
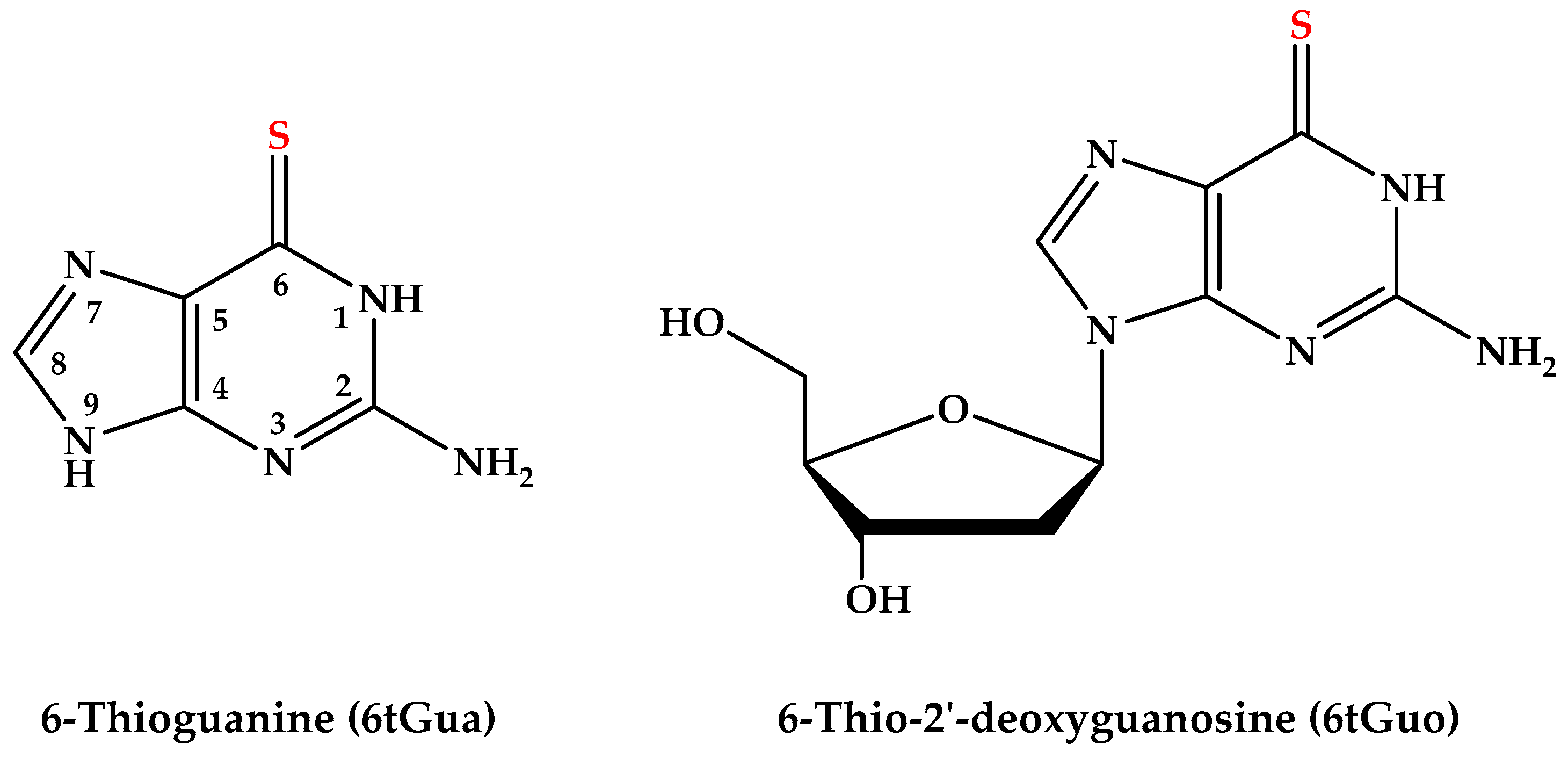
Excited-State Dynamics of the Thiopurine Prodrug 6-Thioguanine: Can N9-Glycosylation Affect Its Phototoxic Activity?


