
Towards Precision Engineering of Canonical Polyketide Synthase Domains: Recent Advances and Future Prospects

Abstract
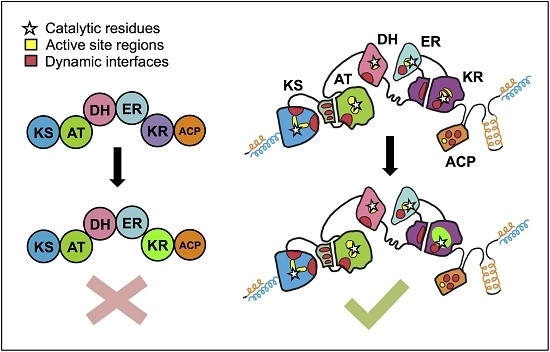
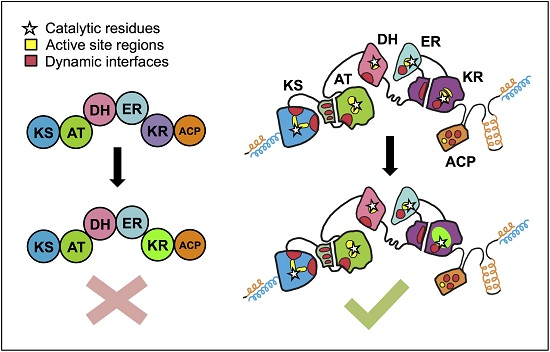
:
1. Introduction
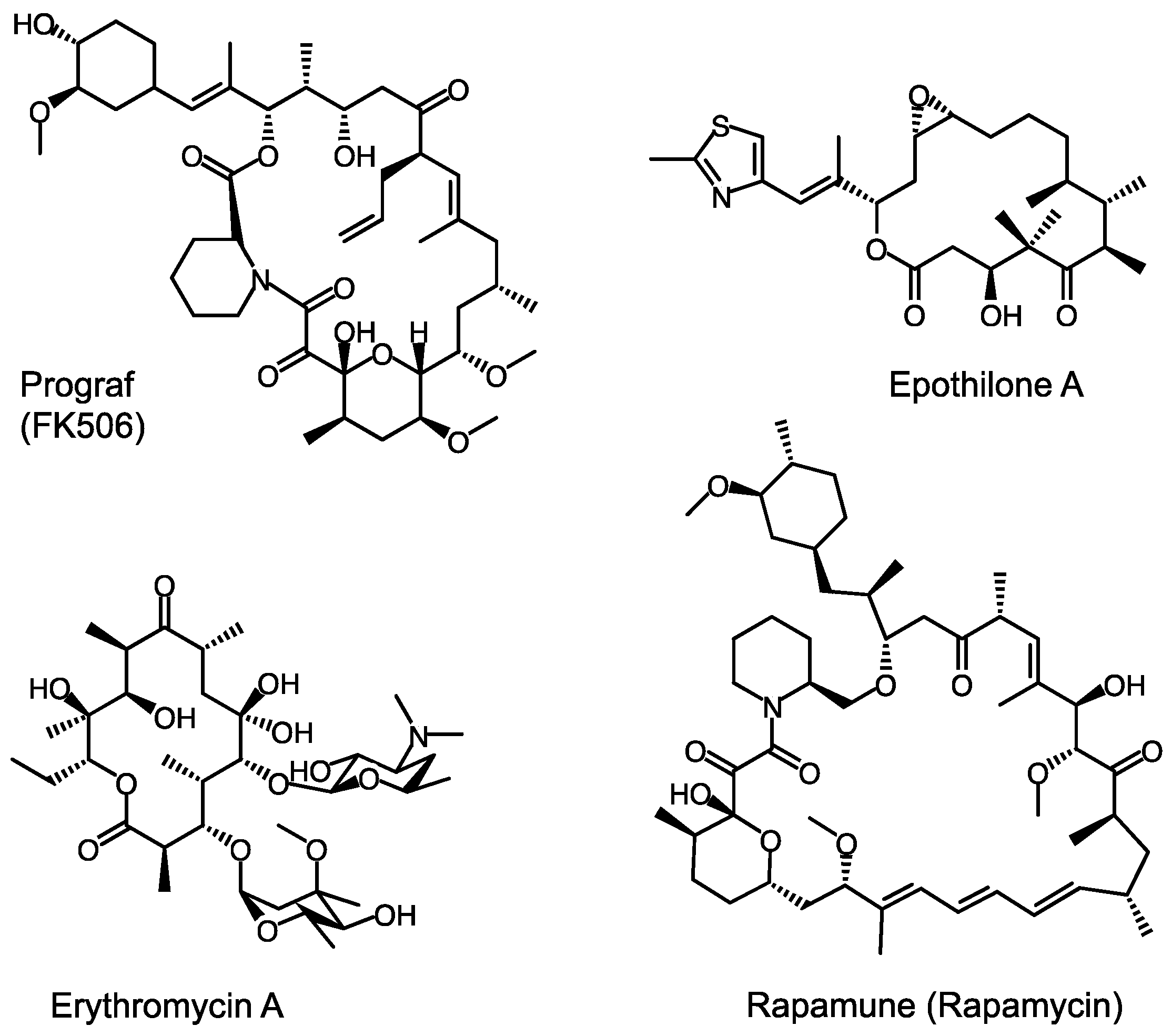
2. A Whirlwind Tour of PKS Research
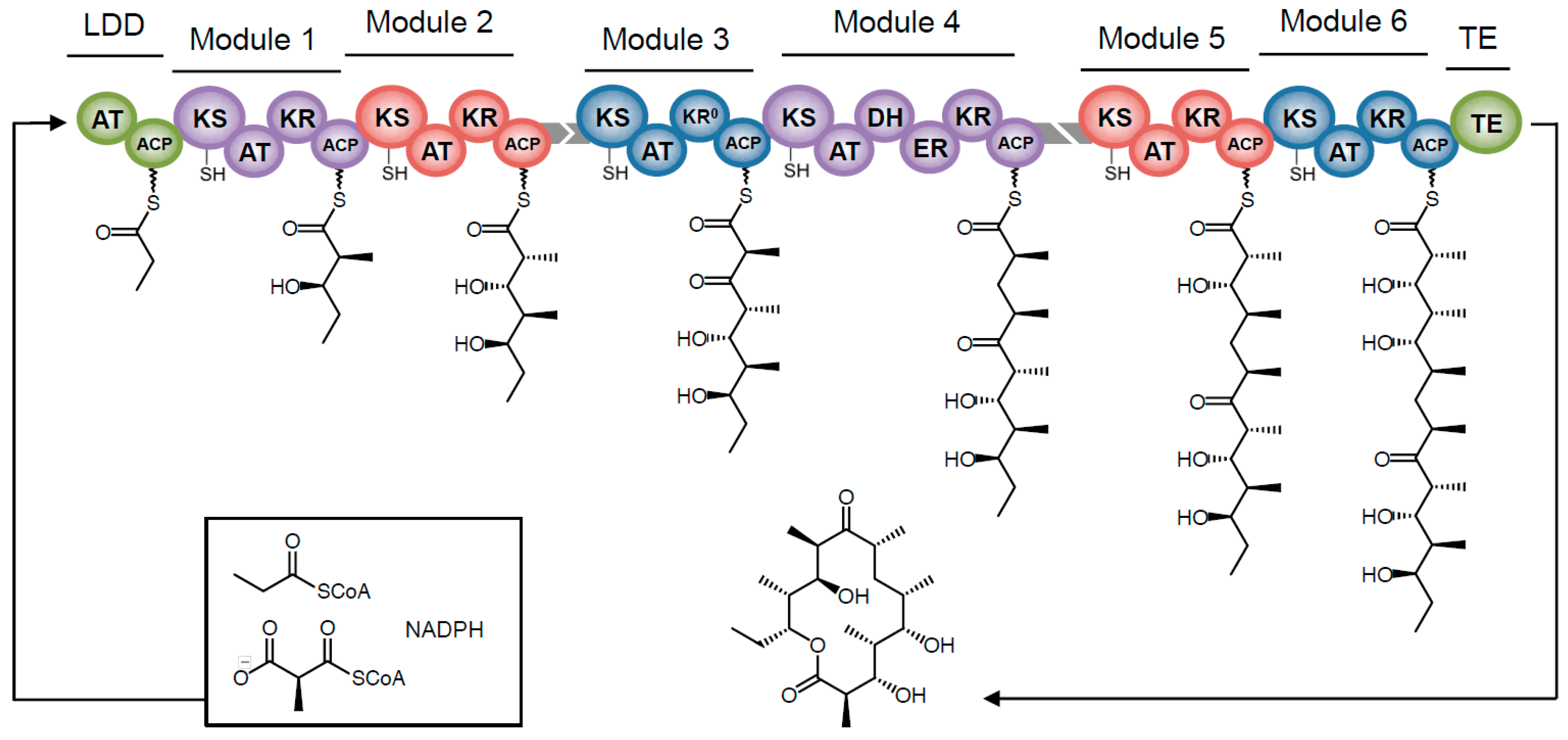
3. Polyketide Synthases: The Nuts and Bolts
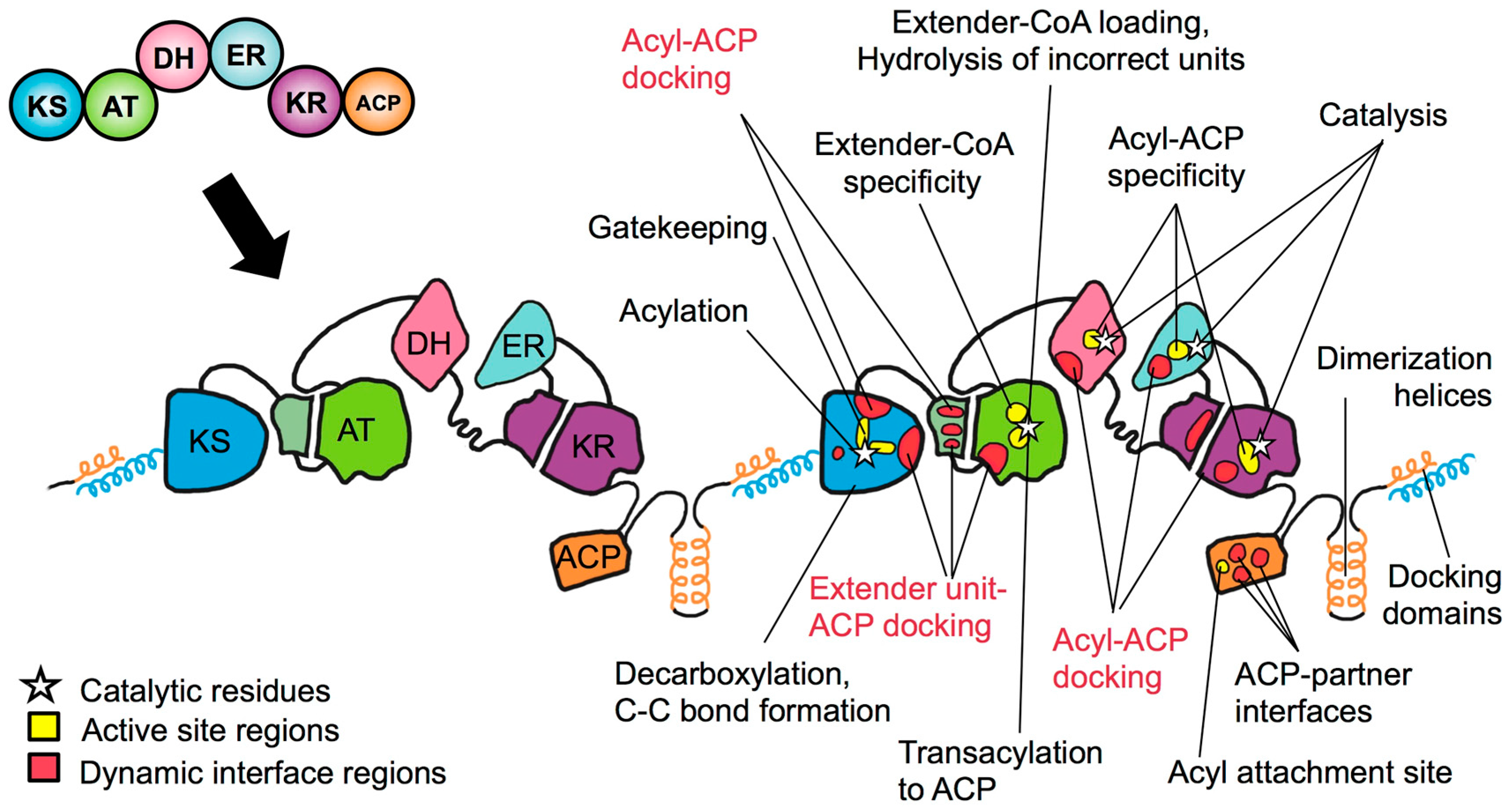
4. The PKS Structure-Function Map: Then and Now
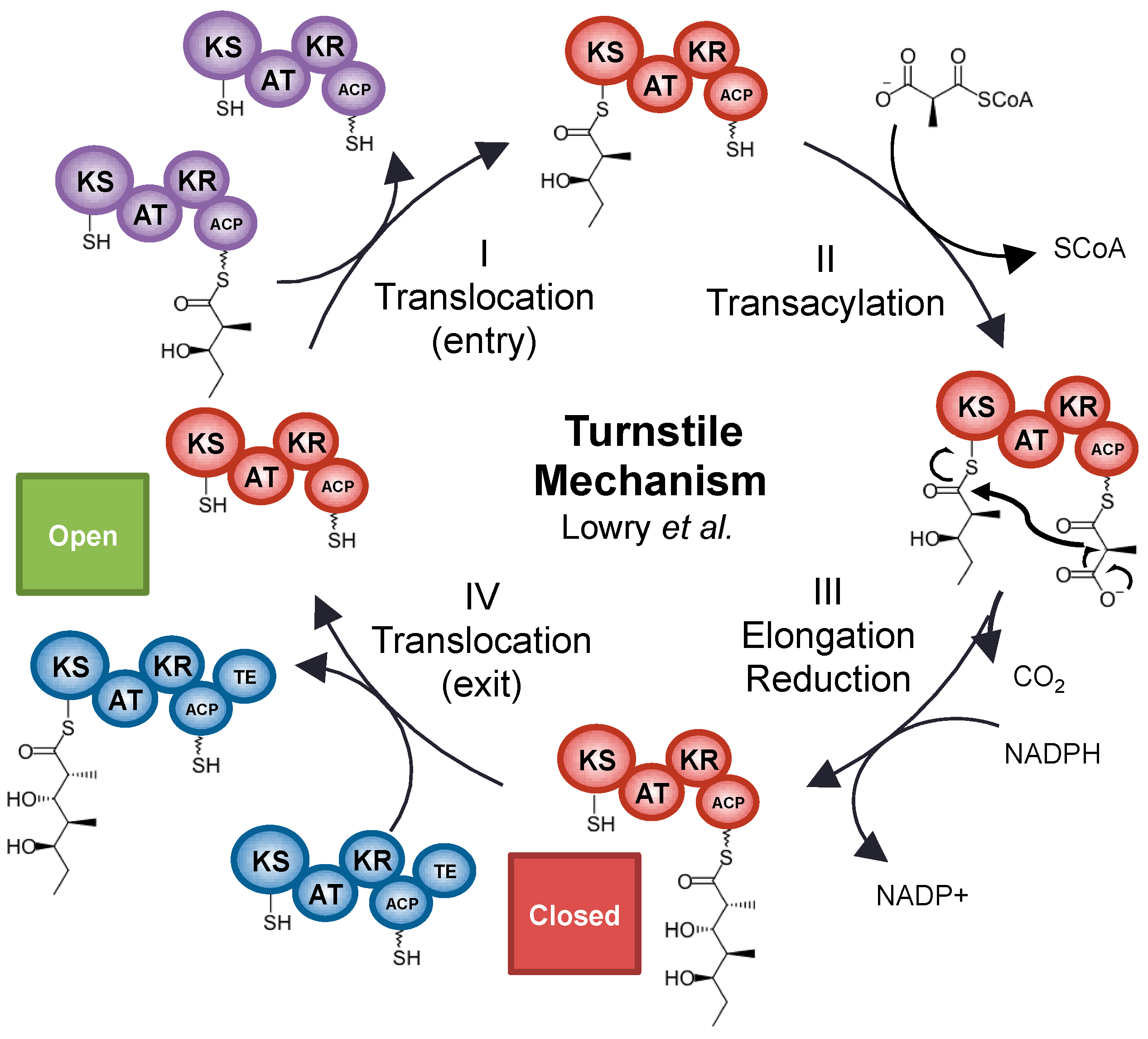
5. Snapshots and Dynamics of Concerted Module Mechanisms
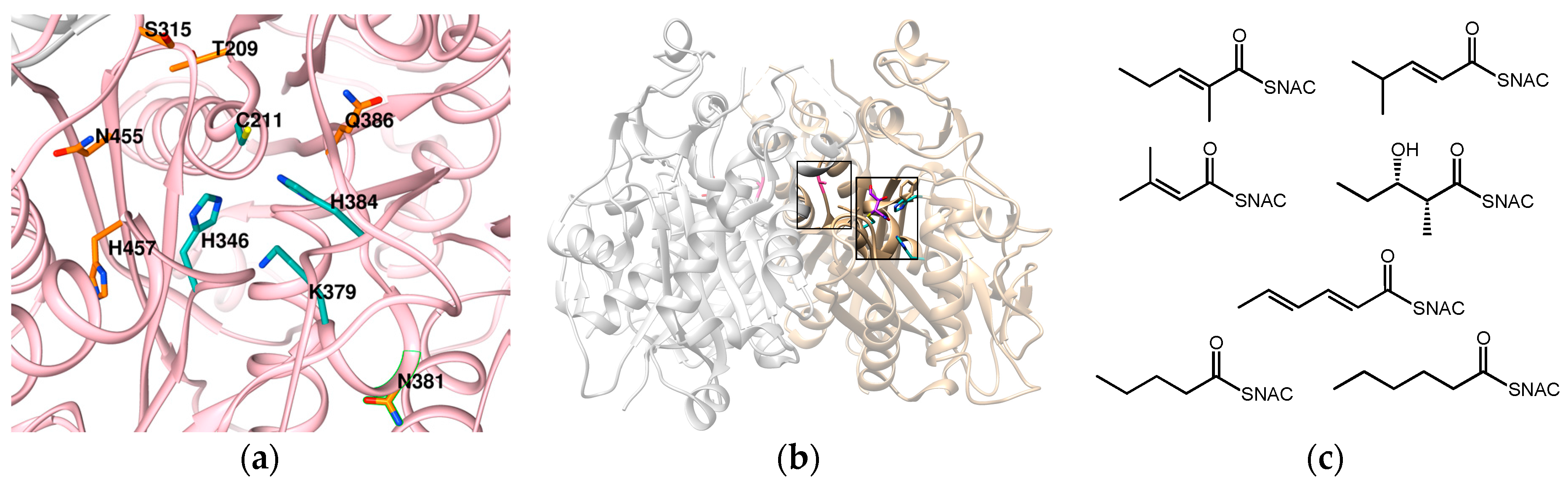
6. Ketosynthase: Key Players in Rate and Promiscuity
7. Acyltransferase: Specificity over Substrate Promiscuity
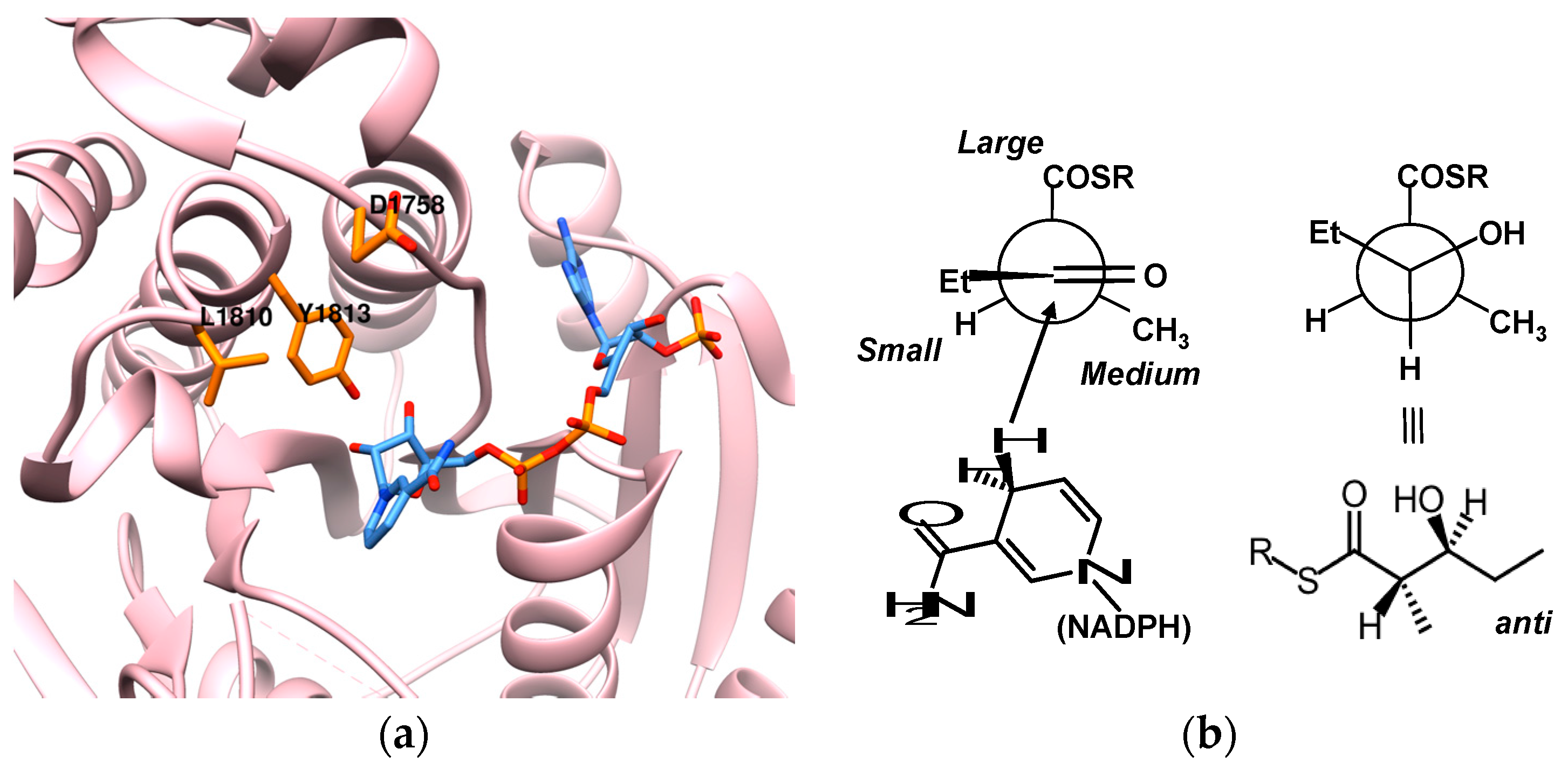
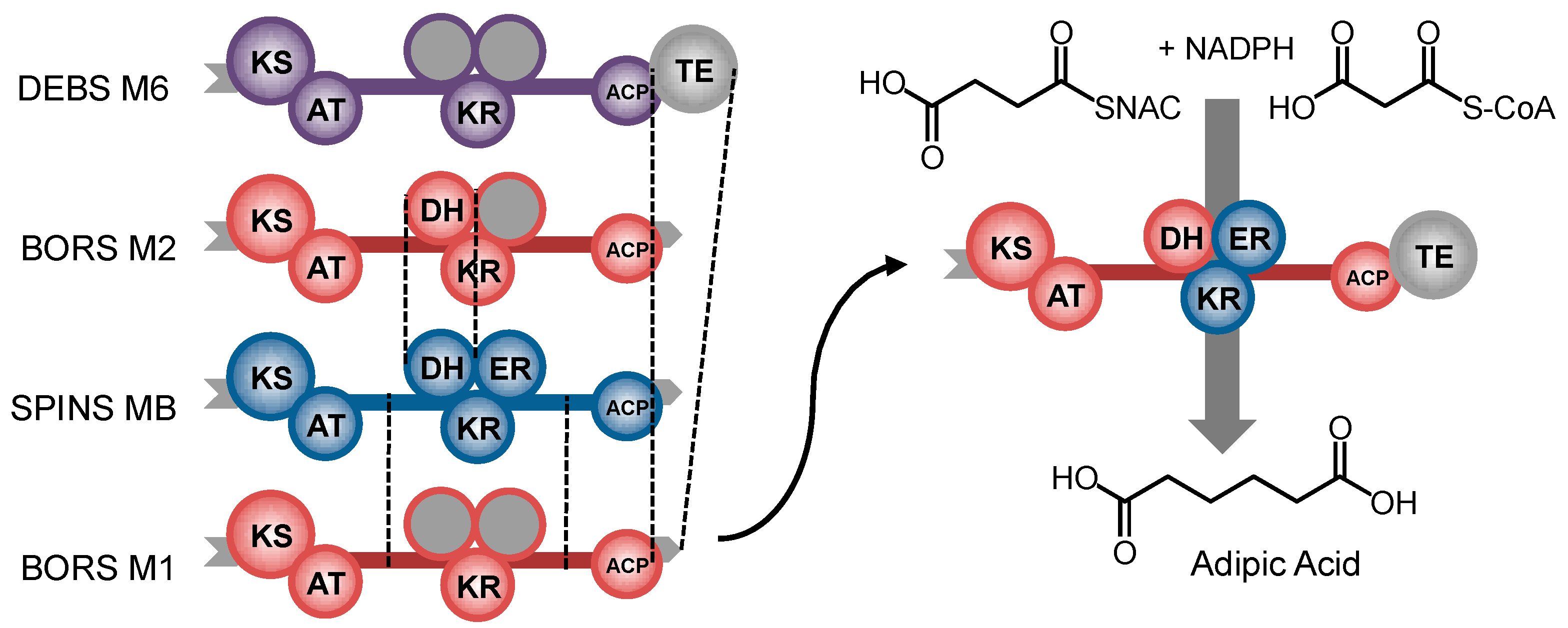
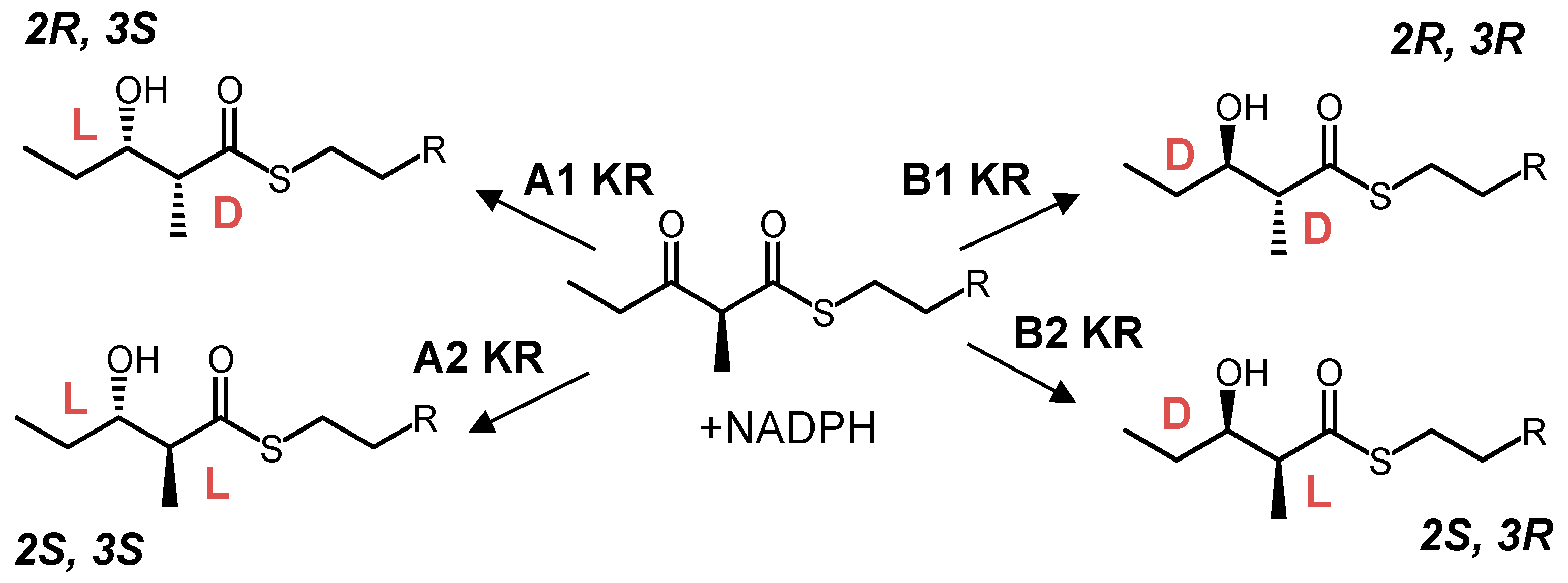
8. Ketoreductase: Modular Context and Catalysis
9. Dehydratase and Enoylreductase: Minimally Controlled
10. Thioesterases: Determinants of Macrocylization
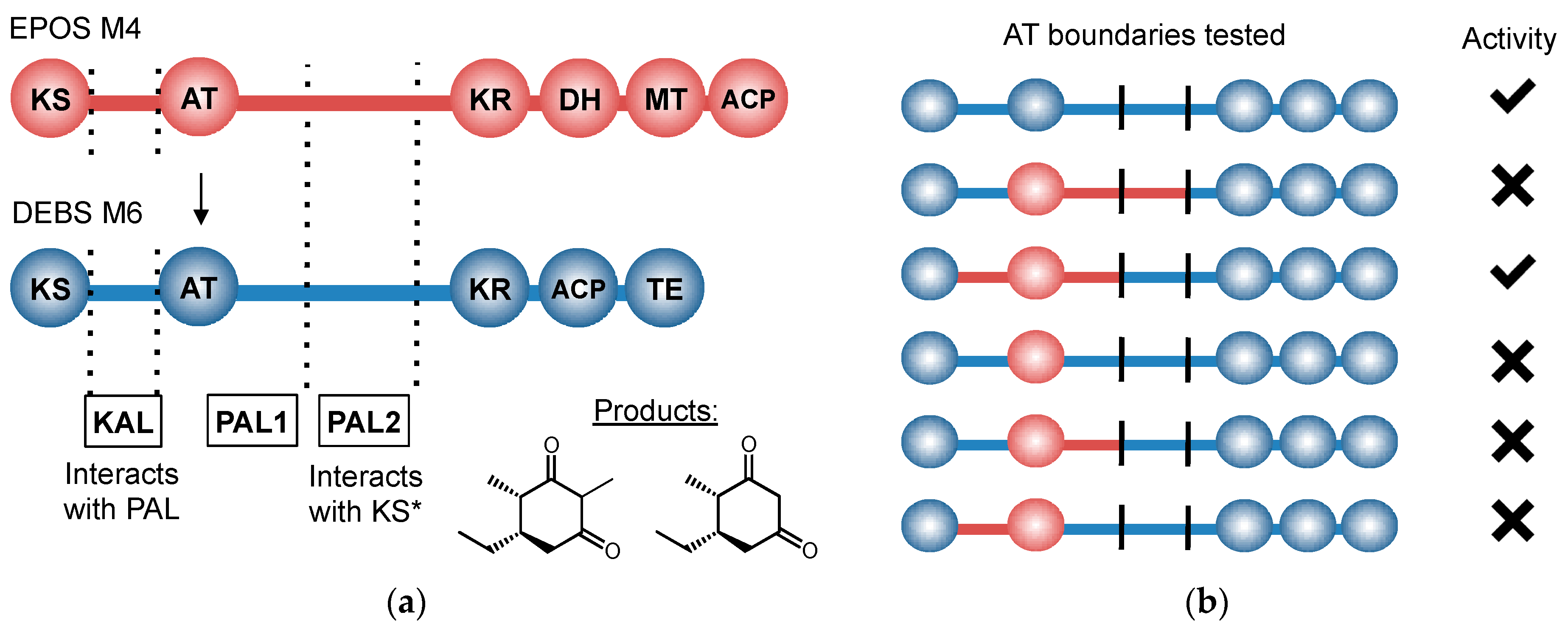
11. Control beyond Single Domains: Extensive, Methodical mPKS Module Engineering
12. Conclusions
Acknowledgments
Conflicts of Interest
References
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J.V. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Ladner, C.C.; Williams, G.J. Harnessing natural product assembly lines: Structure, promiscuity, and engineering. J. Ind. Microbiol. Biotechnol. 2016, 43, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuzawa, S.; Keasling, J.D.; Katz, L. Insights into polyketide biosynthesis gained from repurposing antibiotic-producing polyketide synthases to produce fuels and chemicals. J. Antibiot. 2016, 69, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, Y.; Ishida, K.; Traitcheva, N.; Busch, B.; Dahse, H.M.; Hertweck, C. Freedom and constraint in engineered noncolinear polyketide assembly lines. Chem. Biol. 2015, 22, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Weissman, K.J. Genetic engineering of modular PKSs: From combinatorial biosynthesis to synthetic biology. Nat. Prod. Rep. 2016, 33, 203–230. [Google Scholar] [CrossRef] [PubMed]
- Kwan, D.H.; Schulz, F. The stereochemistry of complex polyketide biosynthesis by modular polyketide synthases. Molecules 2011, 16, 6092–6115. [Google Scholar] [CrossRef] [PubMed]
- Shen, B. Polyketide biosynthesis beyond the type I, II and III polyketide synthase paradigms. Curr. Opin. Chem. Biol. 2003, 7, 285–295. [Google Scholar] [CrossRef]
- Helfrich, E.J.; Piel, J. Biosynthesis of polyketides by trans-AT polyketide synthases. Nat. Prod. Rep. 2016, 33, 231–316. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, Y.; Ji, J.; Zhou, Z.; Yu, J.; Zhu, H.; Su, Z.; Zhang, L.; Zheng, J. Structural and functional analysis of the loading acyltransferase from avermectin modular polyketide synthase. ACS Chem. Biol. 2015, 10, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, Z.; Shao, C.L.; Wang, J.L.; Bai, H.; Wang, C.Y. Bioinformatical analysis of the sequences, structures and functions of fungal polyketide synthase product template domains. Sci. Rep. 2015, 5, 10463. [Google Scholar] [CrossRef] [PubMed]
