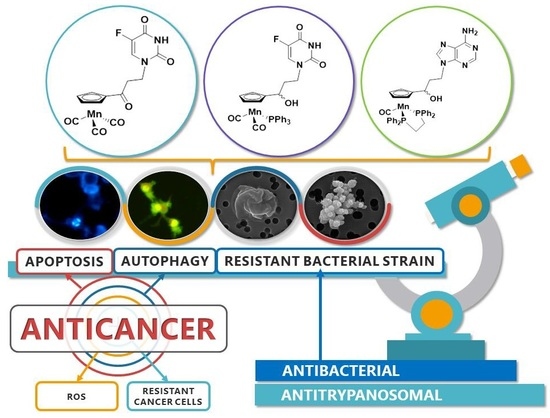
Cymantrenyl-Nucleobases: Synthesis, Anticancer, Antitrypanosomal and Antimicrobial Activity Studies

 , , , ,
, , , ,  ,
,
Abstract
:
1. Introduction
2. Results and Discussion
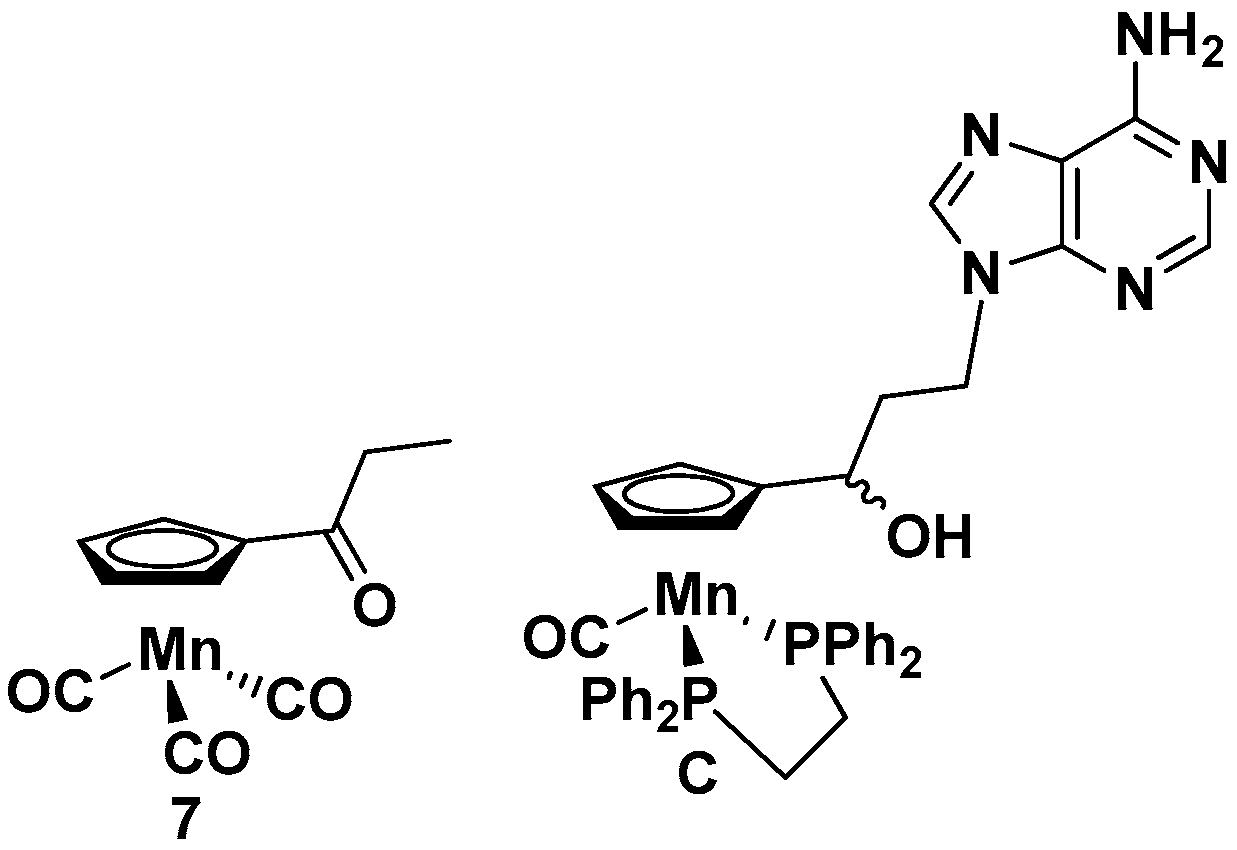
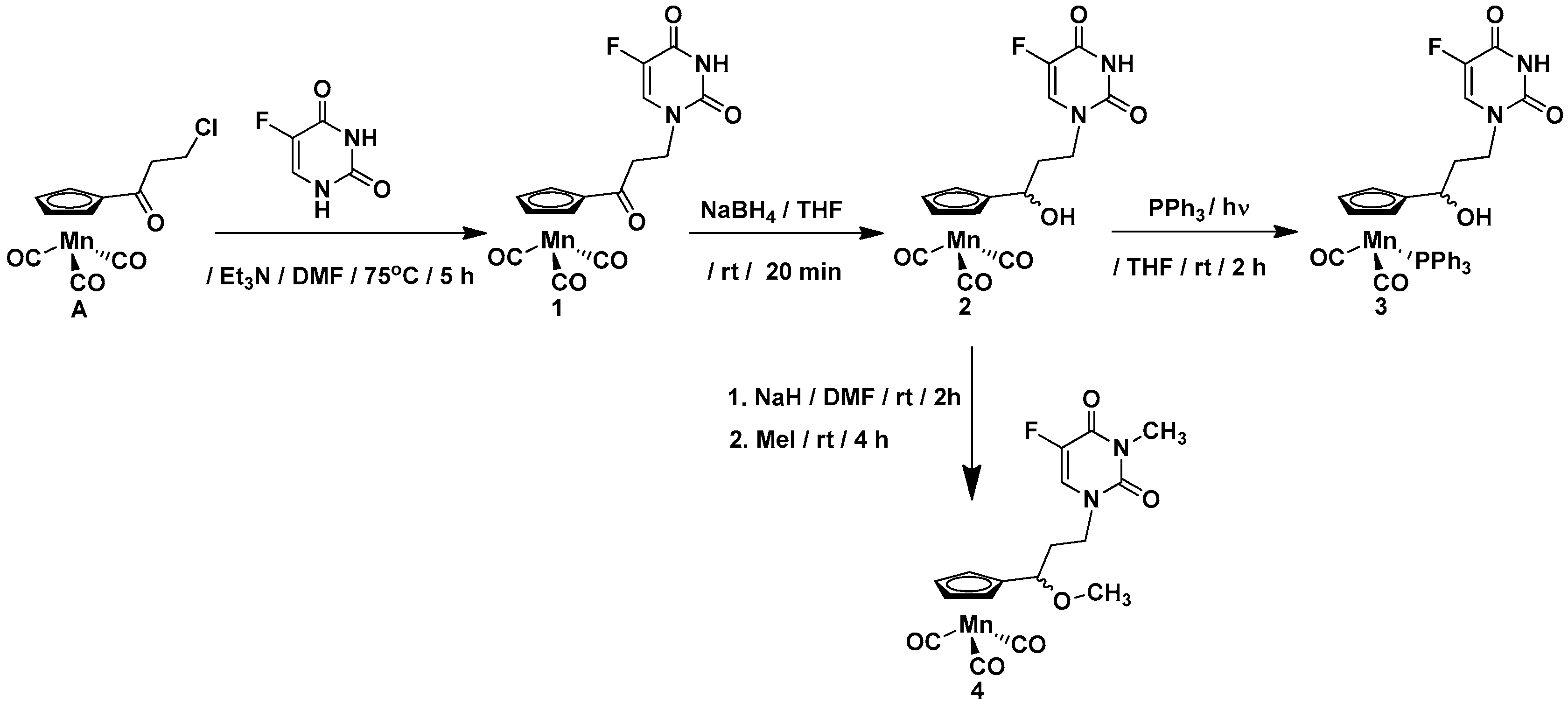
2.1. Synthesis of Compounds 1–7
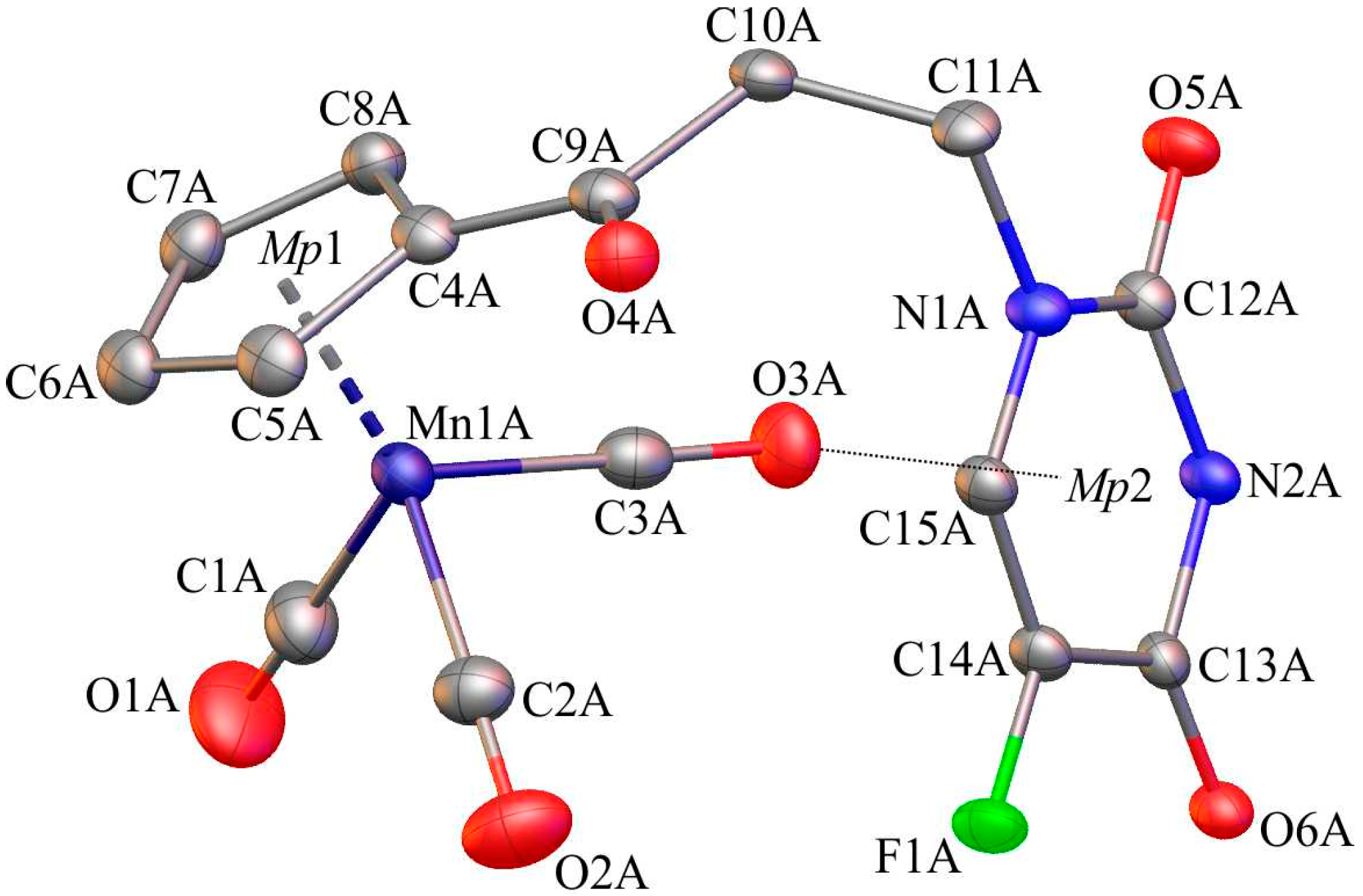
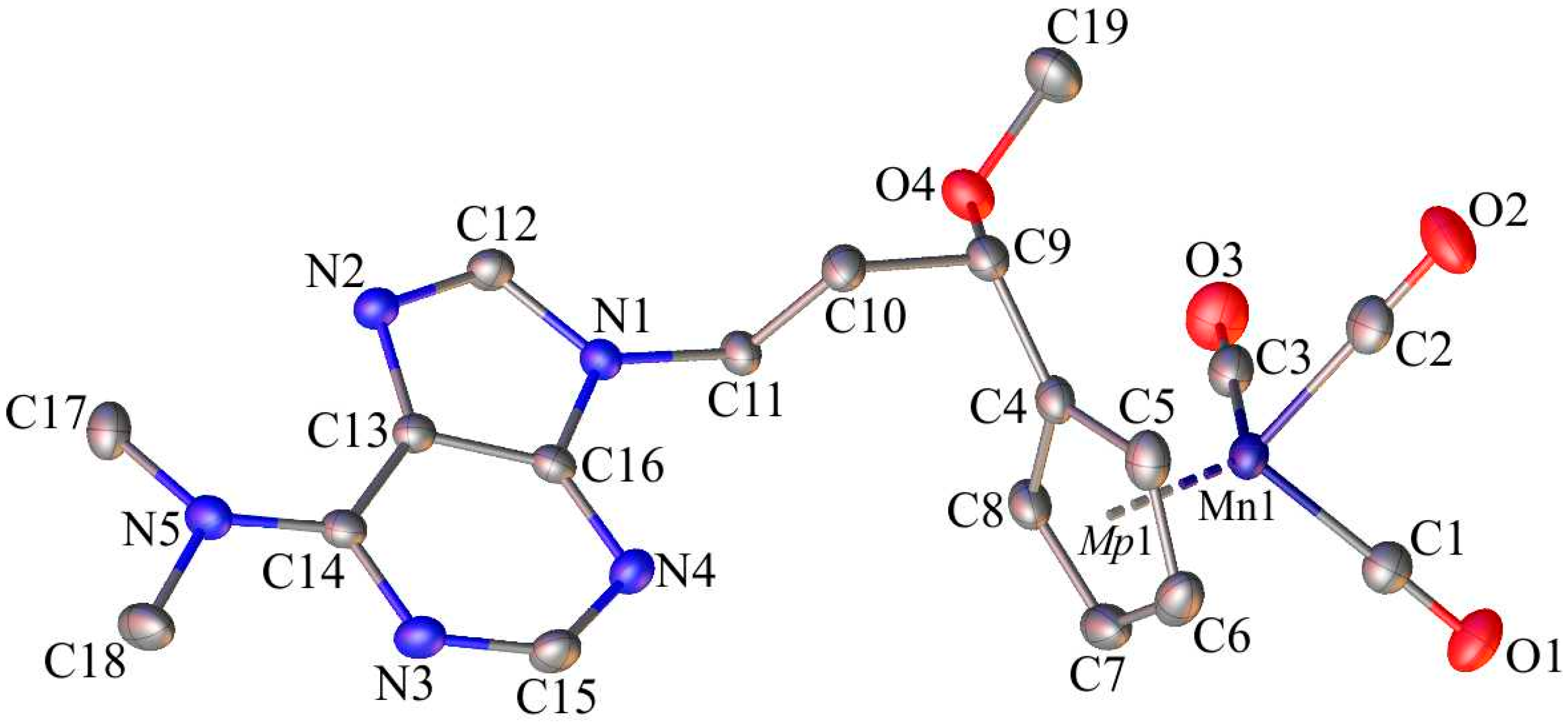
2.2. X-ray Crystal Structures of 1, 6 and C
2.3. Determination of log Po/w
3. Biological Studies
3.1. Evaluation of Anticancer Activity
3.2. Determination of Cytotoxic Mechanism of Action
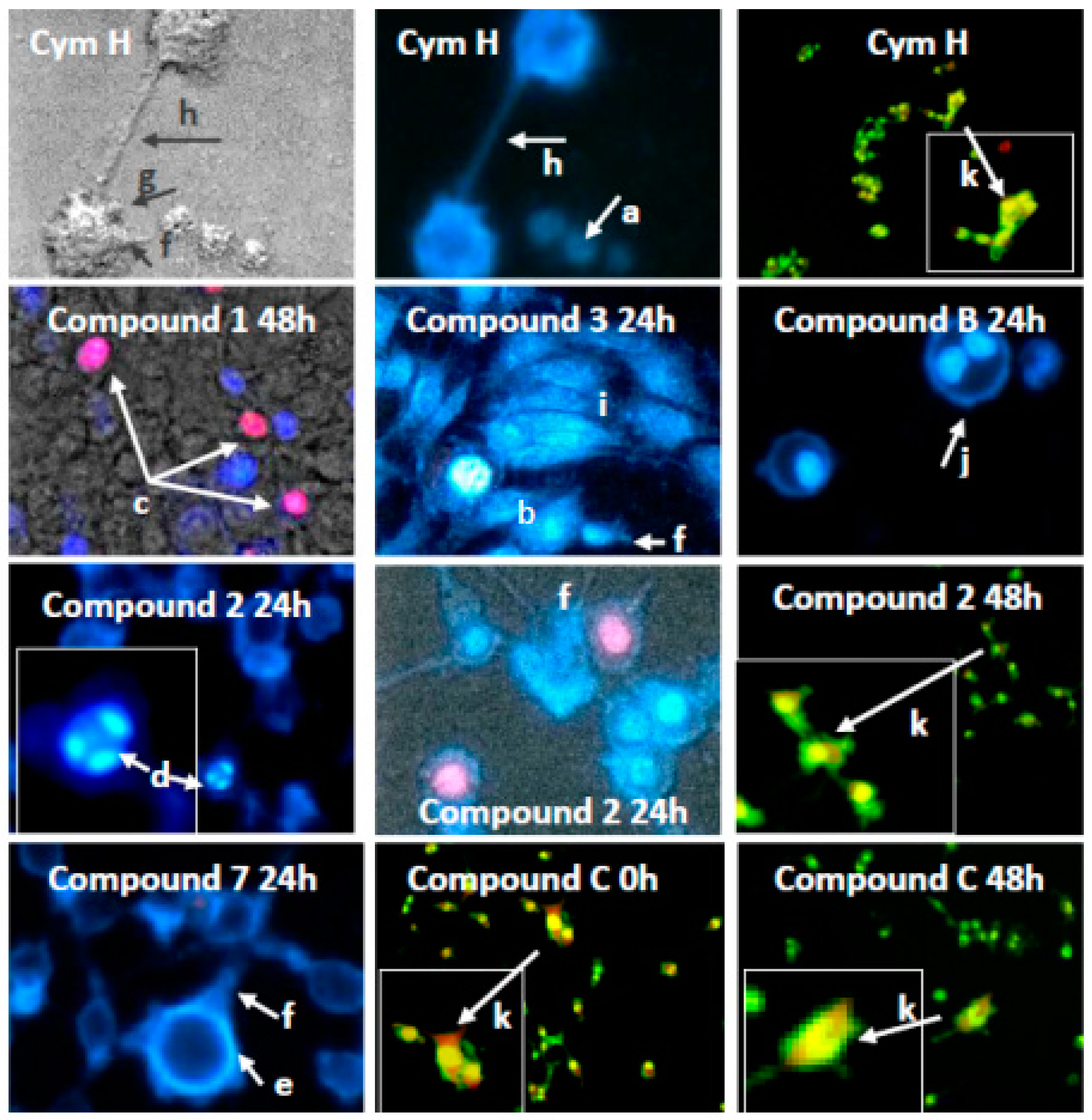
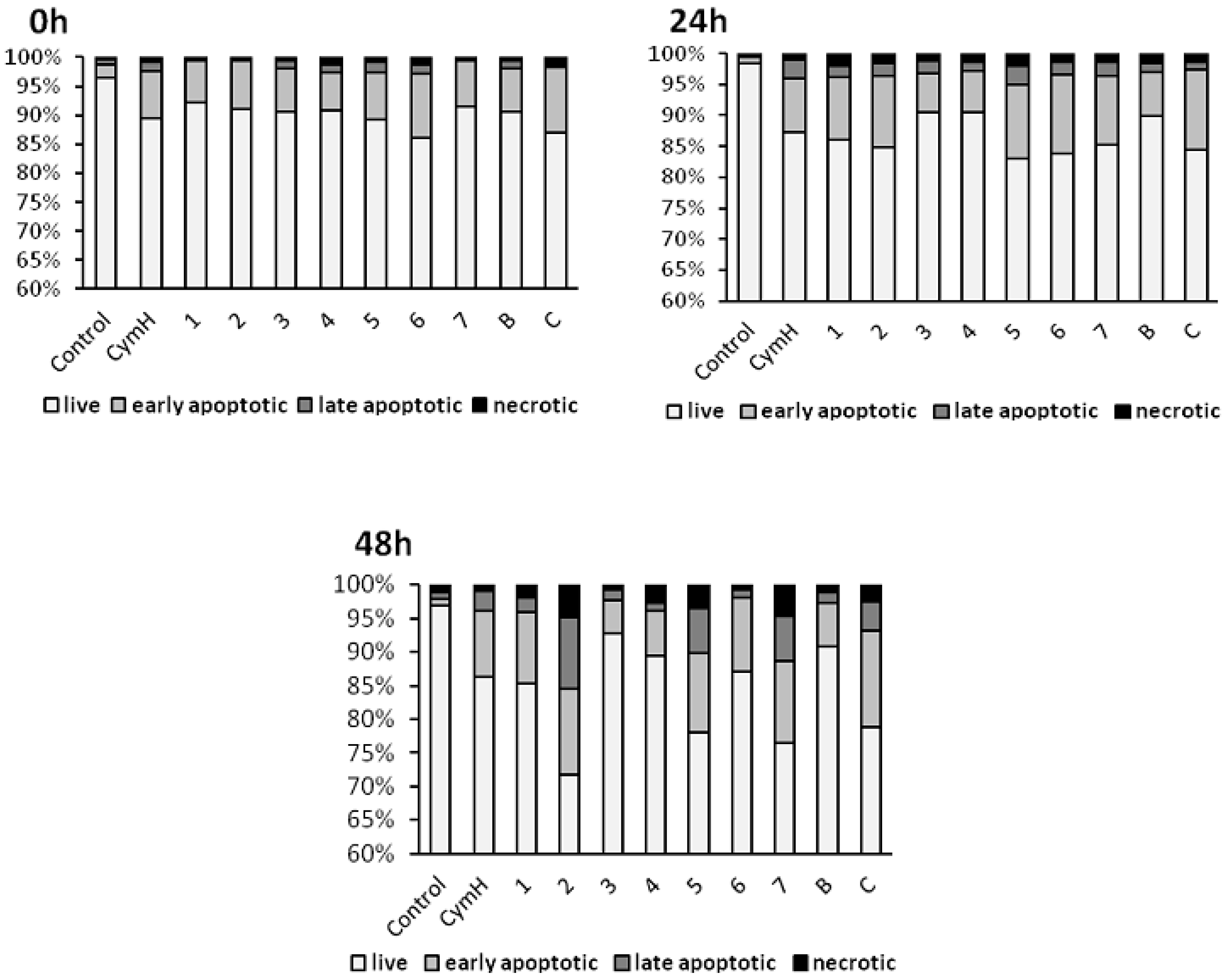
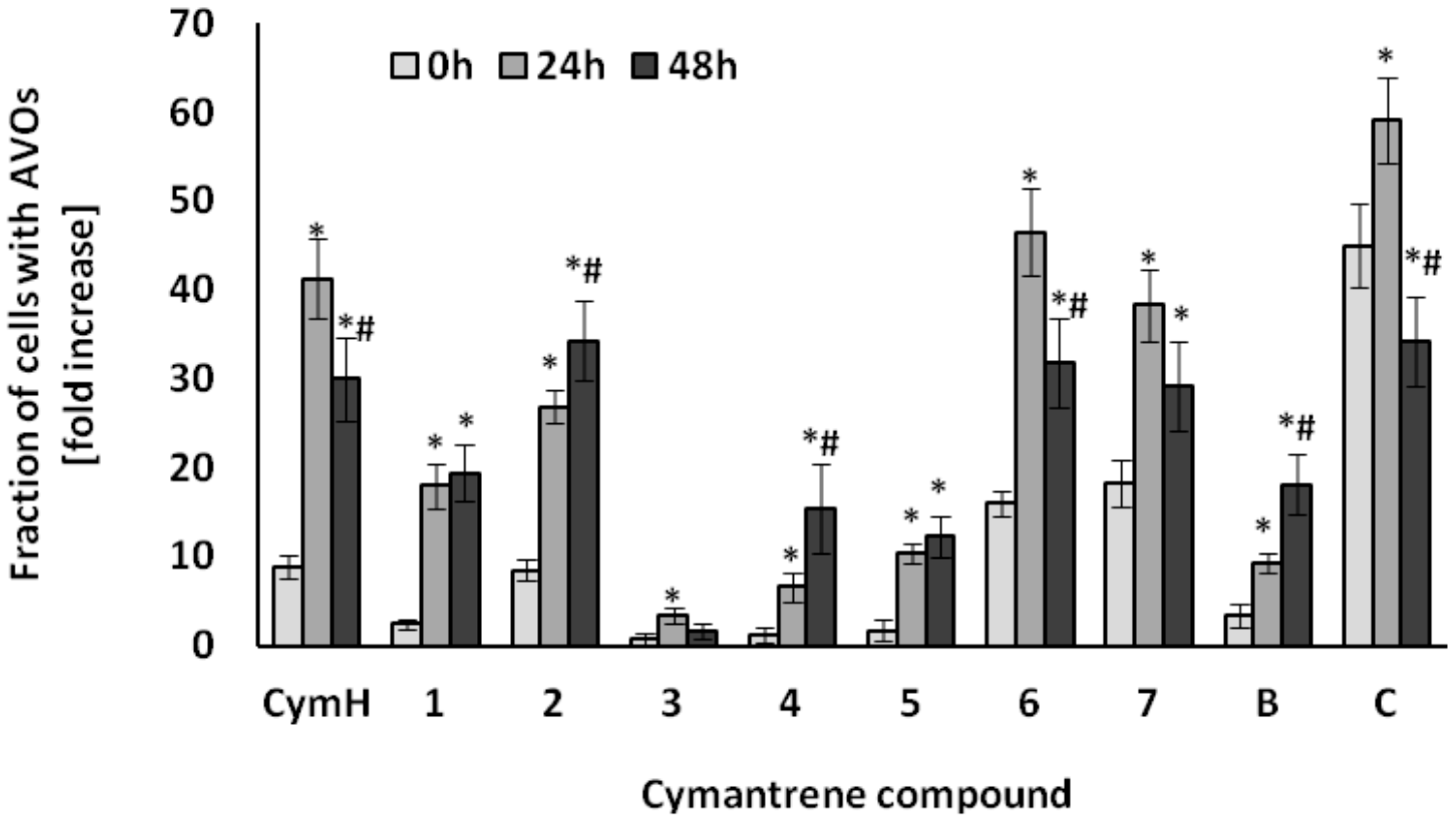
3.2.1. Induction of Apoptosis and Autophagy
3.2.2. Induction of Oxidative Stress
3.3. Evaluation of Antitrypanosomal Activity
3.4. Evaluation of Antibacterial Activity
4. Experimental Section
4.1. General Information
4.2. Chemistry
4.3. Single-Crystal X-ray Structure Analysis of 1, 6 and C
4.4. Determination of the Log Po/w
4.5. Biology
4.5.1. Anticancer Activity
Cell Lines
Cell Culture
Cytotoxicity Assay
Determination of Autophagy and Apoptotic/Necrotic Cell Death
Determination of Intracellular ROS
4.5.2. Statistical Analysis
4.5.3. Trypanocidal Assay
4.5.4. Microbiology
4.5.5. Deposited Data
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Baggaley, E.; Weinstein, J.A.; Gareth Williams, J.A. Lighting the way to see inside the live cell with luminescent transition metal complexes. Coord. Chem. Rev. 2012, 256, 1762–1785. [Google Scholar] [CrossRef]
- Merlino, A. Interactions between proteins and Ru compounds of medicinal interest: A structural perspective. Coord. Chem. Rev. 2016, 326, 111–134. [Google Scholar] [CrossRef]
- Jaouen, G.; Salmain, M. Bioorganometallic Chemistry: Applications in Dug Discovery, Biocatalysis, and Imaging; Wiley-VCH: Weinheim, Germany, 2015. [Google Scholar]
- Kowalski, K. Ferrocenyl-nucleobase complexes: Synthesis, chemistry and applications. Coord. Chem. Rev. 2016, 317, 132–156. [Google Scholar] [CrossRef]
- Patra, M.; Gasser, G. The medicinal chemistry of ferrocene and its derivatives. Nat. Rev. Chem. 2017, 1. [Google Scholar] [CrossRef]
- Gautier, A.; Cisnetti, F. Advances in metal-carbene complexes as potent anti-cancer agents. Metallomics 2012, 4, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Ott, I. On the medicinal chemistry of gold complexes as anticancer drugs. Coord. Chem. Rev. 2009, 253, 1670–1681. [Google Scholar] [CrossRef]
- Jaouen, G.; Vessières, A.; Top, S. Ferrocifen type anti cancer drugs. Chem. Soc. Rev. 2015, 44, 8802–8817. [Google Scholar] [CrossRef] [PubMed]