Abstract
:1. Introduction
2. Results and Discussion
2.1. Steady-State Absorption of 6tGua
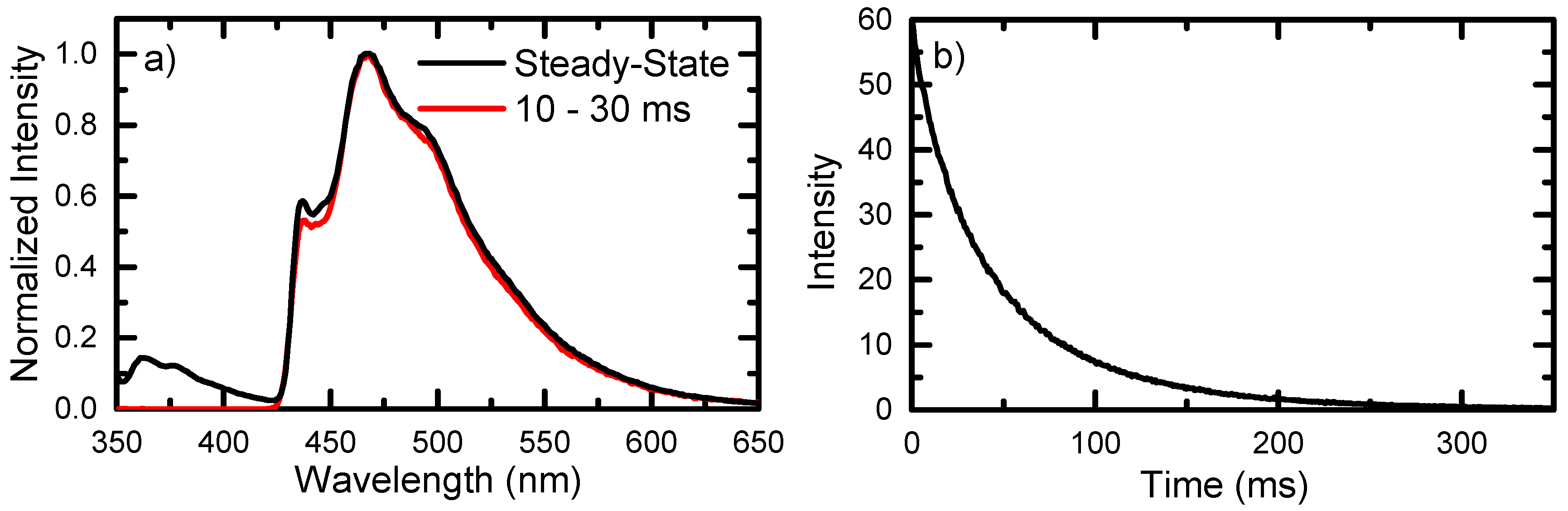
2.2. Steady-State and Time-Resolved Emission of 6tGua
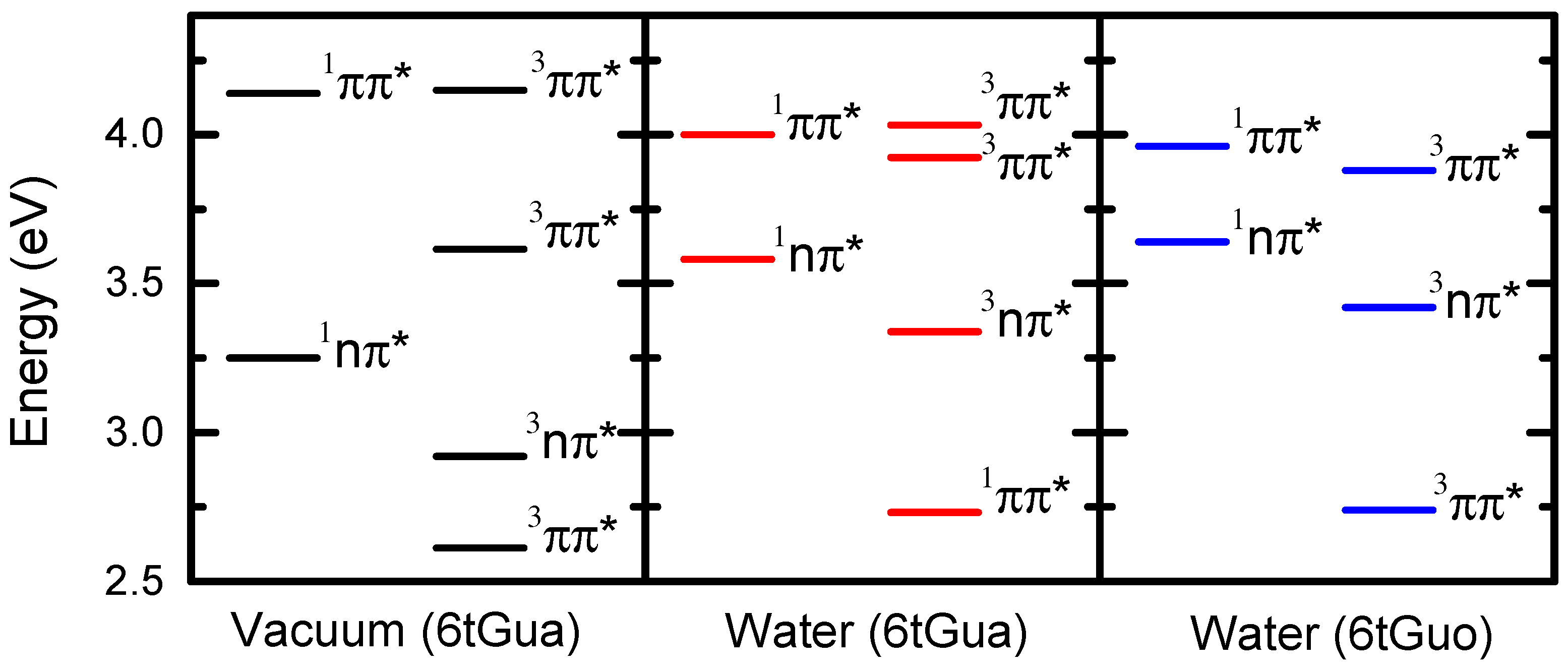
2.3. Quantum-Chemical Calculations of 6tGua
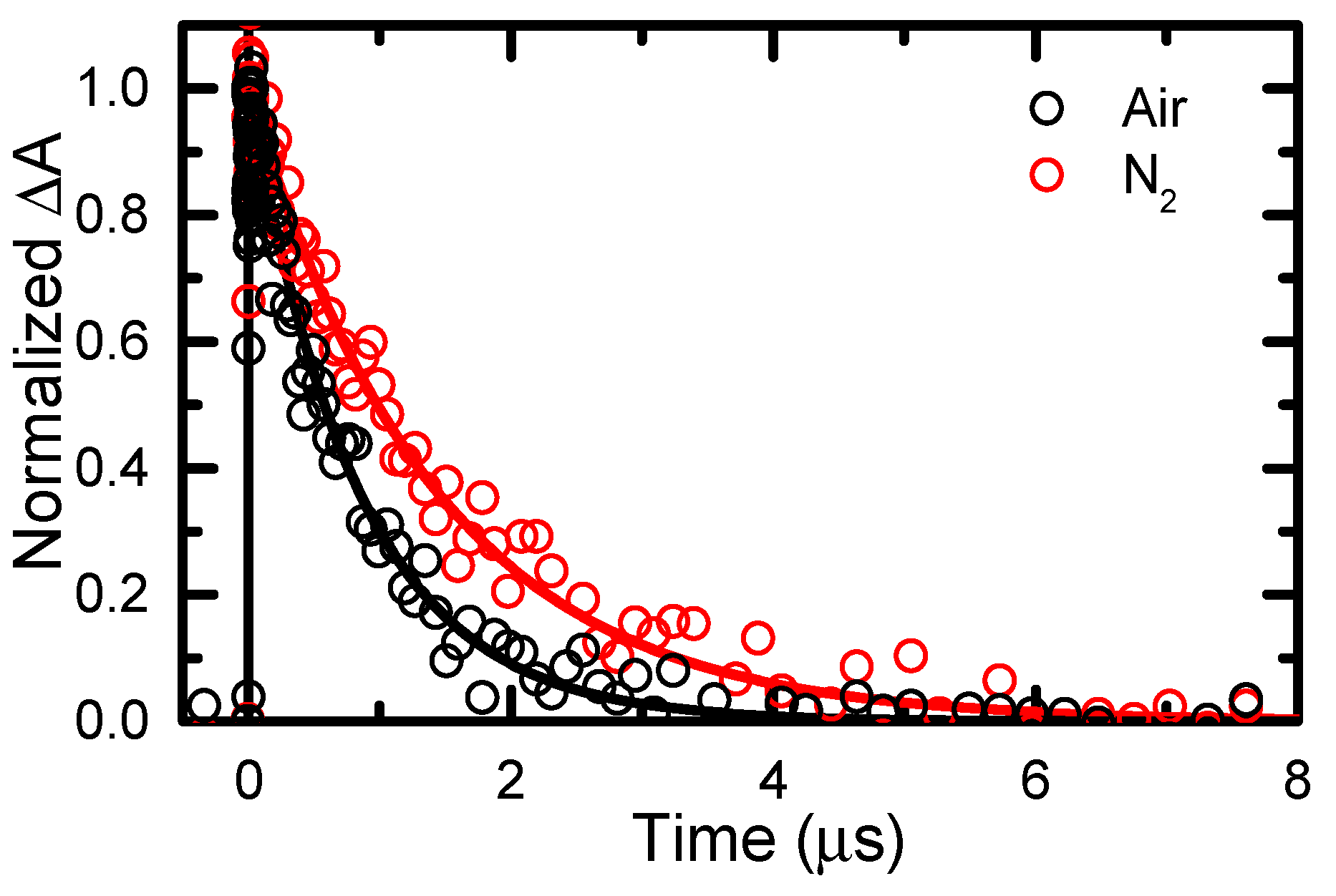
2.4. Transient Absorption Spectroscopy of 6tGua
2.5. Excited-State Relaxation Mechanism of 6tGua
2.6. Effects of N9-Glycosylation on the Photophysics of 6tGua
3. Materials and Methods
3.1. Chemicals
3.2. Steady-State Absorption and Emission Spectra
3.3. Steady-State and Time-Resolved Emission Spectra
3.4. Time-Dependent Density Functional Theory Calculations
3.5. Transient Absorption Spectroscopy
3.6. Determination of Singlet Oxygen Yields
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Swann, P.F.; Waters, T.R.; Moulton, D.C.; Xu, Y.-Z. Role of postreplicative DNA mismatch repair in the cytotoxic action of thioguanine. Science 1996, 273, 1109–1111. [Google Scholar] [CrossRef] [PubMed]
- Flowers, C.R.; Melmon, K.L. Clinical investigators as critical determinants in pharmaceutical innovation. Nat. Med. 1997, 3, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, C.N.; Blanchard, J.F.; Kliewer, E.; Wajda, A. Cancer risk in patients with inflammatory bowel disease. Cancer 2001, 91, 854–862. [Google Scholar] [CrossRef]
- Weinshilboum, R. Thiopurine pharmacogenetics: Clinical and molecular studies of thiopurine methyltransferase. Drug Metab. Dispos. 2001, 29, 601–605. [Google Scholar] [PubMed]
- Euvrard, S.; Kanitakis, J.; Claudy, A. Skin cancers after organ transplantation. N. Engl. J. Med. 2003, 348, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Sahasranaman, S.; Howard, D.; Roy, S. Clinical pharmacology and pharmacogenetics of thiopurines. Eur. J. Clin. Pharmacol. 2008, 64, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Warren, D.J.; Andersen, A.; Slørdal, L. Quantitation of 6-thioguanine residues in peripheral blood leukocyte DNA obtained from patients receiving 6-mercaptopurine-based maintenance therapy. Cancer Res. 1995, 55, 1670–1674. [Google Scholar] [PubMed]
- Cuffari, C.; Seidman, E.; Latour, S.; Theoret, Y. Quantitation of 6-thioguanine in peripheral blood leukocyte DNA in crohn’s disease patients on maintenance 6-mercaptopurine therapy. Can. J. Physiol. Pharmacol. 1996, 74, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhu, X.; Smith, J.; Haygood, M.T.; Gao, R. Direct observation and quantitative characterization of singlet oxygen in aqueous solution upon UVA excitation of 6-thioguanines. J. Phys. Chem. B 2011, 115, 1889–1894. [Google Scholar] [CrossRef] [PubMed]