- Weissman, K.J. The structural biology of biosynthetic megaenzymes. Nat. Chem. Biol. 2015, 11, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Moss, S.J.; Martin, C.J.; Wilkinson, B. Loss of co-linearity by modular polyketide synthases: A mechanism for the evolution of chemical diversity. Nat. Prod. Rep. 2004, 21, 575–593. [Google Scholar] [CrossRef] [PubMed]
- Pang, B.; Wang, M.; Liu, W. Cyclization of polyketides and non-ribosomal peptides on and off their assembly lines. Nat. Prod. Rep. 2016, 33, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, S.; Hertweck, C. On-line enzymatic tailoring of polyketides and peptides in thiotemplate systems. Curr. Opin. Chem. Biol. 2016, 31, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Weber, T.; Blin, K.; Duddela, S.; Krug, D.; Kim, H.U.; Bruccoleri, R.; Lee, S.Y.; Fischbach, M.A.; Muller, R.; Wohlleben, W.; et al. Antismash 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015, 43, W237–W243. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.R.; Boddy, C.N. Clustermine360: A database of microbial pks/nrps biosynthesis. Nucleic Acids Res. 2013, 41, D402–D407. [Google Scholar] [CrossRef] [PubMed]
- Skinnider, M.A.; Dejong, C.A.; Rees, P.N.; Johnston, C.W.; Li, H.; Webster, A.L.; Wyatt, M.A.; Magarvey, N.A. Genomes to natural products prediction informatics for secondary metabolomes (PRISM). Nucleic Acids Res. 2015, 43, 9645–9662. [Google Scholar] [CrossRef] [PubMed]
- Dejong, C.A.; Chen, G.M.; Li, H.; Johnston, C.W.; Edwards, M.R.; Rees, P.N.; Skinnider, M.A.; Webster, A.L.H.; Magarvey, N.A. Polyketide and nonribosomal peptide retro-biosynthesis and global gene cluster matching. Nat. Chem. Biol. 2016, 12, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Ridley, C.P.; Lee, H.Y.; Khosla, C. Evolution of polyketide synthases in bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 4595–4600. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Cimermancic, P.; Sali, A.; Takano, E.; Fischbach, M.A. A systematic computational analysis of biosynthetic gene cluster evolution: Lessons for engineering biosynthesis. PLoS Comput. Biol. 2014, 10, e1004016. [Google Scholar] [CrossRef] [PubMed]
- Wilkening, I.; Gazzola, S.; Riva, E.; Parascandolo, J.S.; Song, L.; Tosin, M. Second-generation probes for biosynthetic intermediate capture: Towards a comprehensive profiling of polyketide assembly. Chem. Commun. 2016, 52, 10392–10395. [Google Scholar] [CrossRef] [PubMed]
- Franke, J.; Hertweck, C. Biomimetic thioesters as probes for enzymatic assembly lines: Synthesis, applications, and challenges. Cell Chem. Biol. 2016, 23, 1179–1192. [Google Scholar] [CrossRef] [PubMed]
- Weissman, K.J. Uncovering the structures of modular polyketide synthases. Nat. Prod. Rep. 2015, 32, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Poust, S.; Hagen, A.; Katz, L.; Keasling, J.D. Narrowing the gap between the promise and reality of polyketide synthases as a synthetic biology platform. Curr. Opin. Biotechnol. 2014, 30, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Keatinge-Clay, A.T. The structural relationship between iterative and modular pkss. Cell Chem. Biol. 2016, 23, 540–542. [Google Scholar] [CrossRef] [PubMed]