- Patra, M.; Gasser, G.; Metzler-Nolte, N. Small organometallic compounds as antibacterial agents. Dalton Trans. 2012, 41, 6350–6358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewandowski, E.M.; Szczupak, Ł.; Wong, S.; Skiba, J.; Guśpiel, A.; Solecka, J.; Vrček, V.; Kowalski, K.; Chen, Y. Antibacterial Properties of Metallocenyl-7-ADCA Derivatives and Structure in Complex with CTX-M β-Lactamase. Organometallics 2017, 36, 1673–1676. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.W.; Hyland, C.J.T. Medicinal organometallic chemistry—An emerging strategy for the treatment of neglected tropical diseases. MedChemComm 2015, 6, 1230–1243. [Google Scholar] [CrossRef]
- Simpson, P.V.; Nagel, C.; Bruhn, H.; Schatzschneider, U. Antibacterial and Antiparasitic Activity of Manganese(I) Tricarbonyl Complexes with Ketoconazole, Miconazole, and Clotrimazole Ligands. Organometallics 2015, 34, 3809–3815. [Google Scholar] [CrossRef]
- Elschenbroich, C.; Salzed, A. Organometallics: A Concise Introduction; Wiley-VCH: Weinheim, Germany, 1992. [Google Scholar]
- Kowalski, K.; Szczupak, Ł.; Saloman, S.; Steverding, D.; Jabłoński, A.; Vrček, V.; Hildebrandt, A.; Lang, H.; Rybarczyk-Pirek, A. Cymantrene, Cyrhetrene and Ferrocene Nucleobase Conjugates: Synthesis, Structure, Computational Study, Electrochemistry and Antitrypanosomal Activity. ChemPlusChem 2017, 82, 303–314. [Google Scholar] [CrossRef]
- Kur, S.A.; Heeg, M.J.; Winter, C.H. Pentamercuration of cyclopentadienylmanganese tricarbonyl and cyclopentadienylrhenium tricarbonyl. Crystal structure of (pentaiodocyclopentadienyl)manganese tricarbonyl. Organometallics 1994, 13, 1865–1869. [Google Scholar] [CrossRef]
- Lynch, T.J.; Dominguez, R.; Helvenston, M.C. Synthesis of the trimetallic manganese and rhenium complexes: N[CH2(η5-C5H4)M(CO)3]3. Derivatives of the nonadentate “TCp” ligand tris(cyclopentadienylmethyl)amine trianion. Organometallics 1988, 7, 2566–2567. [Google Scholar] [CrossRef]
- Heilweil, E.J.; Johnson, J.O.; Mosley, K.L.; Lubet, P.P.; Webster, C.E.; Burkey, T.J. Engineering Femtosecond Organometallic Chemistry: Photochemistry and Dynamics of Ultrafast Chelation of Cyclopentadienylmanganese Tricarbonyl Derivatives with Pendant Benzenecarbonyl and Pyridinecarbonyl Groups. Organometallics 2011, 30, 5611–5619. [Google Scholar] [CrossRef]
- Caulton, K.G. Coordination chemistry of the manganese and rhenium fragments (C5H5)M(CO)2. Coord. Chem. Rev. 1981, 38, 1–43. [Google Scholar] [CrossRef]
- Ogasawara, M.; Tseng, Y.-Y.; Liu, Q.; Chang, N.; Yang, X.; Takahashi, T.; Kamikawa, K. Kinetic Resolution of Planar-Chiral (η5-Bromocyclopentadienyl)manganese(I) Complexes by Molybdenum-Catalyzed Asymmetric Ring-Closing Metathesis. Organometallics 2017, 36, 1430–1435. [Google Scholar] [CrossRef]
