Practical and Sustainable Synthesis of Optically Pure Levocabastine, a H1 Receptor Antagonist

Abstract
:
1. Introduction
2. Results and Discussion
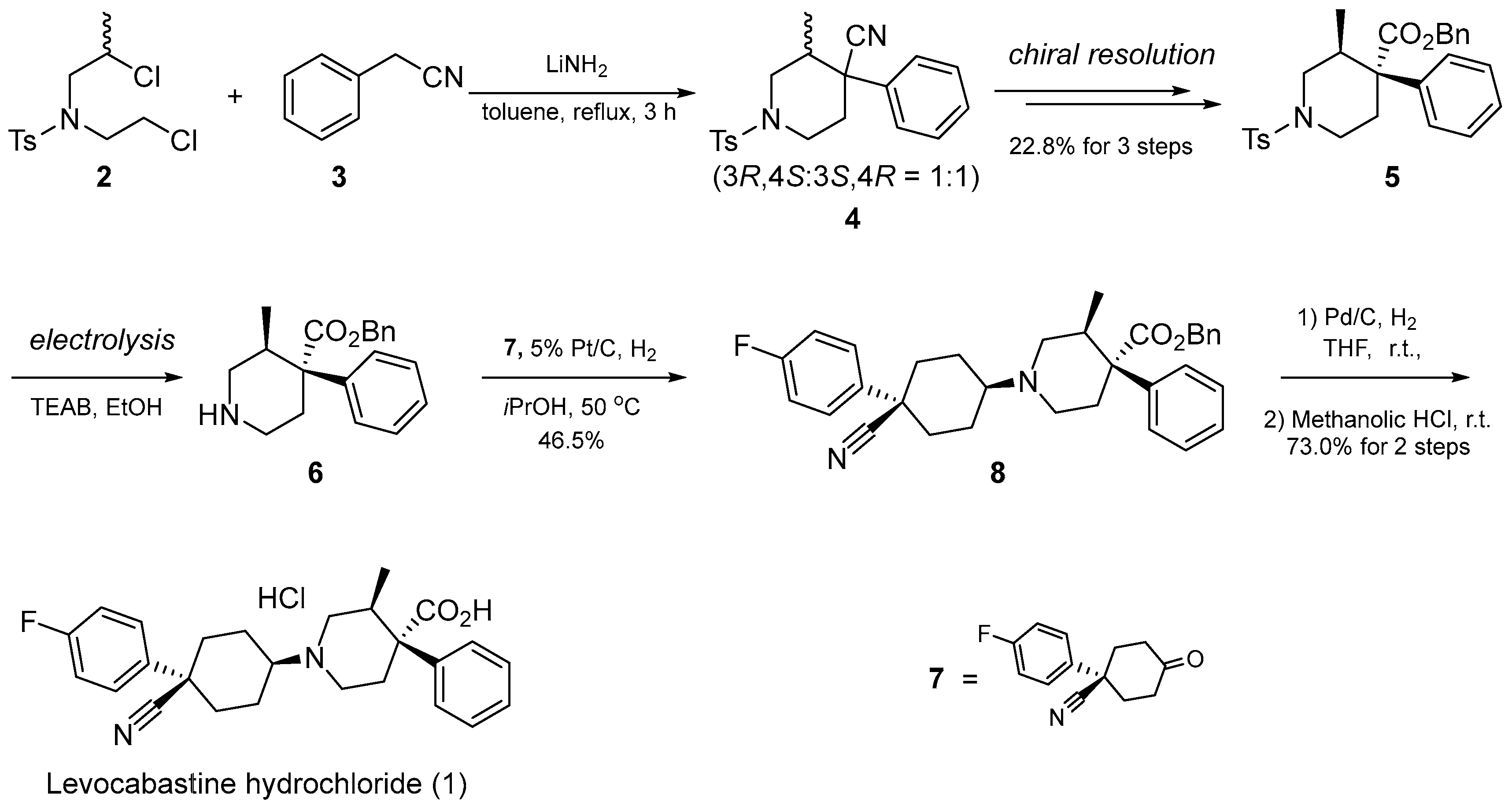
2.1. Synthesis of Optically Pure Intermediate 5
2.2. Deprotection of Tosylate 5
2.3. Completion of the Levocabastine Hydrochloride Synthesis
3. Materials and Methods
3.1. General Information
3.2. Experimental Part
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Maslinski, C.; Fogel, W.A. Catabolism of histamine. Handb. Exp. Pharmacol. 1991, 97, 165–189. [Google Scholar]
- Pearce, F.L. Biological effects of histamine: An overview. Agents Actions 1991, 33, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.J. Distribution, properties, and functional characteristics of three classes of histamine receptor. Pharmacol. Rev. 1990, 42, 45–83. [Google Scholar] [PubMed]
- Leurs, R.; Church, M.K.; Taglialatela, M. H1-antihistamines: Inverse agonism, anti-inflammatory actions and cardiac effects. Clin. Exp. Allergy 2002, 32, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Wade, L.; Bielory, L.; Rudner, S. Ophthalmic antihistamines and H1–H4 receptors. Curr. Opin. Allergy Clin. Immunol. 2012, 12, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Simons, F.E.R.; Simons, K.J. The pharmacology and use of H1-receptor-antagonist drugs. N. Engl. J. Med. 1994, 330, 1663–1670. [Google Scholar] [PubMed]
- Akdis, C.A.; Blaser, K. Histamine in the immune regulation of allergic inflammation. J. Allergy Clin. Immunol. 2003, 112, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.; Rolli-Derkinderen, M.; Arock, M.; Dy, M. Trends in histamine research: New functions during immune responses and hematopoiesis. Trends Immunol. 2002, 23, 255–263. [Google Scholar] [CrossRef]
- Le Coniat, M.; Traiffort, E.; Ruat, M.; Arrang, J.M.; Berger, R. Chromosomal localization of the human histamine H1-receptor gene. Am. J. Hum. Genet. 1994, 94, 186–188. [Google Scholar] [CrossRef]
- Oda, T.; Morikawa, N.; Saito, Y.; Masuho, Y.; Matsumoto, S. Molecular Cloning and Characterization of a Novel Type of Histamine Receptor Preferentially Expressed in Leukocytes. J. Biol. Chem. 2000, 275, 36781–36786. [Google Scholar] [CrossRef] [PubMed]
- Lovenberg, T.W.; Roland, B.L.; Wilson, S.; Jiang, X.; Pyati, J.; Huvar, A.; Jackson, M.R.; Erlander, M.G. Cloning and functional expression of the human histamine H3 receptor. Mol. Pharmacol. 1999, 55, 1101–1107. [Google Scholar] [PubMed]
- Bakker, R.A.; Schoonus, S.B.; Smit, M.J.; Timmerman, H.; Leurs, R. Histamine H1-Receptor Activation of Nuclear Factor-κB: Roles for Gβγ- and Gαq/11-Subunits in Constitutive and Agonist-Mediated Signaling. Mol. Pharmacol. 2001, 60, 1133–1142. [Google Scholar] [PubMed]
