D.M.; Hamilton, J.A. Carbon 13 NMR studies of saturated fatty acids bound to bovine serum albumin. II. Electrostatic interactions in individual fatty acid binding sites. J. Biol. Chem. 1987, 262, 10980–10985. [Google Scholar] [PubMed]
- Cistola, D.P.; Small, D.M.; Hamilton, J.A. Carbon 13 NMR studies of saturated fatty acids bound to bovine serum albumin. I. The filling of individual fatty acid binding sites. J. Biol. Chem. 1987, 262, 10971–10979. [Google Scholar] [PubMed]
- Kenyon, M.A.; Hamilton, J.A. 13C NMR studies of the binding of medium-chain fatty acids to human serum albumin. J. Lipid Res. 1994, 35, 458–467. [Google Scholar] [PubMed]
- Simard, J.R.; Zunszain, P.A.; Hamilton, J.A.; Curry, S. Location of high and low affinity fatty acid binding sites on human serum albumin revealed by NMR drug-competition analysis. J. Mol. Biol. 2006, 361, 336–351. [Google Scholar] [CrossRef] [PubMed]
- Krenzel, E.S.; Chen, Z.; Hamilton, J.A. Correspondence of fatty acid and drug binding sites on human serum albumin: A two-dimensional nuclear magnetic resonance study. Biochemistry 2013, 52, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.A. NMR reveals molecular interactions and dynamics of fatty acid binding to albumin. Biochim. Biophys. Acta. 2013, 1830, 5418–5426. [Google Scholar] [CrossRef] [PubMed]
- Claasen, B.; Axmann, M.; Meinecke, R.; Meyer, B. Direct observation of ligand binding to membrane proteins in living cells by a saturation transfer double difference (STDD) NMR spectroscopy method shows a significantly higher affinity of integrin alpha(IIb)beta3 in native platelets than in liposomes. J. Am. Chem. Soc. 2005, 127, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Viegas, A.; Manso, J.; Nobrega, F.L.; Cabrita, E.J. Saturation-transfer difference (STD) NMR: A simple and fast method for ligand screening and characterization of protein binding. J. Chem. Educ. 2011, 88, 990–994. [Google Scholar] [CrossRef]
- Tanoli, A.K.S.; Tanoli, U.N.; Bondancia, M.T.; Usmani, S.; Ul-Haq, Z.; Fernandes, B.J.; Thomasi, S.S.; Ferreira, G.A. Human serum albumin-specific recognition of the natural herbal extract of Stryphnodendron polyphyllum through STD NMR, hyphenations and docking simulation studies. RSC Adv. 2015, 5, 23431–23442. [Google Scholar] [CrossRef]
- Mari, S.; Invernizzi, C.; Spitaleri, A.; Alberici, L.; Ghitti, M.; Bordignon, C.; Traversari, C.; Rizzardi, G.P.; Musco, G. 2D Tr-NOESY experiments interrogate and rank ligand-receptor interactions in living human cancer cells. Angew. Chem. Int. Ed. 2010, 49, 1071–1074. [Google Scholar] [CrossRef] [PubMed]
- Primikyri, A.; Sayyad, N.; Quilici, G.; Vrettos, E.I.; Lim, K.; Chi, S.-W.; Musco, G.; Tzakos, A.; Gerothanassis, I.P. Probing 3′ quercetin-alanine binding to Bcl-2 protein in living human cells with in-cell NMR spectroscopy. 2017; in preparation. [Google Scholar]
- Kaiser, K.A.; Barding, G.A., Jr.; Lavine, C.K. A comparison of metabolite extraction strategies for 1H-NMR-based metabolic profiling using mature leaf tissue from the model plant Arabidopsis thaliana. Mang. Reson. Chem. 2009, 47, S147–S156. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Wu, H.; Tjeerdema, R.S.; Viant, M.R. Evaluation of metabolite extraction strategies from tissue samples using NMR metabolomics. Metabolomics 2007, 3, 55–67. [Google Scholar] [CrossRef]