- Roemer, M.; Schmiel, P.; Lentz, D. Cymantrene- and Ferrocene-Based Complexes with Perfluorinated Bridging Moieties. Organometallics 2011, 30, 2063–2066. [Google Scholar] [CrossRef]
- Kunz, K.; Bolte, M.; Lerner, H.-W.; Wagner, M. Photochemistry of Cymantrenyl Scorpionates: Formation of a Novel Tritopic Cyclopentadienyl/Scorpionate Hybrid Ligand. Organometallics 2009, 28, 3079–3087. [Google Scholar] [CrossRef]
- Hromadová, M.; Salmain, M.; Fischer-Durand, N.; Pospíšil, L.; Jaouen, G. Electrochemical Microbead-Based Immunoassay Using an (η5-Cyclopentadienyl)tricarbonylmanganese Redox Marker Bound to Bovine Serum Albumin. Langmuir 2006, 22, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Top, S.; Kaloun, E.B.; Toppi, S.; Herrbach, A.; McGlinchey, M.J.; Jaouen, G. Decomplexation of Cyclopentadienylmanganese Tricarbonyls under Very Mild Conditions: A Novel Route to Substituted Cyclopentadienes and Their Application in Organometallic Synthesis. Organometallics 2001, 20, 4554–4561. [Google Scholar] [CrossRef]
- Wu, K.; Top, S.; Hillard, E.A.; Jaouen, G.; Geiger, W.E. Anodic properties of diarylethene derivatives having organometallic piano-stool tags. Chem. Commun. 2011, 47, 10109–10111. [Google Scholar] [CrossRef] [PubMed]
- Splith, K.; Neundorf, I.; Hu, W.; N’Dongo, H.W.P.; Vasylyeva, V.; Merz, K.; Schatzschneider, U. Influence of the metal complex-to-peptide linker on the synthesis and properties of bioactive CpMn(CO)3 peptide conjugates. Dalton Trans. 2010, 39, 2536–2545. [Google Scholar] [CrossRef] [PubMed]
- Neundorf, I.; Hoyer, J.; Splith, K.; Rennert, R.; Peindy N’Dongo, H.W.; Schatzschneider, U. Cymantrene conjugation modulates the intracellular distribution and induces high cytotoxicity of a cell-penetrating peptide. Chem. Commun. 2008, 43, 5604–5606. [Google Scholar] [CrossRef] [PubMed]
- Splith, K.; Hu, W.; Schatzschneider, U.; Gust, R.; Ott, I.; Onambele, L.A.; Prokop, A.; Neundorf, I. Protease-Activatable Organometal-Peptide Bioconjugates with Enhanced Cytotoxicity on Cancer Cells. Bioconj. Chem. 2010, 21, 1288–1296. [Google Scholar] [CrossRef] [PubMed]
- Concha, C.; Quintana, C.; Klahn, A.H.; Artigas, V.; Fuentealba, M.; Biot, C.; Halloum, I.; Kremer, L.; López, R.; Romanos, J.; et al. Organometallic tosyl hydrazones: Synthesis, characterization, crystal structures and in vitro evaluation for anti-Mycobacterium tuberculosis and antiproliferative activities. Polyhedron 2017, 131, 40–45. [Google Scholar] [CrossRef]
- Tirkey, V.; Mishra, S.; Dash, H.R.; Das, S.; Nayak, B.P.; Mobin, S.M.; Chatterjee, S. Synthesis, characterization and antibacterial studies of ferrocenyl and cymantrenyl hydrazone compounds. J. Organomet. Chem. 2013, 732, 122–129. [Google Scholar] [CrossRef]