- Sherman, D.H.; Smith, J.L. Clearing the skies over modular polyketide synthases. ACS Chem. Biol. 2006, 1, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.J. Engineering polyketide synthases and nonribosomal peptide synthetases. Curr. Opin. Struct. Biol. 2013, 23, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wang, P.; Tang, Y. Engineered polyketide biosynthesis and biocatalysis in Escherichia coli. Appl. Microbiol. Biotechnol. 2010, 88, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Chemler, J.A.; Tripathi, A.; Hansen, D.A.; O’Neil-Johnson, M.; Williams, R.B.; Starks, C.; Park, S.R.; Sherman, D.H. Evolution of efficient modular polyketide synthases by homologous recombination. J. Am. Chem. Soc. 2015, 137, 10603–10609. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.G. Biosynthonics: Charting the future role of biocatalysis and metabolic engineering in drug discovery. Ind. Eng. Chem. Res. 2014, 53, 18597–18610. [Google Scholar] [CrossRef]
- McDaniel, R.; Thamchaipenet, A.; Gustaffson, C.; Fu, H.; Betlach, M.; Betlach, M.; Ashley, G. Multiple genetic modifications of the erythromycin polyketide synthase to produce a library of novel “unnatural” natural products. Proc. Natl. Acad. Sci. USA 1999, 96, 1846–1851. [Google Scholar] [CrossRef]
- Menzella, H.G.; Carney, J.R.; Santi, D.V. Rational design and assembly of synthetic trimodular polyketide synthases. Chem. Biol. 2007, 14, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Ashley, G.; Hutchinson, C.R.; Santi, D.V. A multiplasmid approach to preparing large libraries of polyketides. Proc. Natl. Acad. Sci. USA 1999, 96, 11740–11745. [Google Scholar] [CrossRef] [PubMed]
- Till, M.; Race, P.R. The assembly line enzymology of polyketide biosynthesis. Methods Mol. Biol. 2016, 1401, 31–49. [Google Scholar] [PubMed]
- Edwards, A.L.; Matsui, T.; Weiss, T.M.; Khosla, C. Architectures of whole-module and bimodular proteins from the 6-deoxyerythronolide b synthase. J. Mol. Biol. 2014, 426, 2229–2245. [Google Scholar] [CrossRef] [PubMed]
- Whicher, J.R.; Dutta, S.; Hansen, D.A.; Hale, W.A.; Chemler, J.A.; Dosey, A.M.; Narayan, A.R.; Hakansson, K.; Sherman, D.H.; Smith, J.L.; et al. Structural rearrangements of a polyketide synthase module during its catalytic cycle. Nature 2014, 510, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Qiao, K.; Tang, Y. Structural analysis of protein-protein interactions in type I polyketide synthases. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 98–122. [Google Scholar] [CrossRef] [PubMed]
- Keatinge-Clay, A.T. The structures of type I polyketide synthases. Nat. Prod. Rep. 2012, 29, 1050–1073. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, Y.; Ding, L.; Ishida, K.; Hertweck, C. Rational design of modular polyketide synthases: Morphing the aureothin pathway into a luteoreticulin assembly line. Angew. Chem. 2014, 53, 1560–1564. [Google Scholar] [CrossRef] [PubMed]