- Venskutonis, P.R. Effect of drying on the volatile constituents of thyme (Thymus vulgaris L.) and sage (Salvia officinalis L.). Food Chem. 1997, 59, 219–227. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1959, 226, 497–509. [Google Scholar]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Akoka, S.; Barantin, L.; Trierweiler, M. Concentration measurement by proton NMR using the ERETIC method. Anal. Chem. 1999, 71, 2554–2557. [Google Scholar] [CrossRef] [PubMed]
- Castañar, L.; Parella, T. Broadband 1H homodecoupled NMR experiments: Recent developments, method and applications. Magn. Reson. Chem. 2015, 53, 399–426. [Google Scholar] [CrossRef] [PubMed]
- Zangger, K. Pure shift NMR. Prog. Nucl. Magn. Spectrosc. 2015, 86–87, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.H.; Zangge, Κ. Simplifying proton NMR spectra by instant homonuclear broadband decoupling. Angew. Chem. Int. Ed. 2013, 52, 7143–7146. [Google Scholar] [CrossRef] [PubMed]
- Paudel, L.; Adams, R.W.; Kiraly, P.; Aguilar, J.A.; Foroozandeh, M.; Cliff, M.J.; Nilsson, M.; Sandor, P.; Waltho, J.P.; Morris, G.A. Simultaneously enhancing spectral resolution and sensitivity in heteronuclear correlation NMR spectroscopy. Angew. Chem. Int. Ed. 2013, 52, 11616–11619. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Trujillo, M.; Castañar, L.; Monteagudo, E.; Kuhn, L.T.; Nolis, P.; Virgili, A.; Williamsonc, R.T.; Parella, T. Simplifying proton NMR spectra by instant homonuclear broadband decoupling. Chem. Commun. 2014, 50, 10214–10217. [Google Scholar] [CrossRef] [PubMed]
- Glanzer, S.; Zangger, K. Directly decoupled diffusion-ordered NMR spectroscopy for the analysis of compound mixtures. Chem. Eur. J. 2014, 20, 11171–11175. [Google Scholar] [CrossRef] [PubMed]
- Poggetto, D.G.; Castañar, L.; Morris, G.A.; Nillson, M. A new tool for NMR analysis of complex systems: Selective pure shift TOCSY. RSC Adv. 2016, 6, 100063–100066. [Google Scholar] [CrossRef]
- Jeannerat, D. Human- and computer-accessible 2D correlation data for a more reliable structure determination of organic compounds. Future roles of researchers, software developers, spectrometer managers, journal editors, reviewers, publisher and database managers toward artificial-intelligence analysis of NMR spectra. Magn. Reson Chem. 2017, 55, 7–14. [Google Scholar] [PubMed]
- Zani, C.L.; Carroll, A.R. Database for rapid dereplication of known natural products using data from MS and fast NMR experiments. J. Nat. Prod. 2017, 80, 1758–1766. [Google Scholar] [CrossRef] [PubMed]
- Navon, G.; Song, Y.-Q.; Room, T.; Appelt, S.; Taylor, R.E.; Pines, A. Enhancement of solution NMR and MRI with laser-polarized xenon. Science 1996, 271, 1848–1851. [Google Scholar] [CrossRef]
- Ardenkjaer-Larsen, J.H.; Fridlund, B.; Gram, A.; Hansson, G.; Hansson, L.; Lerche, M.H.; Servin, R.; Thaning, M.; Golman, K. Increase in signal-to-noise ratio of >10,000 times in liquid state NMR. Proc. Natl. Acad. Sci. USA 2003, 100, 10158–10163. [Google Scholar] [CrossRef] [PubMed]
- Donovan, K.J.; Frydman, L. HyperBIRD: A sensitivity-enhanced approach to collecting homonuclear-decoupled proton NMR spectra. Angew. Chem. Int. Ed. 2014, 54, 594–598. [Google Scholar] [CrossRef] [PubMed]
- Schlotterbeck, G.; Ceccarelli, S.M. LC-SPE-NMR-MS: A total analysis system for bioanalysis. Bioanalysis 2009, 1, 549–559. [Google Scholar]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational prediction of 1H and 13C chemical shifts: A useful tool for natural product, mechanistic, and synthetic organic chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef] [PubMed]