- Pipkorn, U.; Bende, M.; Hedner, J.; Hedner, T. A Double-Blind Evaluation of Topical Levocabastine, a New Specific H1 Antagonist in Patients with Allergic Conjunctivitis. Allergy 1985, 40, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, M. Topical levocabastine—A review of therapeutic efficacy compared with topical sodium cromoglycate and oral terfenadine on days with high pollen counts. Mediat. Inflamm. 1995, 4, S21–S25. [Google Scholar] [CrossRef] [PubMed]
- Mösges, R.; Spaeth, J.; Klimek, L. Efficacy and tolerability of levocabastine and azelastine nasal sprays for the treatment of allergic rhinitis. Mediat. Inflamm. 1995, 4, S11–S15. [Google Scholar] [CrossRef] [PubMed]
- Raymond, A.S.; Marcel, G.M.L.; Joannes, J.M.W. 1-(Cyclohexyl)-4-aryl-4-piperidinecarboxylic acid derivatives. U.S. Patent 4,369,184, 29 September 1980. [Google Scholar]
- Jonczyk, A.; Fedorynski, M.; Zdrojewski, T.; Kielbasinska, W. (−)-3S-[1(cis)-3,4]]-1-[4-cyjano-4-(4-fluorofenylo)cykloheksylo]-3-methyl-f-fenylo-4-piperydynokarboksylowego. Poland Patent 176214. 31 May 1999. [Google Scholar]
- Senboku, H.; Nakahara, K.; Fukuhara, T.; Hara, S. Hg cathode-free electrochemical detosylation of N,N-disubstituted p-toluenesulfonamides: Mild, efficient, and selective removal of N-tosyl group. Tetrahedron Lett. 2010, 51, 435–438. [Google Scholar] [CrossRef]
- Yasuhara, A.; Kameda, M.; Sakamoto, T. Selective monodesulfonylation of N,N-disulfonylarylamines with tetrabutylammonium fluoride. Chem. Pharm. Bull. 1999, 47, 809–812. [Google Scholar] [CrossRef]
- Sophia, S.M.; Panayiotis, A.K. Detosylation of 3-amino-1-tosylindole-2-carbonitriles using DBU and thiophenol. Tetrahedron 2010, 66, 3016–3023. [Google Scholar]
- Knowles, H.S.; Parsons, A.F.; Pettifer, R.M.; Rickling, S. Desulfonylation of amides using tributyltin hydride, samarium diiodide or zinc/titanium tetrachloride. A comparison of methods. Tetrahedron 2000, 56, 979–988. [Google Scholar] [CrossRef]
- Shohji, N.; Kawaji, T.; Okamoto, S. Ti(O-i-Pr)4/Me3SiCl/Mg-Mediated Reductive Cleavage of Sulfonamides and Sulfonates to Amines and Alcohols. Org. Lett. 2011, 13, 2626–2629. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, M.; Kumar, B.A.; Narender, R. Expedient and simple method for regeneration of alcohols from toluenesulfonates using Mg-MeOH. Tetrahedron Lett. 1998, 39, 2847–2850. [Google Scholar] [CrossRef]
- Nyasse, B.; Grehn, L.; Ragnarsson, U. Mild, efficient cleavage of arenesulfonamides by magnesium reduction. Chem. Commun. 1997, 11, 1017–1018. [Google Scholar] [CrossRef]