- Brem, R.; Guven, M.; Karran, P. Oxidatively-generated damage to DNA and proteins mediated by photosensitized UVA. Free Radic. Biol. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.S.; Duarte, T.L.; Cooper, D.; Chen, J.; Nandagopal, S.; Evans, M.D. Combination of azathioprine and UVA irradiation is a major source of cellular 8-oxo-7, 8-dihydro-2′-deoxyguanosine. DNA Repair 2008, 7, 1982–1989. [Google Scholar] [CrossRef] [PubMed]
- Karran, P.; Attard, N. Thiopurines in current medical practice: Molecular mechanisms and contributions to therapy-related cancer. Nat. Rev. Cancer 2008, 8, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Brem, R.; Karran, P. Multiple forms of DNA damage caused by UVA photoactivation of DNA 6-thioguanine. Photochem. Photobiol. 2012, 88, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Karran, P. Thiopurines, DNA damage, DNA repair and therapy-related cancer. Br. Med. Bull. 2006, 79, 153–170. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Jeffs, G.; Ren, X.; O’Donovan, P.; Montaner, B.; Perrett, C.M.; Karran, P.; Xu, Y.-Z. Novel DNA lesions generated by the interaction between therapeutic thiopurines and UVA light. DNA Repair 2007, 6, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Li, F.; Jeffs, G.; Zhang, X.; Xu, Y.-Z.; Karran, P. Guanine sulphinate is a major stable product of photochemical oxidation of DNA 6-thioguanine by UVA irradiation. Nucleic Acids Res. 2010, 38, 1832–1840. [Google Scholar] [CrossRef] [PubMed]
- Gueranger, Q.; Kia, A.; Frith, D.; Karran, P. Crosslinking of DNA repair and replication proteins to DNA in cells treated with 6-thioguanine and UVA. Nucleic Acids Res. 2011, 39, 5057–5066. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Barnes, A.N.; Zhu, X.; Campbell, N.F.; Gao, R. Quantification of thiopurine/UVA-induced singlet oxygen production. J. Photochem. Photobiol. A 2011, 224, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Pollum, M.; Ortiz-Rodríguez, L.A.; Jockusch, S.; Crespo-Hernández, C.E. The triplet state of 6-thio-2′-deoxyguanosine: Intrinsic properties and reactivity toward molecular oxygen. Photochem. Photobiol. 2016, 92, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Bishop, S.M.; Malone, M.; Phillips, D.; Parker, A.W.; Symons, M.C. Singlet oxygen sensitisation by excited state DNA. J. Chem. Soc. Chem. Commun. 1994, 871–872. [Google Scholar] [CrossRef]
- Hemmens, V.J.; Moore, D.E. Photo-oxidation of 6-mercaptopurine in aqueous solution. J. Chem. Soc. Perkin Trans. II 1984, 209–211. [Google Scholar] [CrossRef]