- Přecechtělová, J.; Novák, P.; Munzarová, M.L.; Kaupp, Μ.; Sklenář, V. Phosphorus chemical shifts in a nucleic acid backbone from combined molecular dynamics and density functional calculations. J. Am. Chem. Soc. 2010, 132, 17139–17148. [Google Scholar] [CrossRef] [PubMed]
- Siskos, M.; Kontogianni, V.G.; Tsiafoulis, C.; Tzakos, A.; Gerothanassis, I.P. Investigation of solute-solvent interactions in phenol compounds: Accurate ab initio calculations of solvent effects on 1H-NMR shieldings. Org. Biomol. Chem. 2013, 11, 7400–7411. [Google Scholar] [CrossRef] [PubMed]
- Siskos, M.G.; Tzakos, A.G.; Gerothanassis, I.P. Accurate ab initio calculations of O–H⋯O and O–H⋯-O proton chemical shifts: Towards elucidation of the nature of the hydrogen bond and prediction of hydrogen bond distances. Org. Biomol. Chem. 2015, 13, 8852–8868. [Google Scholar] [CrossRef] [PubMed]
- Siskos, M.G.; Choudhary, M.I.; Gerothanassis, I.P. Refinement of labile hydrogen positions based on DFT calculations of 1H NMR chemical shifts: Comparison with X-ray and neutron diffraction methods. Org. Biomol. Chem 2017, 15, 4655–4666. [Google Scholar] [CrossRef] [PubMed]
- Apperley, D.C.; Harris, R.K.; Hodgkinson, P. (Eds.) Solid State NMR: Basics Principles and Practices; Momentum Press LLC: New York, NY, USA, 2012. [Google Scholar]
- Elizabeth, S.; Ashbrook, M.; McKay, D. Combining solid-state NMR spectroscopy with first-principles calculations - a guide to NMR crystallography. Chem. Commun. 2016, 52, 7186–7204. [Google Scholar]
- Siskos, M.G.; Choudhary, M.C.; Tzakos, A.G.; Gerothanassis, I.P. 1H NMR chemical shift assignment, structure and conformational elucidation of hypericin with the use of DFT calculations—The challenge of accurate labile hydrogens. Tetrahedron 2016, 72, 8287–8293. [Google Scholar] [CrossRef]
- Siskos, M.G.; Choudhary, M.I.; Gerothanassis, I.P. Hydrogen atomic positions of O–H···O hydrogen bonds in solution and in the solid state: The synergy of quantum chemical calculations with 1H-NMR chemical shifts and X-ray diffraction methods. Molecules 2017, 22, 415. [Google Scholar] [CrossRef] [PubMed]



















































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Common Name | Lipid Notation | Δn Notation | Molecular Formula |
|---|---|---|---|
| Oleic acid | 18:1 (ω-9) | Δ9 | CH3(CH2)7CH=CH(CH2)7COOH |
| Linoleic acid | 18:2 (ω-6) | Δ9,12 | CH3(CH2)4CH=CHCH2CH=CH(CH2)7COOH |
| Arachidonic acid (AA) | 20:4 (ω-6) | Δ5,8,11,14 | CH3(CH2)3(CH2CH=CH)4(CH2)3COOH |
| γ-Linolenic acid | 18:3 (ω-6) | Δ6,9,12 | CH3(CH2)3(CH2CH=CH)3(CH2)COOH |
| Dihomo-γ-linolenic acid | 20:3 (ω-6) | Δ8,11,14 | CH3(CH2)3(CH2CH=CH)4(CH2)3COOH |
| Adrenic acid | 22:4 (ω-6) | Δ7,10,13,16 | CH3(CH2)3(CH2CH=CH)4(CH2)5COOH |
| Common Name | Lipid Notation | Chemical Name |
|---|---|---|
| n/a | 16:3 (ω-3) | all-cis-7,10,13-hexadecatrienoic acid |
| α-Linolenic acid (ALA) | 18:3 (ω-3) | all-cis-9,12,15-octadecatrienoic acid |
| Stearidonic acid (SDA) | 18:4 (ω-3) | all-cis-6,9,12,15-octadecatetraenoic acid |
| Eicosatrienoic acid (ETE) | 20:3 (ω-3) | all-cis-11,14,17-eicosatrienoic acid |
| Eicosatetraenoic acid (ETA) | 20:4 (ω-3) | all-cis-8,11,14,17-eicosatetraenoic acid |
| Eicosapentaenoic acid (EPA) | 20:5 (ω-3) | all-cis-5,8,11,14,17-eicosapentaenoic acid |
| Docosapentaenoic acid (DPA) | 22:5 (ω-3) | all-cis-7,10,13,16,19-docosapentaenoic acid |
| Docosahexaenoic acid (DHA) | 22:6 (ω-3) | all-cis-4,7,10,13,16,19-docosahexaenoic acid |