- Sabitha, G.; Reddy, B.V.S.; Abraham, S.; Yadav, J.S. Deprotection of sulfonamides using iodotrimethylsilane. Tetrahedron Lett. 1999, 40, 1569–1571. [Google Scholar] [CrossRef]
- Yoshida, S.; Igawa, K.; Tomooka, K. Nucleophilic substitution reaction at the nitrogen of arylsulfonamides with phosphide anion. J. Am. Chem. Soc. 2012, 134, 19358–19361. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 5, 8, and 1 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Intermediate | Temperature | Time (h) | Solvent | Diastereomeric Ratio of 12, 13 and 5 a | Diastereomeric Ratio of 12, 13 and 5 after Recrystallization a | Yield (%) b |
|---|---|---|---|---|---|---|---|
| 1 | 12 | Reflux to r.t. | 1 | MeOH | 40:60 | 53:47:00 | 70.8 |
| 2 | 12 | 1 | MeOH/CHCl3 | 40:60 | 48:52:00 | 68.3 | |
| 3 | 12 | 1 | Acetone | 40:60 | 50:50:00 | 66.5 | |
| 4 | 12 | R.t. | 6 | MeCN/MeOH | 40:60 | 46:54:00 | 71.2 |
| 5 | 13 | Reflux to r.t. | 1 | IPE | 50:50:00 | 98:02:00 | 23 |
| 6 | 13 | 40 °C | 1.5 | IPE | 50:50:00 | 95:05:00 | 22.9 |
| 7 | 5 | R.t. to 0 °C | 16 | MeOH/IPE | 52:48:00 | 99:01:00 | 41.5 |
| 8 | 5 | 16 | MeOH/HEX/IPE | 52:48:00 | 98:02:00 | 38.2 |

| Entry | Reagents (eq.) | Temperature (°C) | Reaction Time (h) | Solvent | Yield (%) a |
|---|---|---|---|---|---|
| 1 | TBAF (10.0) | Reflux | 5 | THF | NR b |
| 2 | Thiophenol (1.3)/K2CO3 (2.0) | 21 | DMF/MeCN | NR | |
| 3 | TMSCl (1.5)/NaI (1.5) | 17 | MeCN | NR | |
| 4 | TMSI (neat) | 80 | 1 | — | NR |
| 5 | Mg (5.0)/Ti(OiPr)4 (1.0) | 50 | 21 | THF | NR |
| 6 | 0.5 M KPPh2 in THF (1.3) | −78 | 24 | 68 | |
| 7 | 0.5 M KPPh2 in THF (1.3) | −40 | 3 | 69.5 | |
| 8 | −20 | 3 | 43.7 | ||
| 9 | 0 | 3 | 30.4 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, S.K.; Nam, D.H.; Ahn, J.; Lee, J.; Sim, J.; Lee, J.; Suh, Y.-G. Practical and Sustainable Synthesis of Optically Pure Levocabastine, a H1 Receptor Antagonist. Molecules 2017, 22, 1971. https://doi.org/10.3390/molecules22111971
Kang SK, Nam DH, Ahn J, Lee J, Sim J, Lee J, Suh Y-G. Practical and Sustainable Synthesis of Optically Pure Levocabastine, a H1 Receptor Antagonist. Molecules. 2017; 22(11):1971. https://doi.org/10.3390/molecules22111971
Chicago/Turabian StyleKang, Sung Kwon, Dong Hyuk Nam, Jaeseung Ahn, Jaemin Lee, Jaehoon Sim, Jeeyeon Lee, and Young-Ger Suh. 2017. "Practical and Sustainable Synthesis of Optically Pure Levocabastine, a H1 Receptor Antagonist" Molecules 22, no. 11: 1971. https://doi.org/10.3390/molecules22111971
APA StyleKang, S. K., Nam, D. H., Ahn, J., Lee, J., Sim, J., Lee, J., & Suh, Y. -G. (2017). Practical and Sustainable Synthesis of Optically Pure Levocabastine, a H1 Receptor Antagonist. Molecules, 22(11), 1971. https://doi.org/10.3390/molecules22111971