- Hemmens, V.J.; Moore, D.E. Photochemical sensitization by azathioprine and its metabolites—I. 6-mercaptopurine. Photochem. Photobiol. 1986, 43, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Wenska, G.; Filipiak, P.; Taras-Goślińska, K.; Sobierajska, A.; Gdaniec, Z. Orientation-dependent quenching of the triplet excited 6-thiopurine by nucleobases. J. Photochem. Photobiol. A 2011, 217, 55–61. [Google Scholar] [CrossRef]
- Brem, R.; Daehn, I.; Karran, P. Efficient DNA interstrand crosslinking by 6-thioguanine and UVA radiation. DNA Repair 2011, 10, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Rana, T.M. RNA-protein interactions in the tat-trans-activation response element complex determined by site-specific photo-cross-linking. Biochemistry 1998, 37, 4235–4243. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, C.; Guo, C.; Crespo-Hernández, C.E. Excited-state dynamics in 6-thioguanosine from the femtosecond to microsecond time scale. J. Phys. Chem. B. 2011, 115, 3263–3270. [Google Scholar] [CrossRef] [PubMed]
- Taras-Goślińska, K.; Burdziński, G.; Wenska, G. Relaxation of the T1 excited state of 2-thiothymine, its riboside and deoxyriboside-enhanced nonradiative decay rate induced by sugar substituent. J. Photochem. Photobiol. A 2014, 275, 89–95. [Google Scholar] [CrossRef]
- Pollum, M.; Jockusch, S.; Crespo-Hernández, C.E. 2,4-dithiothymine as a potent UVA chemotherapeutic agent. J. Am. Chem. Soc. 2014, 136, 17930–17933. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Zhao, H.; Yu, Y.; Su, H. Formation of guanine-6-sulfonate from 6-thioguanine and singlet oxygen: A combined theoretical and experimental study. J. Am. Chem. Soc. 2013, 135, 4509–4515. [Google Scholar] [CrossRef] [PubMed]
- Rubin, Y.V.; Blagoi, Y.P.; Bokovoy, V. 6-Thioguanine luminescence probe to study DNA and low-molecular-weight systems. J. Fluoresc. 1995, 5, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Rubin, Y.V. Physical properties of anticancer drug 6-thioguanine. In Proceedings of the SPIE 5507; International Society for Optics and Photonics: Bellingham, WA, USA, 2004; pp. 346–357. [Google Scholar]
- Chuan, D.; Wen, Y.; Shaomin, S.; Pin, Y. Determination of thioguanine in pharmaceutical preparations by paper substrate room temperature phosphorimetry. Analyst 2000, 125, 1327–1330. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.J.; Leszczynski, J.; Rubin, Y.V.; Blagoi, Y.P. Tautomerism of thioguanine: From gas phase to DNA. J. Phys. Chem. A 1997, 101, 4753–4760. [Google Scholar] [CrossRef]
- Reichardt, C.; Crespo-Hernández, C.E. Room-temperature phosphorescence of the DNA monomer analogue 4-thiothymidine in aqueous solutions after UVA excitation. J. Phys. Chem. Lett. 2010, 1, 2239–2243. [Google Scholar] [CrossRef]