| Tetracosapentaenoic acid | 24:5 (ω-3) | all-cis-9,12,15,18,21-docosahexaenoic acid |
| Tetracosahexaenoic acid (Nisinic acid) | 24:6 (ω-3) | all-cis-6,9,12,15,18,21-tetracosenoic acid |
| Double Bond | δ-Value | Double Bond | δ-Value |
|---|---|---|---|
| 2-cis | 6.145 5.680 | 2-trans | 6.85 5.72 |
| 3-cis | 5.51 | 3-trans | 5.47 |
| 4-cis | 5.31 | 4-trans | 5.40 |
| 5-cis | 5.32 | 5-trans | 5.34 |
| 6-cis | 5.29 | 6-trans | - |
| 7-cis | 5.28 | 7-trans | - |
| 17-cis | 5.72 4.94 4.88 | 17-trans | - - - |
| Compound | Atom | Functional Group | δ (ppm) | Multiplicity (Hz) |
|---|---|---|---|---|
| (10-cis, 12-cis)-CLA | H11, H12 | –CH= | 6.85 | m |
| (7-trans, 9-cis)-CLA | H8 | –CH= | 6.3 | dd |
| (9-trans, 11-cis)-CLA | H10 | –CH= | 6.29 | t |
| (9-cis, 11-trans)-CLA | H11 | –CH= | 6.28 | dd |
| (9-cis, 11-cis)-CLA | H10, H11 | –CH= | 6.22 | dd |
| (10-trans, 12-cis)-CLA | H11 | –CH= | 6.22 | m |
| (10-cis, 12-cis)-CLA | H10, H13 | –CH= | 6.13 | m |
| (10-trans, 12-trans)-CLA | H11, H12 | –CH= | 5.99 | m |
| (9-trans, 11-trans)-CLA | H10, H11 | –CH= | 5.96 | m |
| (9-cis, 11-trans)-CLA | H10 | –CH= | 5.93 | t |
| (9-trans, 11-cis)-CLA | H9 | –CH= | 5.93 | m |
| (7-trans, 9-cis)-CLA | H9 | –CH= | 5.93 | dd |
| (10-trans, 12-cis)-CLA | H12 | –CH= | 5.87 | t |
| (9-trans, 11-cis)-CLA | H11 | –CH= | 5.66 | t |
| (9-cis, 11-trans)-CLA | H12 | –CH= | 5.65 | m |
| (7-trans, 9-cis)-CLA | H7 | –CH= | 5.64 | m |
| (10-trans, 12-cis)-CLA | H10 | –CH= | 5.58 | m |
| (10-trans, 12-trans)-CLA | H10, H13 | –CH= | 5.56 | m |
| (9-trans, 11-trans)-CLA | H9, H12 | –CH= | 5.54 | m |
| (9-cis, 11-cis)-CLA | H9, H12 | –CH= | 5.40 | m |
| (9-cis, 11-trans)-CLA | H9 | –CH= | 5.33 | m |
| (7-trans, 9-cis)-CLA | H10 | –CH= | 5.31 | m |
| (9-trans, 11-cis)-CLA | H12 | –CH= | 5.30 | m |
| (10-trans, 12-cis)-CLA | H13 | –CH= | 5.23 | m |
| Proton Notation | β-eleostearic Acid: (9-trans, 11-trans, 13-trans) 18:3 | Punicic Acid: (9-cis, 11-trans, 13-cis) 18:3 | α-eleostearic Acid: (9-cis, 11-trans, 13-trans) 18:3 |
|---|---|---|---|
| 9,14-H | 5.66, J = 12.8 Hz | 5.46, J = 10.8 Hz | 5.40 (9-H, J = 10.8 Hz) 5.74 (14-H, J = 14 Hz) |
| 10,13-H | 6.04, J = 12.8 Hz | 6.08, J = 10.8 Hz | 6.01 (10-H, J = 10.8 Hz) 6.12 (13-H, J = 14 Hz) |
| 11,12-H | 6.10, J = 11.6 Hz | 6.48, J = 12.8 Hz | 6.40 (12-H, J = 13.6 Hz) 6.19 (11-H, J = 13.6 Hz) |
| Compound | Functional Group | δ (ppm) | Multiplicity | J Coupling (Hz) |
|---|---|---|---|---|
| Glycerol in TAG | 2′-CHOCO– | 5.26 | m | J1′a,2′ = 5.9 Hz |
| sn-1,2/2,3 DAG | 2′-CHOCO– | 5.08 | m | - |
| sn-1,2 DAG | 1′b-CH2–OCO– | 4.31 | dd | J1′a,1′b = 11.9 Hz J1′a,2′ = 4.5 Hz |
| Glycerol in TAG | 1′a,b-CH2–OCO– | 4.30 | dd | J3′a,3′b = 11.9 Hz J3′a,2′ = 4.4 Hz |
| sn-1,2 DAG | 1′a-CH2–OCO– | 4.23 | dd | J1′a,1′b = 11.9 Hz J3′a,2′ = 5.7 Hz |
| sn-1,3 DAG | 1′b, 3′b-CH2–OCO– | 4.18 | dd | J1′a,1′b = 11.4 Hz J1′a,2′ = 4.4 Hz |
| Glycerol in TAG | –CH2–OCO– | 4.15 | dd | - |
| sn-1,3 DAG | 1′a, 3′a-CH2–OCO– | 4.13 | dd | J1′a,1′b = 11.4 Hz J1′a,2′ = 6.0 Hz |
| sn-1,3 DAG | –CH2–OCO– | 4.03 | m | - |
| sn-1,2/2,3 DAG | HO–CH2–CH– | 3.72 | m | - |
| Glycerol in 1-MAG | 3′a-CH2–OCO- | 3.59 | dd | J3′a,3′b = 11.4 Hz J3′a,2′ = 6.0 Hz |
| Group | δ-Value |
|---|---|
  | 2.72 2.67 2.61 2.76 |
 | (a) 2.78 (b) 2.76 |
 | 2.04 |
 | (a) 1.38 (b) 2.00 |
 | (a) 1.34 (b) 2.00 |
| Group | δ-Value |
|---|---|
 | 3.06–3.07 |
 | 2.98–2.99 |
 | 3.04–3.05 |
 | (a) 3.04 (b) 3.0–3.10 |
 | 2.33 |
| Compound | Functional Group | δ (ppm) | Multiplicity |