- Wenzel, M.; Patra, M.; Senges, C.H.R.; Ott, I.; Stepanek, J.J.; Pinto, A.; Prochnow, P.; Vuong, C.; Langklotz, S.; Metzler-Nolte, N.; et al. Analysis of the Mechanism of Action of Potent Antibacterial Hetero-tri-organometallic Compounds: A Structurally New Class of Antibiotics. ACS Chem. Biol. 2013, 8, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Patra, M.; Gasser, G.; Wenzel, M.; Merz, K.; Bandow, J.E.; Metzler-Nolte, N. Synthesis and Biological Evaluation of Ferrocene-Containing Bioorganometallics Inspired by the Antibiotic Platensimycin Lead Structure. Organometalics 2010, 29, 4312–4319. [Google Scholar] [CrossRef]
- Glans, L.; Hu, W.; Jöst, C.; de Kock, C.; Smith, P.J.; Haukka, M.; Bruhn, H.; Schatzschneider, U.; Nordlander, E. Synthesis and biological activity of cymantrene and cyrhetrene 4-aminoquinoline conjugates against malaria, leishmaniasis, and trypanosomiasis. Dalton Trans. 2012, 41, 6443–6450. [Google Scholar] [CrossRef] [PubMed]
- Day, D.P.; Dann, T.; Hughes, D.L.; Oganesyan, V.S.; Steverding, D.; Wildgoose, G.G. Cymantrene-Triazole “Click” Products: Structural Characterization and Electrochemical Properties. Organometallics 2014, 33, 4687–4696. [Google Scholar] [CrossRef] [Green Version]
- Skiba, J.; Rajnisz, A.; de Oliveira, K.N.; Ott, I.; Solecka, J.; Kowalski, K. Ferrocenyl bioconjugates of ampicillin and 6-aminopenicillinic acid—Synthesis, electrochemistry and biological activity. Eur. J. Med. Chem. 2012, 57, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, K.; Koceva-Chyła, A.; Pieniążek, A.; Bernasińska, J.; Skiba, J.; Rybarczyk-Pirek, A.; Jóźwiak, Z. The synthesis, structure, electrochemistry and in vitro anticancer activity studies of ferrocenyl-thymine conjugates. J. Organomet. Chem. 2012, 700, 58–68. [Google Scholar] [CrossRef]
- Kowalski, K.; Skiba, J.; Oehninger, L.; Ott, I.; Solecka, J.; Rajnisz, A.; Therrien, B. Metallocene-Modified Uracils: Synthesis, Structure, and Biological Activity. Organometallics 2013, 32, 5766–5773. [Google Scholar] [CrossRef]
- Skiba, J.; Schmidt, C.; Lippmann, P.; Ensslen, P.; Wagenknecht, H.-A.; Czerwieniec, R.; Brandl, F.; Ott, I.; Bernaś, T.; Krawczyk, B.; et al. Substitution of Metallocenes with [2.2]Paracyclophane to Enable Confocal Microscopy Imaging in Living Cells. Eur. J. Inorg. Chem. 2017, 2017, 297–305. [Google Scholar] [CrossRef]
- European Commission. Council Regulation No. 440/2008. Off. J. EU 2008, 440, 67–74. [Google Scholar]
- Organisation for Economic Co-operation and Development (OECD). Test No. 117: Partition Coefficient (n-octanol/water), HPLC Method; OECD Guidelines for the Testing of Chemicals, Section 1; OECD Publishing: Paris, France, 2004. [Google Scholar]