- Traitcheva, N.; Jenke-Kodama, H.; He, J.; Dittmann, E.; Hertweck, C. Non-colinear polyketide biosynthesis in the aureothin and neoaureothin pathways: An evolutionary perspective. ChemBioChem 2007, 8, 1841–1849. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Hertweck, C. Iteration as programmed event during polyketide assembly; molecular analysis of the aureothin biosynthesis gene cluster. Chem. Biol. 2003, 10, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Kushnir, S.; Sundermann, U.; Yahiaoui, S.; Brockmeyer, A.; Janning, P.; Schulz, F. Minimally invasive mutagenesis gives rise to a biosynthetic polyketide library. Angew. Chem. 2012, 51, 10664–10669. [Google Scholar] [CrossRef] [PubMed]
- Sherman, D.H. The lego-ization of polyketide biosynthesis. Nat. Biotechnol. 2005, 23, 1083–1084. [Google Scholar] [CrossRef] [PubMed]
- Thomas, I.; Martin, C.J.; Wilkinson, B.; Staunton, J.; Leadlay, P.F. Skipping in a hybrid polyketide synthase: Evidence for acp-to-acp chain transfer. Chem. Biol. 2002, 9, 781–787. [Google Scholar] [CrossRef]
- Khosla, C.; Kapur, S.; Cane, D.E. Revisiting the modularity of modular polyketide synthases. Curr. Opin. Chem. Biol. 2009, 13, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Labonte, J.W.; Townsend, C.A. Active site comparisons and catalytic mechanisms of the hot dog superfamily. Chem. Rev. 2013, 113, 2182–2204. [Google Scholar] [CrossRef] [PubMed]
- Valenzano, C.R.; You, Y.-O.; Garg, A.; Keatinge-Clay, A.; Khosla, C.; Cane, D.E. Stereospecificity of the dehydratase domain of the erythromycin polyketide synthase. J. Am. Chem. Soc. 2010, 132, 14697–14699. [Google Scholar] [CrossRef] [PubMed]
- Robbins, T.; Kapilivsky, J.; Cane, D.E.; Khosla, C. Roles of conserved active site residues in the ketosynthase domain of an assembly line polyketide synthase. Biochemistry 2016, 55, 4476–4484. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Whicher, J.R.; Hansen, D.A.; Hale, W.A.; Chemler, J.A.; Congdon, G.R.; Narayan, A.R.H.; Hakansson, K.; Sherman, D.H.; Smith, J.L.; et al. Structure of a modular polyketide synthase. Nature 2014, 510, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Davison, J.; Dorival, J.; Rabeharindranto, H.; Mazon, H.; Chagot, B.; Gruez, A.; Weissman, K.J. Insights into the function of trans-acyl transferase polyketide synthases from the saxs structure of a complete module. Chem. Sci. 2014, 5, 3081–3095. [Google Scholar] [CrossRef]
- Herbst, D.A.; Jakob, R.P.; Zahringer, F.; Maier, T. Mycocerosic acid synthase exemplifies the architecture of reducing polyketide synthases. Nature 2016, 531, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Dorival, J.; Annaval, T.; Risser, F.; Collin, S.; Roblin, P.; Jacob, C.; Gruez, A.; Chagot, B.; Weissman, K.J. Characterization of intersubunit communication in the virginiamycin trans-acyl transferase polyketide synthase. J. Am. Chem. Soc. 2016, 138, 4155–4167. [Google Scholar] [CrossRef] [PubMed]