|---|---|---|---|
| DHA | –OOC–CH2–CH2– | 2.46 | d |
| All FA | –OOC–CH2–CH2– | 2.33 | t |
| ω-3 | CH3–CH2–CH=CH– | 2.07 | m |
| ω-6 | –CH2–CH=CH– | 2.03 | m |
| UFA | –CH2–CH=CH– | 2.02 | m |
| ω-9 | –CH2–CH=CH– | 2.01 | m |
| α-linolenic acid | –CH3 | 0.98 | t (7.5 Hz) |
| Butyric acid | –CH3 | 0.94 | t (7.4 Hz) |
| Linoleic acid | –CH3 | 0.883 | t (7.0 Hz) |
| ω-9 | –CH3 | 0.880 | t (7.2 Hz) |
| SFA | –CH3 | 0.88-0.87 | t (6.9 Hz) |
| 1H Resonances | Chemical Shift (ppm) | T1 (s) |
|---|---|---|
| Glycerol residue | ||
| 1(3) | 4.32 | 0.42 |
| 1′(3′) | 4.17 | 0.44 |
| 2 | 5.26 | 0.79 |
| Acyl residues | ||
| 2 | 2.30 | 0.53 |
| 3 | 1.57 | 0.73 |
| 4–7 | 1.25 | 0.86 |
| 8 | 2.00 | 1.06 |
| 9, 10 | 5.34 | 1.26 |
| 11 | 2.76 | 1.30 |
| 18 | 0.87 | 1.51 |
| Compound | Carbon | Functional Group | δ (ppm) |
|---|---|---|---|
| (9-cis, 11-trans)-CLA, (9-trans, 11-cis)-CLA, (11-cis, 13-trans)-CLA, (10-trans, 12-cis)-CLA | C1 | OOC–CH2- | 180.09 |
| (8-trans, 10-cis)-CLA | C1 | OOC–CH2- | 180.05 |
| (9-cis, 11-cis)-CLA | C1 | OOC–CH2- | 179.98 |
| (9-trans, 11-trans)-CLA | C1 | OOC–CH2- | 179.86 |
| FFA | C1 | OOC–CH2- | 178.04 |
| Unsaturated FA in sn-1,3 of DAG | C1 | –CH2–OOC–CH2- | 173.32 |
| FA in sn-1 (sn-3) of 1,2 (2,3) DAG | C1 | –CH2–OOC–CH2- | 173.18 |
| FA in sn-2 of 1,2 (2,3) DAG | C1 | –CH–OOC–CH2- | 173.08 |
| Unsaturated FA in sn-1,3 of TAG | C1 | –CH2–OOC–CH2- | 172.70 |
| Butyric acid in sn-1,3 of TAG | C1 | –CH2–OOC–CH2- | 172.60 |
| Unsaturated FA in sn-2 of TAG | C1 | –CH–OOC–CH2- | 172.38 |
| Carbon Atom | δ (ppm) a | NOE (1 + η) | Τ1 (s) |
|---|---|---|---|
| C1 sn-1(3)-Tripalmitin | 173.10 | 1.77 | 5.6 |
| Triolein | 173.07 | 1.781 | 5.6 |
| Trilinolein | 173.06 | 1.73 | 5.4 |
| C1 sn-2-Tripalmitin | 172.70 | 1.74 | 3.9 |
| Triolein | 172.67 | 1.67 | 4.5 |
| Trilinolein | 172.66 | 1.68 | 4.4 |
| Compound | Carbon | Functional Group | δ (ppm) |
|---|---|---|---|
| Caproleic acid | C9 | –CH=CH2 | 138.70 |
| (9-cis, 11-trans)-CLA | C12 | –CH=CH | 135.80 |
| (11-cis, 13-trans)-CLA | C14 | –CH=CH | 134.66 |
| (10-trans, 12-cis)-CLA | C10 | –CH=CH | 134.58 |
| (9-trans, 11-cis)-CLA | C9 | –CH=CH | 134.51 |
| (8-trans, 10-cis)-CLA | C8 | –CH=CH | 134.32 |
| (9-trans, 11trans)-CLA | C12 | –CH=CH | 132.53 |
| (9-trans, 11-trans)-CLA | C9 | –CH=CH | 132.21 |
| (9-cis, 11-cis)-CLA | C12 | –CH=CH | 132.19 |
| (9-cis, 11-cis)-CLA | C9 | –CH=CH | 131.87 |
| All ω-3 FA | ω3 | –CH=CH | 131.66 |
| (9-trans, 11-trans)-CLA | C10 | –CH=CH | 130.46 |
| (9-trans, 11trans)-CLA | C11 | –CH=CH | 130.30 |
| (8-trans, 10-cis)-CLA | C11 | –CH=CH | 130.27 |
| (9-trans, 11-cis)-CLA | C12 | –CH=CH | 130.17 |
| (10-trans, 12-cis)-CLA | C13 | –CH=CH | 130.16 |
| (11-cis, 13-trans)-CLA | C11 | –CH=CH | 130.04 |
| Linoleic acid, Linolenic acid | C13, C9 | –CH=CH | 129.89 |
| (9-cis, 11-trans)-CLA | C9 | –CH=CH | 129.89 |
| Linoleic acid | C9 | –CH=CH | 129.51–129.49 |
| (9-cis, 11-trans)-CLA | C10 | –CH=CH | 128.73 |
| (11-cis, 13-trans)-CLA | C12 | –CH=CH | 128.66 |
| (10-trans, 12-cis)-CLA | C12 | –CH=CH | 128.60 |
| (9-trans, 11-cis)-CLA | C11 | –CH=CH | 128.57 |
| (8-trans, 10-cis)-CLA | C10 | –CH=CH | 128.54 |
| Linolenic acid | C13, C12 | –CH=CH | 127.97–127.92 |
| Linoleic acid | C10 | –CH=CH | 127.77–127.76 |
| Linoleic acid | C12 | –CH=CH | 127.59–127.58 |
| Linolenic acid | C10 | –CH=CH | 127.46–127.44 |
| All ω-3 FA | ω4 | –CH=CH | 126.77 |
| (8-trans, 10-cis)-CLA | C9 | –CH=CH | 125.83 |
| (9-trans, 11-cis)-CLA | C10 | –CH=CH | 125.72 |