- Vendrell-Criado, V.; Sáez, J.A.; Lhiaubet-Vallet, V.; Cuquerella, M.C.; Miranda, M.A. Photophysical properties of 5-substituted 2-thiopyrimidines. Photochem. Photobiol. Sci. 2013, 12, 1460–1465. [Google Scholar] [CrossRef] [PubMed]
- Kuramochi, H.; Kobayashi, T.; Suzuki, T.; Ichimura, T. Excited-state dynamics of 6-aza-2-thiothymine and 2-thiothymine: Highly efficient intersystem crossing and singlet oxygen photosensitization. J. Phys. Chem. B 2010, 114, 8782–8789. [Google Scholar] [CrossRef] [PubMed]
- Harada, Y.; Suzuki, T.; Ichimura, T.; Xu, Y.-Z. Triplet formation of 4-thiothymidine and its photosensitization to oxygen studied by time-resolved thermal lensing technique. J. Phys. Chem. B 2007, 111, 5518–5524. [Google Scholar] [CrossRef] [PubMed]
- Lancelot, G.; Helene, C. Spin-orbit coupling in sulphur-containing purine and pyrimidine derivatives. A comparison of caffeine and 6-thiocaffeine. Chem. Phys. Lett. 1971, 9, 327–331. [Google Scholar] [CrossRef]
- Lim, E.; Li, Y.; Li, R. Vibronic interactions between nπ* and ππ* states and radiative and nonradiative T1 → S0 transitions in aromatic carbonyl compounds. J. Chem. Phys. 1970, 53, 2443–2448. [Google Scholar] [CrossRef]
- Shain, A.L.; Sharnoff, M. Static and dynamic paramagnetism of the undistorted triplet state of benzophenone. J. Chem. Phys. 1973, 59, 2335–2343. [Google Scholar] [CrossRef]
- Taherian, M.-R.; Maki, A. Optically detected magnetic resonance study of the phosphorescent states of thiouracils. Chem. Phys. 1981, 55, 85–96. [Google Scholar] [CrossRef]
- Wenska, G.; Taras-Goślińska, K.; Łukaszewicz, A.; Burdziński, G.; Koput, J.; Maciejewski, A. Mechanism and dynamics of intramolecular triplet state decay of 1-propyl-4-thiouracil and its α-methyl-substituted derivatives studied in perfluoro-1, 3-dimethylcyclohexane. Photochem. Photobiol. Sci. 2011, 10, 1294–1302. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Fernández, L.; Pepino, A.J.; Segarra-Martí, J.; Banyasz, A.; Garavelli, M.; Improta, R. Computing the absorption and emission spectra of 5-methylcytidine in different solvents: A test-case for different solvation models. J. Chem. Theory Comput. 2016, 12, 4430–4439. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; Ashwood, B.; Pollum, M.; Marquetand, P.; Crespo-Hernández, C.E.; González, L. Solvatochromic effects on the absorption spectrum of 2-thiocytosine. J. Phys. Chem. 2017. submitted. [Google Scholar]
- Martínez-Fernández, L.; Granucci, G.; Pollum, M.; Crespo-Hernández, C.; Persico, M.; Corral Perez, I. Decoding the molecular basis for the population mechanism of the triplet phototoxic precursors in UVA light-activated pyrimidine anticancer drugs. Chem. Eur. J. 2017, 23, 2619–2627. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Fernández, L.; González, L.; Corral, I. An ab initio mechanism for efficient population of triplet states in cytotoxic sulfur substituted DNA bases: The case of 6-thioguanine. Chem. Commun. 2012, 48, 2134–2136. [Google Scholar] [CrossRef] [PubMed]