- Khosla, C.; Herschlag, D.; Cane, D.E.; Walsh, C.T. Assembly line polyketide synthases: Mechanistic insights and unsolved problems. Biochemistry 2014, 53, 2875–2883. [Google Scholar] [CrossRef] [PubMed]
- Lowry, B.; Li, X.; Robbins, T.; Cane, D.E.; Khosla, C. A turnstile mechanism for the controlled growth of biosynthetic intermediates on assembly line polyketide synthases. ACS Cent. Sci. 2016, 2, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Kapur, S.; Chen, A.Y.; Cane, D.E.; Khosla, C. Molecular recognition between ketosynthase and acyl carrier protein domains of the 6-deoxyerythronolide b synthase. Proc. Natl. Acad. Sci. USA 2010, 107, 22066–22071. [Google Scholar] [CrossRef] [PubMed]
- Charkoudian, L.K.; Liu, C.W.; Capone, S.; Kapur, S.; Cane, D.E.; Togni, A.; Seebach, D.; Khosla, C. Probing the interactions of an acyl carrier protein domain from the 6-deoxyerythronolide b synthase. Protein Sci. 2011, 20, 1244–1255. [Google Scholar] [CrossRef] [PubMed]
- Robbins, T.; Liu, Y.C.; Cane, D.E.; Khosla, C. Structure and mechanism of assembly line polyketide synthases. Curr. Opin. Struct. Biol. 2016, 41, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Chandran, S.S.; Menzella, H.G.; Carney, J.R.; Santi, D.V. Activating hybrid modular interfaces in synthetic polyketide synthases by cassette replacement of ketosynthase domains. Chem. Biol. 2006, 13, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Menzella, H.G.; Reid, R.; Carney, J.R.; Chandran, S.S.; Reisinger, S.J.; Patel, K.G.; Hopwood, D.A.; Santi, D.V. Combinatorial polyketide biosynthesis by de novo design and rearrangement of modular polyketide synthase genes. Nat. Biotechnol. 2005, 23, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Klaus, M.; Ostrowski, M.P.; Austerjost, J.; Robbins, T.; Lowry, B.; Cane, D.E.; Khosla, C. Protein–protein interactions, not substrate recognition, dominates the turnover of chimeric assembly line polyketide synthases. J. Biol. Chem. 2016. [Google Scholar] [CrossRef] [PubMed]
- Vance, S.; Tkachenko, O.; Thomas, B.; Bassuni, M.; Hong, H.; Nietlispach, D.; Broadhurst, W. Sticky swinging arm dynamics: Studies of an acyl carrier protein domain from the mycolactone polyketide synthase. Biochem. J. 2016, 473, 1097–1110. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Kinoshita, K.; Khosla, C.; Cane, D.E. Biochemical analysis of the substrate specificity of the β-ketoacyl-acyl carrier protein synthase domain of module 2 of the erythromycin polyketide synthase. Biochemistry 2004, 43, 16301–16310. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and swiss-pdbviewer: A historical perspective. Electrophoresis 2009, 30 (Suppl. 1), S162–S173. [Google Scholar] [CrossRef] [PubMed]
- Stinear, T.P.; Mve-Obiang, A.; Small, P.L.; Frigui, W.; Pryor, M.J.; Brosch, R.; Jenkin, G.A.; Johnson, P.D.; Davies, J.K.; Lee, R.E.; et al. Giant plasmid-encoded polyketide synthases produce the macrolide toxin of mycobacterium ulcerans. Proc. Natl. Acad. Sci. USA 2004, 101, 1345–1349. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.C.; Hong, H.; Vance, S.; Broadhurst, R.W.; Leadlay, P.F. Broadening substrate specificity of a chain-extending ketosynthase through a single active-site mutation. Chem. Commun. 2016, 52, 8373–8376. [Google Scholar] [CrossRef] [PubMed]