| (10-trans, 12-cis)-CLA | C11 | –CH=CH | 125.70 |
| (11-cis, 13-trans)-CLA | C13 | –CH=CH | 125.65 |
| (9-cis, 11-trans)-CLA | C11 | –CH=CH | 125.58 |
| (9-cis, 11-cis)-CLA | C10 | –CH=CH | 123.72 |
| (9-cis, 11-cis)-CLA | C11 | –CH=CH | 123.55 |
| Caproleic acid | C10 | –CH=CH2 | 114.05 |
| Carbon Notation | β-eleostearic Acid: (trans-9, trans-11, trans-13) 18:3 | Punicic Acid: (cis-9, trans-11, cis-13) 18:3 | α-eleostearic Acid: (cis-9, trans-11, trans-13) 18:3 |
|---|---|---|---|
| C-9 | 134.46 | 132.69 | 131.75 |
| C-10 | 130.87 | 128.82 | 128.72 |
| C-11 | 130.51 | 127.94 | 132.83 |
| C-12 | 130.41 | 127.79 | 126.00 |
| C-13 | 130.73 | 128.71 | 130.53 |
| C-14 | 134.23 | 132.46 | 135.17 |
| Δδ | 7-cis, 9-trans | 8-cis, 10-trans | 9-cis, 11-trans | 10-cis, 12-trans | 11-cis, 13-trans |
|---|---|---|---|---|---|
| C1ol–C2ol | 0.45 | 0.83 | 1.07 | 1.22 | 1.30 |
| C4ol–C3ol | 9.43 | 9.25 | 9.14 | 9.06 | 8.93 |
| Compound | Carbon | Functional Group | δ (ppm) |
|---|---|---|---|
| Glycerol in 1,2/2,3 DAG | –CH–OOC– | 71.85 | |
| Glycerol in TAG | –CH–OOC– | 68.72 | |
| Glycerol in 1,3 DAG | HO-CH–(CH2)2 | 67.81 | |
| Glycerol in 1,3 DAG | –CH2–OOC– | 64.75 | |
| Glycerol in 1,2/2,3 DAG | HO–CH2–CH– | 62.02 | |
| Glycerol in TAG | –CH2–OOC– | 61.83 | |
| Glycerol in 1,2 DAG | –CH2–OOC– | 60.83 |
| Compound | Carbon | Functional Group | δ (ppm) |
|---|---|---|---|
| Butyric acid | C2 | –OOC–CH2–CH2– | 35.62 |
| (9-trans, 11-cis)-CLA | C2 | –OOC–CH2–CH2– | 34.10 |
| (9-cis, 11-cis)-CLA, (11-cis, 13-trans)-CLA, (10-trans, 12-cis)-CLA | C2 | –OOC–CH2–CH2– | 34.06 |
| (9-cis, 11-trans)-CLA | C2 | –OOC–CH2–CH2– | 34.05 |
| (18-trans, 10-cis)-CLA | C2 | –OOC–CH2–CH2– | 34.03 |
| (9-trans, 11-tans)-CLA | C2 | –OOC–CH2–CH2– | 34.00 |
| All FA except butyric in sn-2 of TAG | C2 | –OOC–CH2–CH2– | 33.92 |
| All FA except butyric in sn-1,3 of TAG | C2 | –OOC–CH2–CH2– | 33.76 |
| (9-cis, 11-trans)-CLA | C13 | –CH2–CH=CH– | 32.90 |
| (10-trans, 12-cis)-CLA | C9 | –CH2–CH=CH– | 32.87 |
| (9-trans, 11-cis)-CLA | C8 | –CH2–CH=CH– | 32.86 |
| (8-trans, 10-cis)-CLA | C7 | –CH2–CH=CH– | 32.78 |
| (9-trans, 11-trans)-CLA | C13 | –CH2–CH=CH– | 32.63 |
| (11-cis, 13-trans)-CLA | C15 | –CH2–CH=CH– | 32.57 |
| (9-trans, 11-trans)-CLA | C8 | –CH2–CH=CH– | 32.56 |
| (8-trans, 10-cis)-CLA | C16 | –CH2–CH2–CH3 | 31.87 |
| (9-trans, 11-trans)-CLA, (9-trans, 11-cis)-CLA | C16 | –CH2–CH2–CH3 | 31.77 |
| (9-cis, 11-cis)-CLA, (9-cis, 11-trans)-CLA | C16 | –CH2–CH2–CH3 | 31.76 |
| (11-cis, 13-trans)-CLA | C16 | –CH2–CH2–CH3 | 31.59 |
| (10-trans, 12-cis)-CLA | C16 | –CH2–CH2–CH3 | 31.50 |
| Linoleic acid | ω3 | –CH2–CH2–CH3 | 31.32 |
| (8-trans, 10-cis)-CLA | C13 | –CH2– | 29.76 |
| (11-cis, 13-trans)-CLA | C9 | –CH2– | 29.73 |
| (9-trans, 11-cis)-CLA | C14 | –CH2– | 29.73 |
| (9-cis, 11-cis)-CLA | C14 | –CH2– | 29.64 |
| (9-cis, 11-cis)-CLA | C7 | –CH2– | 29.57 |
| All FA | –(CH2)n– | 29.56–28.73 | |
| (10-trans, 12-cis)-CLA | C15 | –CH2– | 29.44 |
| (9-trans, 11-trans)-CLA | C14 | –CH2– | 29.42 |
| (9-cis, 11-trans)-CLA | C14 | –CH2– | 29.40 |
| (10-trans, 12-cis)-CLA | C8 | –CH2– | 29.39 |
| (9-trans, 11-trans)-CLA | C7 | –CH2– | 29.36 |
| (10-trans, 12-cis)-CLA | C5, C6, C7 | –CH2– | 29.16–29.29 |
| (9-trans, 11-cis)-CLA | C5, C6, C7 | –CH2– | 29.13–29.45 |
| (9-cis, 11-trans)-CLA | C5, C6, C7 | –CH2– | 29.12–29.67 |
| (11-cis, 13-trans)-CLA | C4, C5, C6, C7, C8 | –CH2– | 29.07–29.45 |
| (10-trans, 12-cis)-CLA | C4 | –CH2– | 29.06 |