- Jacquemin, D.; Duchemin, I.; Blondel, A.; Blase, X. Benchmark of bethe-salpeter for triplet excited-states. J. Chem. Theory Comput. 2017, 13, 767–783. [Google Scholar] [CrossRef] [PubMed]
- Silva-Junior, M.R.; Schreiber, M.; Sauer, S.P.; Thiel, W. Benchmarks for electronically excited states: Time-dependent density functional theory and density functional theory based multireference configuration interaction. J. Chem. Phys. 2008, 129, 104103. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; Pollum, M.; Martínez-Fernández, L.; Dunn, N.; Marquetand, P.; Corral, I.; Crespo-Hernández, C.E.; González, L. The origin of efficient triplet state population in sulfur-substituted nucleobases. Nat. Commun. 2016, 7, 13077. [Google Scholar] [CrossRef] [PubMed]
- Pollum, M.; Martínez-Fernández, L.; Crespo-Hernández, C.E. Photochemistry of nucleic acid bases and their thio-and aza-analogues in solution. In Photoinduced Phenomena in Nucleic Acids I; Springer: Berlin, Germany, 2015; Volume 355, pp. 245–327. [Google Scholar]
- El-Sayed, M. Spin—Orbit coupling and the radiationless processes in nitrogen heterocyclics. J. Chem. Phys. 1963, 38, 2834–2838. [Google Scholar] [CrossRef]
- Schmidt, R.; Tanielian, C.; Dunsbach, R.; Wolff, C. Phenalenone, a universal reference compound for the determination of quantum yields of singlet oxygen O2 (1Δg) sensitization. J. Photochem. Photobiol. A 1994, 79, 11–17. [Google Scholar] [CrossRef]
- Pollum, M.; Crespo-Hernández, C.E. Communication: The dark singlet state as a doorway state in the ultrafast and efficient intersystem crossing dynamics in 2-thiothymine and 2-thiouracil. J. Chem. Phys. 2014, 140, 071101. [Google Scholar] [CrossRef] [PubMed]
- Pollum, M.; Jockusch, S.; Crespo-Hernández, C.E. Increase in the photoreactivity of uracil derivatives by doubling thionation. Phys. Chem. Chem. Phys. 2015, 17, 27851–27861. [Google Scholar] [CrossRef] [PubMed]
- Harada, Y.; Okabe, C.; Kobayashi, T.; Suzuki, T.; Ichimura, T.; Nishi, N.; Xu, Y.-Z. Ultrafast intersystem crossing of 4-thiothymidine in aqueous solution. J. Phys. Chem. Lett. 2009, 1, 480–484. [Google Scholar] [CrossRef]