- Dunn, B.J.; Khosla, C. Engineering the acyltransferase substrate specificity of assembly line polyketide synthases. J. R. Soc. Interface 2013, 10, 20130297. [Google Scholar] [CrossRef] [PubMed]
- Hans, M.; Hornung, A.; Dziarnowski, A.; Cane, D.E.; Khosla, C. Mechanistic analysis of acyl transferase domain exchange in polyketide synthase modules. J. Am. Chem. Soc. 2003, 125, 5366–5374. [Google Scholar] [CrossRef] [PubMed]
- Yuzawa, S.; Deng, K.; Wang, G.; Baidoo, E.E.; Northen, T.R.; Adams, P.D.; Katz, L.; Keasling, J.D. Comprehensive in vitro analysis of acyltransferase domain exchanges in modular polyketide synthases and its application for short-chain ketone production. ACS Synth. Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Yuzawa, S.; Kapur, S.; Cane, D.E.; Khosla, C. Role of a conserved arginine residue in linkers between the ketosynthase and acyltransferase domains of multimodular polyketide synthases. Biochemistry 2012, 51, 3708–3710. [Google Scholar] [CrossRef] [PubMed]
- Koryakina, I.; Kasey, C.; McArthur, J.B.; Lowell, A.N.; Chemler, J.A.; Li, S.; Hansen, D.A.; Sherman, D.H.; Williams, G.J. Inversion of extender unit selectivity in the erythromycin polyketide synthase by acyltransferase domain engineering. ACS Chem. Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Dunn, B.J.; Cane, D.E.; Khosla, C. Mechanism and specificity of an acyltransferase domain from a modular polyketide synthase. Biochemistry 2013, 52, 1839–1841. [Google Scholar] [CrossRef] [PubMed]
- Bali, S.; O’Hare, H.M.; Weissman, K.J. Broad substrate specificity of ketoreductases derived from modular polyketide synthases. ChemBioChem 2006, 7, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Bali, S.; Weissman, K.J. Ketoreduction in mycolactone biosynthesis: Insight into substrate specificity and stereocontrol from studies of discrete ketoreductase domains in vitro. ChemBioChem 2006, 7, 1935–1942. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, H.M.; Baerga-Ortiz, A.; Popovic, B.; Spencer, J.B.; Leadlay, P.F. High-throughput mutagenesis to evaluate models of stereochemical control in ketoreductase domains from the erythromycin polyketide synthase. Chem. Biol. 2006, 13, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Siskos, A.P.; Baerga-Ortiz, A.; Bali, S.; Stein, V.; Mamdani, H.; Spiteller, D.; Popovic, B.; Spencer, J.B.; Staunton, J.; Weissman, K.J.; et al. Molecular basis of Celmer’s rules: Stereochemistry of catalysis by isolated ketoreductase domains from modular polyketide synthases. Chem. Biol. 2005, 12, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Piasecki, S.K.; Keatinge-Clay, A.T. Structural studies of an A2-type modular polyketide synthase ketoreductase reveal features controlling alpha-substituent stereochemistry. ACS Chem. Biol. 2013, 8, 1964–1971. [Google Scholar] [CrossRef] [PubMed]