| (9-cis, 11-trans)-CLA | C4 | –CH2– | 29.03 |
| (9-cis, 11-cis)-CLA | C4, C5, C6, C15 | –CH2– | 28.99–29.11 |
| (9-trans, 11-cis)-CLA | C4, C15 | –CH2– | 28.97 |
| (9-cis, 11-trans)-CLA | C15 | –CH2– | 28.93 |
| (9-trans, 11-trans)-CLA | C4, C5, C6, C15 | –CH2– | 28.92–29.10 |
| (8-trans, 10-cis)-CLA | C4, C5, C6, C14, C15 | –CH2– | 28.81–29.25 |
| (9-trans, 11-cis)-CLA | C13 | –CH2–CH=CH– | 27.72 |
| (8-trans, 10-cis)-CLA | C12 | –CH2–CH=CH– | 27.71 |
| (11-cis, 13-trans)-CLA | C10 | –CH2–CH=CH– | 27.69 |
| (10-trans, 12-cis)-CLA | C14 | –CH2–CH=CH– | 27.68 |
| (9-cis, 11-trans)-CLA | C8 | –CH2–CH=CH– | 27.65 |
| (9-cis, 11-cis)-CLA | C8 | –CH2–CH=CH– | 27.52 |
| (9-cis, 11-cis)-CLA | C13 | –CH2–CH=CH– | 27.43 |
| Unsaturated FA | –CH2–CH=CH– | 26.91–26.88 | |
| PUFA | –CH=CH–CH2–CH=CH– | 25.34 | |
| (9-trans, 11-cis)-CLA | C3 | –CH2– | 24.95 |
| (11-cis, 13-trans)-CLA | C3 | –CH2– | 24.69 |
| (9-cis, 11-cis)-CLA | C3 | –CH2– | 24.68 |
| (9-trans, 11-trans)-CLA, (9-cis, 11-trans)-CLA, (10-trans,12-cis)-CLA | C3 | –CH2– | 24.67 |
| (8-trans, 10-cis)-CLA | C3 | –CH2– | 24.63 |
| All FA except butyric | –OOC–CH2–CH2– | 24.58 | |
| (8-trans, 10-cis)-CLA | C17 | –CH2–CH3 | 22.68 |
| (9-trans, 11-cis)-CLA | C17 | –CH2–CH3 | 22.65 |
| (9-cis, 11-cis)-CLA | C17 | –CH2–CH3 | 22.64 |
| (9-trans, 11-trans)-CLA, (9-cis, 11-trans)-CLA | C17 | –CH2–CH3 | 22.63 |
| (10-trans, 12-cis)-CLA | C17 | –CH2–CH3 | 22.57 |
| All FA except ω-3 | ω2 | –CH2–CH3 | 22.48 |
| (11-cis, 13-trans)-CLA | C17 | –CH2–CH3 | 22.29 |
| ω-3 | ω2 | –CH2–CH3 | 20.13 |
| Butyric acid | C3 | –CH2–CH3 | 18.10 |
| (9-trans, 11-cis)-CLA | C18 | –CH3 | 14.12 |
| ω-3 | ω1 | –CH3 | 14.11 |
| (8-trans, 10-cis)-CLA | C18 | –CH3 | 14.11 |
| (9-trans, 11-trans)-CLA, (9-cis, 11-trans)-CLA | C18 | –CH3 | 14.10 |
| (9-cis, 11-cis)-CLA | C18 | –CH3 | 14.09 |
| (10-trans, 12-cis)-CLA | C18 | –CH3 | 14.06 |
| (11-cis, 13-trans)-CLA | C18 | –CH3 | 13.95 |
| Saturated | ω-1 | –CH3 | 13.96 |
| ω-9 | ω-1 | –CH3 | 13.95 |
| ω-7 | ω-1 | –CH3 | 13.94 |
| ω-6 | ω-1 | –CH3 | 13.91 |
| Functional Group | Atom | δ (ppm) | Multiplicity |
|---|---|---|---|
| HOOC– | H1 | 10.43 | a |
| –CH= | H11 | 6.28 | dd |
| –CH= | H10 | 5.93 | t |
| –CH= | H12 | 5.65 | m |
| –CH= | H9 | 5.33 | m |
| –CH2-COOH | H2 | 2.35 | t |
| –CH2–CH=CH– | H8, H13 | 2.13 | m |
| –CH2-CH2–COOH | H3 | 1.65 | m |
| –CH2– | H4–H7, H14–H17 | 1.25–1.35 | m |
| –CH3 | H18 | 0.87 | t |
| 1H | δΗ (ppm) | 13C | δc (ppm) |
|---|---|---|---|
| H1 | 10.43 | C1 | 179.0 |
| H2 | 2.35 | C2 | 34.2 |
| H3 | 1.65 | C3 | 24.9 |
| H4 | 1.31 | C4 | 29.5 |
| H5 | 1.32 | C5 | 29.4 |
| H6 | 1.34 | C6 | 29.6 |
| H7 | 1.36 | C7 | 29.7 |
| H8 | 2.1 | C8 | 28.1 |
| H9 | 5.33 | C9 | 129.6 |
| H10 | 5.93 | C10 | 128.4 |
| H11 | 6.28 | C11 | 125.3 |
| H12 | 5.65 | C12 | 134.8 |
| H13 | 2.08 | C13 | 33.4 |
| H14 | 1.30 | C14 | 29.1 |
| H15 | 1.28 | C15 | 28.9 |
| H16 | 1.27 | C16 | 32.0 |
| H17 | 1.26 | C17 | 23.1 |
| H18 | 0.87 | C18 | 14.3 |
| Common Name | Linnaean Name | ω-3 (%) |
|---|---|---|
| Chia | Salvia hispanica | 64 |
| Kiwifruit | Actinidia chinensis | 62 |
| Perilla | Perilla frutescens | 58 |
| Flax | Linum usitatissimum | 55 |
| Lingonberry | Vaccinium vitis-idaea | 49 |
| Camelina | Camelina sativa | 36 |
| Purslane | Portulaca oleracea | 35 |
| Black Raspberry | Rubus occidentalis | 33 |
| Sample | Analyte/Units | 1D TOCSY a | 1H-NMR a | Relative Deviation b (%) | GC-MS | Relative Deviation c (%) |
|---|---|---|---|---|---|---|