- Bai, S.; Barbatti, M. Why replacing different oxygens of thymine with sulfur causes distinct absorption and intersystem crossing. J. Phys. Chem. A 2016, 120, 6342–6350. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Bai, S.; Fazzi, D.; Niehaus, T.; Barbatti, M.; Thiel, W. Evaluation of spin-orbit couplings with linear-response TD-DFT, TDA, and TD-DFTB. J. Chem. Theory Comput. 2017, 13, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Fernández, L.; Corral, I.; Granucci, G.; Persico, M. Competing ultrafast intersystem crossing and internal conversion: A time resolved picture for the deactivation of 6-thioguanine. Chem. Sci. 2014, 5, 1336–1347. [Google Scholar] [CrossRef]
- Mai, S.; Marquetand, P.; González, L. Intersystem crossing pathways in the noncanonical nucleobase 2-thiouracil: A time-dependent picture. J. Phys. Chem. Lett. 2016, 7, 1978–1983. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of hartree–fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Vogt, R.A.; Gray, T.G.; Crespo-Hernández, C.E. Subpicosecond intersystem crossing in mono-and di (organophosphine) gold (I) naphthalene derivatives in solution. J. Am. Chem. Soc. 2012, 134, 14808–14817. [Google Scholar] [CrossRef] [PubMed]
- Brister, M.M.; Piñero-Santiago, L.E.; Morel, M.; Arce, R.; Crespo-Hernández, C.E. The photochemical branching ratio in 1,6-dinitropyrene depends on the excitation energy. J. Phys. Chem. Lett. 2016, 7, 5086–5092. [Google Scholar] [CrossRef] [PubMed]
- Jacquemin, D.; Mennucci, B.; Adamo, C. Excited-state calculations with TD-DFT: From benchmarks to simulations in complex environments. Phys. Chem. Chem. Phys. 2011, 13, 16987–16998. [Google Scholar] [CrossRef] [PubMed]
- Improta, R.; Barone, V. Absorption and fluorescence spectra of uracil in the gas phase and in aqueous solution: A TD-DFT quantum mechanical study. J. Am. Chem. Soc. 2004, 126, 14320–14321. [Google Scholar] [CrossRef] [PubMed]
- Shukla, M.; Leszczynski, J. Electronic transitions of thiouracils in the gas phase and in solutions: Time-dependent density functional theory (TD-DFT) study. J. Phys. Chem. A. 2004, 108, 10367–10375. [Google Scholar] [CrossRef]
- Varsano, D.; Di Felice, R.; Marques, M.A.; Rubio, A. A TD-DFT study of the excited states of DNA bases and their assemblies. J. Phys. Chem. B. 2006, 110, 7129–7138. [Google Scholar] [CrossRef] [PubMed]
- Cances, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Reichardt, C.; Vogt, R.A.; Crespo-Hernández, C.E. On the origin of ultrafast nonradiative transitions in nitro-polycyclic aromatic hydrocarbons: Excited-state dynamics in 1-nitronaphthalene. J. Chem. Phys. 2009, 131, 224518. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, C.; Wen, C.; Vogt, R.A.; Crespo-Hernández, C.E. Role of intersystem crossing in the fluorescence quenching of 2-aminopurine 2′-deoxyriboside in solution. Photochem. Photobiol. Sci. 2013, 12, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Capellos, C.; Bielski, B.H. Mathematical Description of Chemical Kinetics in Solution Kinetic Systems; Wiley Interscience: New York, NY, USA, 1972. [Google Scholar]