- Kellenberger, L.; Galloway, I.S.; Sauter, G.; Bohm, G.; Hanefeld, U.; Cortes, J.; Staunton, J.; Leadlay, P.F. A polylinker approach to reductive loop swaps in modular polyketide synthases. ChemBioChem 2008, 9, 2740–2749. [Google Scholar] [CrossRef] [PubMed]
- Keatinge-Clay, A.T. Stereocontrol within polyketide assembly lines. Nat. Prod. Rep. 2016, 33, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Kwan, D.H.; Tosin, M.; Schlager, N.; Schulz, F.; Leadlay, P.F. Insights into the stereospecificity of ketoreduction in a modular polyketide synthase. Org. Biomol. Chem. 2011, 9, 2053–2056. [Google Scholar] [CrossRef] [PubMed]
- Annaval, T.; Paris, C.; Leadlay, P.F.; Jacob, C.; Weissman, K.J. Evaluating ketoreductase exchanges as a means of rationally altering polyketide stereochemistry. ChemBioChem 2015, 16, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Garg, A.; Keatinge-Clay, A.T.; Khosla, C.; Cane, D.E. Epimerase and reductase activities of polyketide synthase ketoreductase domains utilize the same conserved tyrosine and serine residues. Biochemistry 2016, 55, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Baerga-Ortiz, A.; Popovic, B.; Siskos, A.P.; O’Hare, H.M.; Spiteller, D.; Williams, M.G.; Campillo, N.; Spencer, J.B.; Leadlay, P.F. Directed mutagenesis alters the stereochemistry of catalysis by isolated ketoreductase domains from the erythromycin polyketide synthase. Chem. Biol. 2006, 13, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.B.; Pasman, M.E.; Keatinge-Clay, A.T. Substrate structure-activity relationships guide rational engineering of modular polyketide synthase ketoreductases. Chem. Commun. 2016, 52, 792–795. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Taylor, C.A.; Piasecki, S.K.; Keatinge-Clay, A.T. Structural and functional analysis of A-type ketoreductases from the amphotericin modular polyketide synthase. Structure 2010, 18, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.H.; Yuzawa, S.; Wang, G.; Baidoo, E.E.; Katz, L.; Keasling, J.D. Alteration of polyketide stereochemistry from anti to syn by a ketoreductase domain exchange in a type I modular polyketide synthase subunit. Biochemistry 2016, 55, 1677–1680. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, M.P.; Cane, D.E.; Khosla, C. Recognition of acyl carrier proteins by ketoreductases in assembly line polyketide synthases. J. Antibiot. 2016, 69, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Gay, D.; You, Y.O.; Keatinge-Clay, A.; Cane, D.E. Structure and stereospecificity of the dehydratase domain from the terminal module of the rifamycin polyketide synthase. Biochemistry 2013, 52, 8916–8928. [Google Scholar] [CrossRef] [PubMed]