| 1 | Caproleic acid/mM | 1.02 ± 0.03 | 0.97 ± 0.02 a | 5.15 | ||
| Caproleic acid/% of the lipid fraction | 0.26 ± 0.01 | 0.28 | −7.14 | |||
| 2 | Caproleic acid/mM | 1.92 ± 0.06 | 1.83 ± 0.06 a | 1.75 | ||
| % of the lipid fraction | 0.74 ± 0.01 | 0.68 | 8.82 |
| Signal | Chemical Shift (ppm) | Multiplicity | Functional Group |
|---|---|---|---|
| Primary Oxidation Compounds | |||
| –CH=CH–CH=CH– | 6.58 | dddd | (Z,E)-conjugated double |
| 6.00 | ddtd | bonds associated with | |
| 5.56 | ddm | hydroperoxides (OOH) | |
| 5.51 | dtm | ||
| –CH=CH–CH=CH– | 6.45 | ddd | (Z,E)-conjugated double |
| 5.94 | dd | bonds associated with | |
| 5.64 | dd | hydroxides (OH) | |
| 5.40 | ddt | ||
| –CH=CH–CH=CH– | 6.27 | ddm | (E,E)-conjugated double |
| 6.06 | ddtd | bonds associated with | |
| 5.76 | dtm | hydroperoxides (OOH) | |
| 5.47 | ddm | ||
| –CHOOH–CH=CH– | 5.72 | m | Double bond associated with |
| hydroperoxides (OOH) | |||
| –OOH | 8.3 to 8.9 | - | Hydroperoxide group |
| Secondary or Further Oxidation Compounds | |||
| Aldehydes | |||
| –CHO | 9.49 | d | (E)-2-alkenals |
| –CHO | 9.52 | d | (E,E)-2,4-alkadienals |
| –CHO | 9.55 | d | 4,5-epoxy-2-alkenals |
| –CHO | 9.57 | d | 4-hydroxy-(E)-2-alkenals |
| –CHO | 9.58 | d | 4-hydroperoxy-(E)-2-alkenals |
| –CHO | 9.60 | d | (Z,E)-2,4-alkadienals |
| –CHO | 9.75 | t | n-alkanals |
| –CHO | 9.78 | t | 4-oxo-alkanals |
| –CHO | 9.79 | t | n-alkanals of low molecular |
| weight (ethanal and propanal) | |||
| Alcohols | |||
| –CHOH–CHOH– | 3.43 | m | 9,10-dihydroxy-12-octadecenoate |
| (leukotoxindiol) | |||
| –CHOH– | 3.54–3.59 | m | secondary alcohols |
| –CH2OH– | 3.62 | t | primary alcohols |
| Epoxides | |||
| –CHOHC– | 2.63 | m | (E)-9,10-epoxystearate |
| –CHOHC– | 2.88 | m | (Z)-9,10-epoxystearate |
| –CHOHC– | 2.90 | m | 9,10-epoxy-octadecanoate; |
| 9,10-epoxy-12- | |||
| octadecenoate | |||
| (leukotoxin); 12,13-epoxy- | |||
| 9-octadecenoate | |||
| (isoleukotoxin) | |||
| –CHOHC–CHOHC– | 2.90 | m | 9,10–12,13-diepoxy |
| octadecanoate | |||
| –CHOHC–CH2–CHOHC– | 3.10 | m | 9,10–12,13-diepoxy |
| octadecanoate | |||
| Ketones and Unidentified | |||
| O=C<CH=CH– | 6.08 | dt | Double bond conjugated with a keto group |
| 6.82 | m | ||
| Unidentified | 7.50 | - | Unidentified |
| Unidentified | 8.10 | - | Unidentified |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alexandri, E.; Ahmed, R.; Siddiqui, H.; Choudhary, M.I.; Tsiafoulis, C.G.; Gerothanassis, I.P. High Resolution NMR Spectroscopy as a Structural and Analytical Tool for Unsaturated Lipids in Solution. Molecules 2017, 22, 1663. https://doi.org/10.3390/molecules22101663
Alexandri E, Ahmed R, Siddiqui H, Choudhary MI, Tsiafoulis CG, Gerothanassis IP. High Resolution NMR Spectroscopy as a Structural and Analytical Tool for Unsaturated Lipids in Solution. Molecules. 2017; 22(10):1663. https://doi.org/10.3390/molecules22101663
Chicago/Turabian StyleAlexandri, Eleni, Raheel Ahmed, Hina Siddiqui, Muhammad I. Choudhary, Constantinos G. Tsiafoulis, and Ioannis P. Gerothanassis. 2017. "High Resolution NMR Spectroscopy as a Structural and Analytical Tool for Unsaturated Lipids in Solution" Molecules 22, no. 10: 1663. https://doi.org/10.3390/molecules22101663
APA StyleAlexandri, E., Ahmed, R., Siddiqui, H., Choudhary, M. I., Tsiafoulis, C. G., & Gerothanassis, I. P. (2017). High Resolution NMR Spectroscopy as a Structural and Analytical Tool for Unsaturated Lipids in Solution. Molecules, 22(10), 1663. https://doi.org/10.3390/molecules22101663