- Van Stokkum, I.H.; Larsen, D.S.; van Grondelle, R. Global and target analysis of time-resolved spectra. Biochim. Biophys. Acta 2004, 1657, 82–104. [Google Scholar] [CrossRef] [PubMed]
- Rasmusson, M.; Tarnovsky, A.N.; Åkesson, E.; Sundström, V. On the use of two-photon absorption for determination of femtosecond pump–probe cross-correlation functions. Chem. Phys. Lett. 2001, 335, 201–208. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are not available from the authors.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| State | Vacuum (eV) | Vacuum (eV) [46] | Water (eV) | 6tGuo Water (eV) [26] |
|---|---|---|---|---|
| S2(ππ*) | 4.14 (0.26) | 4.05 (0.54) | 4.00 (0.40) | 3.96 (0.45) |
| S1 (nπ*) | 3.25 (0.00) | 3.36 (0.00) | 3.58 (0.00) | 3.64 (0.00) |
| T3 (ππ*) a | 3.62 | 4.24 | 3.92 | 3.88 |
| T2 (nπ*) | 2.92 | 3.31 | 3.34 | 3.42 |
| T1 (ππ*) | 2.61 | 3.10 | 2.73 | 2.74 |
| ΔE(S2-S1) | 0.89 | 0.69 | 0.42 | 0.32 |
| ΔE(S2-T3) | 0.52 | −0.19 | 0.08 | 0.08 |
| ΔE(S2-T2) | 1.22 | 0.74 | 0.66 | 0.54 |
| ΔE(S1-T2) | 0.33 | 0.05 | 0.24 | 0.22 |
| ΔE(S1-T1) | 0.64 | 0.26 | 0.85 | 0.90 |
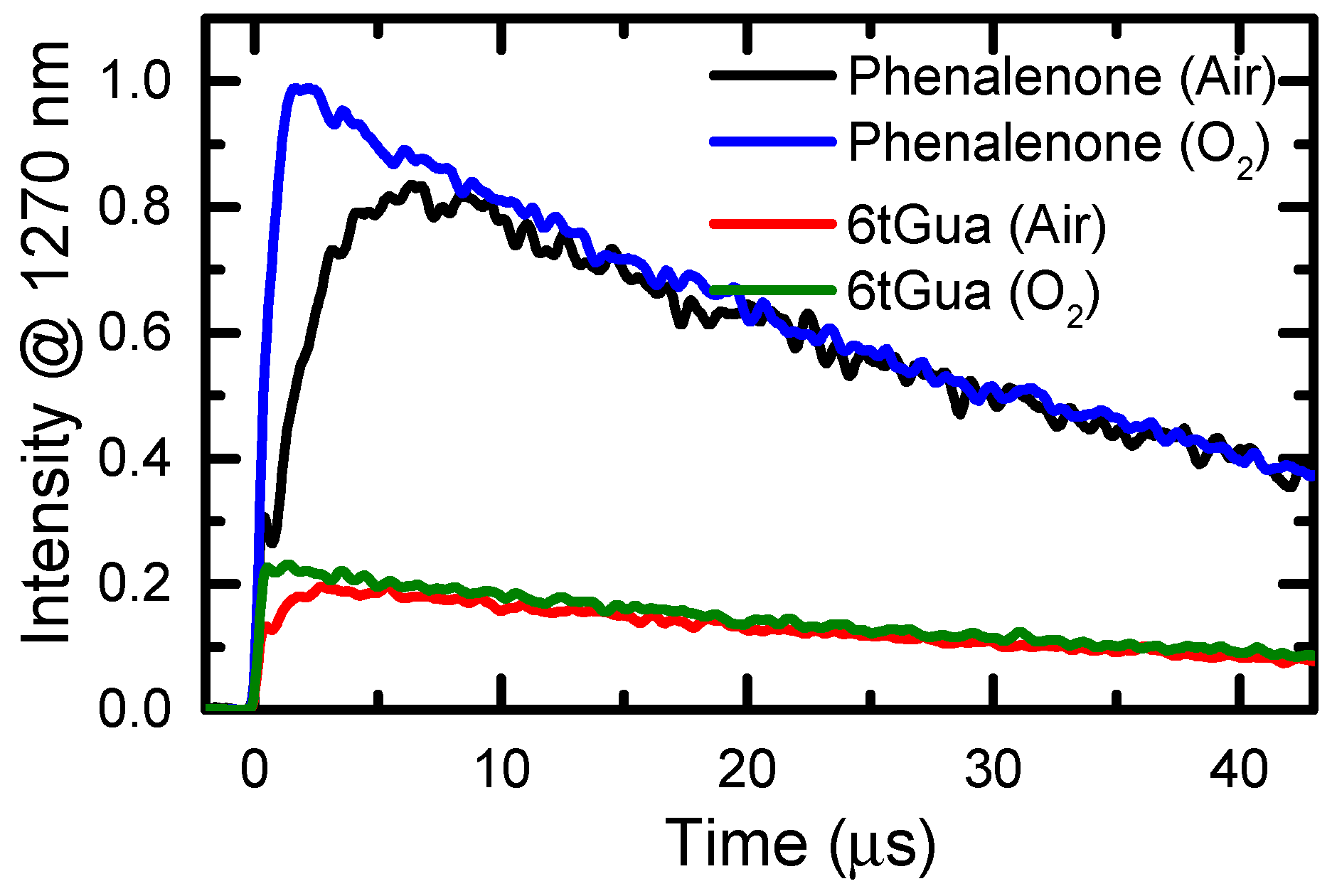
| Base | τ1 (ps) | τ2 (ps) | τ3 (ns) (Air) | τ3 (ns) (N2) | ΦΔ (Air) | ΦΔ (O2) |
|---|---|---|---|---|---|---|
| 6tGua | 0.56 ± 0.06 | 26 ± 3 | 830 ± 70 | 1420 ± 180 | 0.21 ± 0.02 | 0.23 ± 0.02 |
| 6tGuo | 0.31 ± 0.05 [26] | 80 ± 15 [26] | 460 ± 15 [26] | 720 ± 10 [26] | 0.13 ± 0.02 [19] | 0.24 ± 0.02 [19] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashwood, B.; Jockusch, S.; Crespo-Hernández, C.E. Excited-State Dynamics of the Thiopurine Prodrug 6-Thioguanine: Can N9-Glycosylation Affect Its Phototoxic Activity? Molecules 2017, 22, 379. https://doi.org/10.3390/molecules22030379
Ashwood B, Jockusch S, Crespo-Hernández CE. Excited-State Dynamics of the Thiopurine Prodrug 6-Thioguanine: Can N9-Glycosylation Affect Its Phototoxic Activity? Molecules. 2017; 22(3):379. https://doi.org/10.3390/molecules22030379
Chicago/Turabian StyleAshwood, Brennan, Steffen Jockusch, and Carlos E. Crespo-Hernández. 2017. "Excited-State Dynamics of the Thiopurine Prodrug 6-Thioguanine: Can N9-Glycosylation Affect Its Phototoxic Activity?" Molecules 22, no. 3: 379. https://doi.org/10.3390/molecules22030379
APA StyleAshwood, B., Jockusch, S., & Crespo-Hernández, C. E. (2017). Excited-State Dynamics of the Thiopurine Prodrug 6-Thioguanine: Can N9-Glycosylation Affect Its Phototoxic Activity? Molecules, 22(3), 379. https://doi.org/10.3390/molecules22030379