- Hagen, A.; Poust, S.; Rond, T.; Fortman, J.L.; Katz, L.; Petzold, C.J.; Keasling, J.D. Engineering a polyketide synthase for in vitro production of adipic acid. ACS Synth. Biol. 2016, 5, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Kwan, D.H.; Sun, Y.; Schulz, F.; Hong, H.; Popovic, B.; Sim-Stark, J.C.; Haydock, S.F.; Leadlay, P.F. Prediction and manipulation of the stereochemistry of enoylreduction in modular polyketide synthases. Chem. Biol. 2008, 15, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Tallorin, L.; Finzel, K.; Nguyen, Q.G.; Beld, J.; La Clair, J.J.; Burkart, M.D. Trapping of the enoyl-acyl carrier protein reductase–acyl carrier protein interaction. J. Am. Chem. Soc. 2016, 138, 3962–3965. [Google Scholar] [CrossRef] [PubMed]
- Horsman, M.E.; Hari, T.P.; Boddy, C.N. Polyketide synthase and non-ribosomal peptide synthetase thioesterase selectivity: Logic gate or a victim of fate? Nat. Prod. Rep. 2016, 33, 183–202. [Google Scholar] [CrossRef] [PubMed]
- Tran, L.; Broadhurst, R.W.; Tosin, M.; Cavalli, A.; Weissman, K.J. Insights into protein-protein and enzyme-substrate interactions in modular polyketide synthases. Chem. Biol. 2010, 17, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Argyropoulos, P.; Bergeret, F.; Pardin, C.; Reimer, J.M.; Pinto, A.; Boddy, C.N.; Schmeing, T.M. Towards a characterization of the structural determinants of specificity in the macrocyclizing thioesterase for deoxyerythronolide b biosynthesis. Biochim. Biophys. Acta 2016, 1860, 486–497. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Choi, S.S.; Sherman, D.H.; Kim, E.S. Thioesterase domain swapping of a linear polyketide tautomycetin with a macrocyclic polyketide pikromycin in Streptomyces sp. Ck4412. J. Ind. Microbiol. Biotechnol. 2016, 43, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-P.; Shi, T.; Wang, X.-L.; Wang, J.; Chen, Q.; Bai, L.; Zhao, Y.-L. Theoretical studies on the mechanism of thioesterase-catalyzed macrocyclization in erythromycin biosynthesis. ACS Catal. 2016, 6, 4369–4378. [Google Scholar] [CrossRef]
- Dorrestein, P.C.; Bumpus, S.B.; Calderone, C.T.; Garneau-Tsodikova, S.; Aron, Z.D.; Straight, P.D.; Kolter, R.; Walsh, C.T.; Kelleher, N.L. Facile detection of acyl and peptidyl intermediates on thiotemplate carrier domains via phosphopantetheinyl elimination reactions during tandem mass spectrometry. Biochemistry 2006, 45, 12756–12766. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bayly, C.L.; Yadav, V.G. Towards Precision Engineering of Canonical Polyketide Synthase Domains: Recent Advances and Future Prospects. Molecules 2017, 22, 235. https://doi.org/10.3390/molecules22020235
Bayly CL, Yadav VG. Towards Precision Engineering of Canonical Polyketide Synthase Domains: Recent Advances and Future Prospects. Molecules. 2017; 22(2):235. https://doi.org/10.3390/molecules22020235
Chicago/Turabian StyleBayly, Carmen L., and Vikramaditya G. Yadav. 2017. "Towards Precision Engineering of Canonical Polyketide Synthase Domains: Recent Advances and Future Prospects" Molecules 22, no. 2: 235. https://doi.org/10.3390/molecules22020235
APA StyleBayly, C. L., & Yadav, V. G. (2017). Towards Precision Engineering of Canonical Polyketide Synthase Domains: Recent Advances and Future Prospects. Molecules, 22(2), 235. https://doi.org/10.3390/molecules22020235