- Eadsforth, C.V.; Moser, P. Assessment of reverse-phase chromatographic methods for determining partition coefficients. Chemosphere 1983, 12, 1459–1475. [Google Scholar] [CrossRef]
- Soczewiński, E.; Wachtmeister, C.A. The relation between the composition of certain ternary two-phase solvent systems and RM values. J. Chromatogr. 1962, 7, 311–320. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [PubMed]
- Law, B.Y.; Chan, W.K.; Xu, S.W.; Wang, J.R.; Bai, L.P.; Liu, L.; Wong, V.K. Natural small-molecule enhancers of autophagy induce autophagic cell death in apoptosis-defective cells. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Fimia, G.M.; Piacentini, M. Regulation of autophagy in mammals and its interplay with apoptosis. Cell. Mol. Life Sci. 2010, 67, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Tang, D. Autophagy in pancreatic cancer pathogenesis and treatment. Am. J. Cancer Res. 2012, 2, 383–396. [Google Scholar] [PubMed]
- Jabs, T. Reactive oxygen intermediates as mediators of programmed cell death in plants and animals. Biochem. Pharmacol. 1999, 57, 231–245. [Google Scholar] [CrossRef]
- Wang, J.; Yi, J. Cancer cell killing via ROS: To increase or decrease, that is the question. Cancer Biol. Ther. 2008, 7, 1875–1884. [Google Scholar] [CrossRef] [PubMed]
- Szwed, M.; Wrona, D.; Kania, K.D.; Koceva-Chyła, A.; Marczak, A. Doxorubicin-transferrin conjugate triggers pro-oxidative disorders in solid tumor cells. Toxicol. In Vitro 2016, 31, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Phang, C.-W.; Karsani, S.A.; Nurestri, S.; Malek, A. Induction of apoptosis and cell cycle arrest by flavokawain C on HT-29 human colon adenocarcinoma via enhancement of reactive oxygen species generation, upregulation of p21, p27, and Gadd153, and inactivation of inhibitor of apoptosis proteins. Pharmacogn. Mag. 2017, 13, S321–S328. [Google Scholar] [PubMed]
- Raza, M.H.; Siraj, S.; Arshad, A.; Waheed, U.; Aldakheel, F.; Alduraywish, S.; Arshad, M. ROS-modulated therapeutic approaches in cancer treatment. J. Cancer Res. Clin. Oncol. 2017, 143, 1789–1809. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Helot, J.-M.; Guille-Collignon, M.; Lemaitre, F.; Jaouen, G.; Vessieres, A.; Amatore, C. Quantitative Analyses of ROS and RNS Production in Breast Cancer Cell Lines Incubated with Ferrocifens. ChemMedChem 2014, 9, 1286–1293. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Poillet-Perez, L.; Despouy, G.; Delage-Mourroux, R.; Boyer-Guittaut, M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2015, 4, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Hirumi, H.; Hirumi, K.; Doyle, J.J.; Cross, G.A.M. In vitro cloning of animal-infective bloodstream forms of Trypanosoma brucei. Parasitology 1980, 80, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.J.; Gallo, R.C.; Gallagher, R.E. Continuous growth and differentiation of human myeloid leukaemic cells in suspension culture. Nature 1977, 270, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Merschjohann, K.; Sporer, F.; Steverding, D.; Wink, M. In Vitro Effect of Alkaloids on Bloodstream Forms of Trypanosoma brucei and T. congolense. Planta Med. 2001, 67, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Barrett, S.V.; Barrett, M.P. Anti-sleeping Sickness Drugs and Cancer Chemotherapy. Parasitol. Today 2000, 16, 7–9. [Google Scholar] [CrossRef]
- Steverding, D.; Rushworth, S.A. Front-line glioblastoma chemotherapeutic temozolomide is toxic to Trypanosoma brucei and potently enhances melarsoprol and eflornithine. Exp. Parasitol. 2017, 178, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.F.; Drown, B.S.; Riley, A.P.; Garcia, A.; Shirai, T.; Svec, R.L.; Hergenrother, P.J. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 2017, 545, 299–304. [Google Scholar] [CrossRef] [PubMed]
- CrysAlis CCD and CrysAlis RED, Oxford Diffraction; Oxford Diffraction Ltd.: Yarnton, UK, 2008.
- Clark, R.C.; Reid, J.S. The Analytical Calculation of Absorption in Multifaceted Crystals. Acta Crystallogr. Sect. A 1994, 51, 887–897. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Koceva-Chyła, A.; Jedrzejczak, M.; Skierski, J.; Kania, K.; Józwiak, Z. Mechanisms of induction of apoptosis by anthraquinone anticancer drugs aclarubicin and mitoxantrone in comparison with doxorubicin: Relation to drug cytotoxicity and caspase-3 activation. Apoptosis 2005, 10, 1497–1514. [Google Scholar] [CrossRef] [PubMed]
- Koceva-Chyła, A.; Matczak, K.; Hikisz, P.; Durka, K.; Kochel, K.; Süss-Fink, G.; Furrer, J.; Kowalski, K. Insights into the in vitro Anticancer Effects of Diruthenium-1. ChemMedChem 2016, 11, 2171–2187. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.G.; Cross, G.A.M. Selective cleavage of variant surface glycoproteins from Trypanosoma brucei. Biochem J. 1979, 178, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Baltz, T.; Baltz, D.; Giroud, C.; Crockett, J. Cultivation in a semi-defined medium of animal infective forms of Trypanosoma brucei, T. equiperdum, T. evansi, T. rhodesiense and T. gambiense. EMBO J. 1985, 4, 1273–1277. [Google Scholar] [PubMed]
- Steverding, D.; Wang, X. Trypanocidal activity of the proteasome inhibitor and anti-cancer drug bortezomib. Parasites Vectors 2009, 2. [Google Scholar] [CrossRef] [PubMed]
- Huber, W.; Koella, J.C. A comparison of three methods of estimating EC50 in studies of drug resistance of malaria parasites. Acta Trop. 1993, 55, 257–262. [Google Scholar] [CrossRef]
- Clinical & Laboratory Standards Institute (CLSI). M07-A8 Method for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, 8th ed.; Clinical & Laboratory Standards Institute (CLSI): Wayne, PA, USA, 2009; ISBN 1-56238-689-1. [Google Scholar]
- Mendoza, G.; Regiel-Futyra, A.; Andreu, V.; Sebastián, V.; Kyzioł, A.; Stochel, G.; Arruebo, M. Bactericidal Effect of Gold-Chitosan Nanocomposites in Coculture Models of Pathogenic Bacteria and Human Macrophages. ACS Appl. Mater. Interfaces 2017, 9, 17693–17701. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1–6 are available from the authors. |













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Log Po/w | SKOV-3 | MCF-7 | MCF-7/DX | MDA-MB-231 | A549 | HepG2 | U87-MG |
|---|---|---|---|---|---|---|---|---|
| CymH | 3.5 | 121 ± 3.05 | 86.01 ± 1.30 | 143.55 ± 2.24 | 68.60 ± 1.74 | 87.05 ± 1.35 | 74.66 ± 3.73 | 84.44 ± 4.22 |
| 1 | 2.6 | 75.14 ± 3.76 | 70.48 ± 2.52 | 137.49 ± 3.41 | 27.86 ± 1.11 | 25.05 ± 1.25 | 114.45 ± 2.72 | 64.47 ± 3.22 |
| 2 | 2.8 | 97.48 ± 2.87 | 119.45 ± 2.97 | nd * | >150 | 7.24 ± 0.34 | >150 | 100.29 ± 1.01 |
| 3 | 5.2 | 53.01 ± 2.65 | 57.78 ± 2.89 | 39.92 ± 1.41 | 35.73 ± 1.44 | 20.35 ± 1.09 | 65.76 ± 3.29 | 24.89 ± 1.24 |
| 4 | 3.5 | >150 | >150 | nd | >150 | >150 | >150 | >150 |
| 5 | 4.8 | 63.83 ± 3.19 | 102.16 ± 1.11 | nd | 55.95 ± 2.07 | 23.72 ± 0.86 | 106.23 ± 2.31 | 54.19 ± 2.71 |
| 6 | 3.5 | 77.1 ± 3.86 | 111.34 ± 3.57 | nd | >150 | 101.51 ± 1.08 | 86.89 ± 2.34 | 130.61 ± 3.53 |
| 7 | 3.2 | 69.84 ± 3.49 | >150 | nd | 63.01 ± 1.32 | 30.61 ± 1.35 | 109.57 ± 2.48 | 83.62 ± 1.18 |
| B | 2.6 | >150 | >150 | nd | >150 | >150 | >150 | >150 |
| C | 5.7 | 7.11 ± 0.36 | 103.74 ± 1.19 | nd | 43.07 ± 1.95 | 16.47 ± 0.82 | 66.59 ± 3.33 | 38.26 ± 1.91 |
| DOX | nd | 0.858 ± 0.044 | 0.439 ± 0.026 | 48.50 ± 2.78 | 1.073 ± 0.062 | 0.300 ± 0.018 | 0.569 ± 0.021 | 0.610 ± 0.029 |
| T. brucei | HL-60 | Selectivity | ||||
|---|---|---|---|---|---|---|
| Compound | MIC a (μM) | GI50 b (μM) | MIC (μM) | GI50 (μM) | MIC Ratio | GI50 Ratio |
| CymH | 100 | 44.6 ± 12.3 | >100 | >100 | >1 | >2.2 |
| 1 | 10 | 3.02 ± 0.14 | 100 | 33.1 ± 3.4 | 10 | 11.0 |
| 2 | >100 | >100 | >100 | >100 | 1 | 1 |
| 3 | 10 | 4.27 ± 0.13 | 100 | 30.6 ± 2.6 | 10 | 7.2 |
| 4 | 100 | 29.6 ± 6.8 | >100 | >100 | >1 | >3.4 |
| 5 | 10 | 3.14 ± 0.31 | 100 | 28.7 ± 3.0 | 10 | 9.1 |
| 6 | 100 | 26.1 ± 1.9 | >100 | 95.2 ± 44.4 | >1 | 3.6 |
| B c | >100 | 30.8 ± 3.5 | >100 | >100 | 1 | >3.2 |
| C c | 10 | 1.67 ± 0.37 | 100 | 21.8 ± 8.6 | 10 | 13.1 |
| DFMO d | >125 | 23.9 | n.d. | n.d. | n.d. | n.d. |
| Suramin | 0.1–1 | 0.039 ± 0.003 | >100 | >100 | >1000–100 | >2564 |
| Compound | MIC (μg/mL) | ||||
|---|---|---|---|---|---|
| S. aureus ATCC® 29213™ (MSSA) | S. aureus ATCC® 43300™ (MRSA) | S. aureus ATCC® 700787™ (VISA) | S. epidermidis ATCC® 12228™ | E. coli ATCC® BAA-198™ | |
| C | 32 | 32 | 32 | 16 | >128 |
| 1 | 64 | 32 | 32 | 8 | >512 |
| 2 | >512 | >512 | >512 | >512 | >512 |
| 3 | >256 | >256 | >256 | >256 | >256 |
| 4 | >512 | >512 | >512 | >512 | >512 |
| 5 | >256 | >256 | >256 | >256 | >256 |
| 6 | >512 | >512 | >512 | >512 | >512 |
| 7 | >512 | >512 | >512 | >512 | >512 |
| CymH | >512 | >512 | >512 | >512 | >512 |
| penicillin G | 2 | 16 | 32 | 2 | >256 |
| polymyxin B | 256 | 256 | >256 | 64 | 0.125 |
| Compound | MIC (μg/mL) | |||||
|---|---|---|---|---|---|---|
| Clinical S. aureus (MSSA) 26/11 | Clinical S. aureus (MSSA) 30/11 | Clinical S. aureus (MRSA) 31/12 | Clinical S. aureus (MRSA) 44/12 | Clinical S. epidermidis (MSSA) 37/12 | Clinical S. epidermidis (MSSA) 43/12 | |
| C | 32 | 32 | 32 | 32 | 16 | 16 |
| 1 | 32 | 64 | 64 | 32 | 4 | 8 |
| penicillin G | 0.5 | 2 | 32 | 8 | 16 | 8 |
| polymyxin B | 256 | 256 | 256 | 256 | 128 | 64 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jabłoński, A.; Matczak, K.; Koceva-Chyła, A.; Durka, K.; Steverding, D.; Jakubiec-Krześniak, K.; Solecka, J.; Trzybiński, D.; Woźniak, K.; Andreu, V.; et al. Cymantrenyl-Nucleobases: Synthesis, Anticancer, Antitrypanosomal and Antimicrobial Activity Studies. Molecules 2017, 22, 2220. https://doi.org/10.3390/molecules22122220
Jabłoński A, Matczak K, Koceva-Chyła A, Durka K, Steverding D, Jakubiec-Krześniak K, Solecka J, Trzybiński D, Woźniak K, Andreu V, et al. Cymantrenyl-Nucleobases: Synthesis, Anticancer, Antitrypanosomal and Antimicrobial Activity Studies. Molecules. 2017; 22(12):2220. https://doi.org/10.3390/molecules22122220
Chicago/Turabian StyleJabłoński, Artur, Karolina Matczak, Aneta Koceva-Chyła, Kamil Durka, Dietmar Steverding, Katarzyna Jakubiec-Krześniak, Jolanta Solecka, Damian Trzybiński, Krzysztof Woźniak, Vanesa Andreu, and et al. 2017. "Cymantrenyl-Nucleobases: Synthesis, Anticancer, Antitrypanosomal and Antimicrobial Activity Studies" Molecules 22, no. 12: 2220. https://doi.org/10.3390/molecules22122220
APA StyleJabłoński, A., Matczak, K., Koceva-Chyła, A., Durka, K., Steverding, D., Jakubiec-Krześniak, K., Solecka, J., Trzybiński, D., Woźniak, K., Andreu, V., Mendoza, G., Arruebo, M., Kochel, K., Krawczyk, B., Szczukocki, D., & Kowalski, K. (2017). Cymantrenyl-Nucleobases: Synthesis, Anticancer, Antitrypanosomal and Antimicrobial Activity Studies. Molecules, 22(12), 2220. https://doi.org/10.3390/molecules22122220