
The Formyl Peptide Receptors: Diversity of Ligands and Mechanism for Recognition

Abstract
:1. Introduction
2. The Formyl Peptide Receptor Family
2.1. The Human Formyl Peptide Receptors
2.2. The Mouse Formyl Peptide Receptors
3. Agonists for the Formyl Peptide Receptors
3.1. Conventional Formyl Peptide Agonists for FPRs
3.2. Other Peptide Agonists for FPRs
3.3. Non-peptide Agonists Screened from Small Molecule Library
3.4. Host-derived Lipid and Lipopeptide Agonists
4. Antagonists for the Formyl Peptide Receptors
4.1. Natural Peptides and Analogs
4.2. Other Peptide Antagonists
4.3. Lipopeptide Antagonists
4.4. Non-Peptide Molecules and Derivatives
5. Structural Basis for Ligand Detection
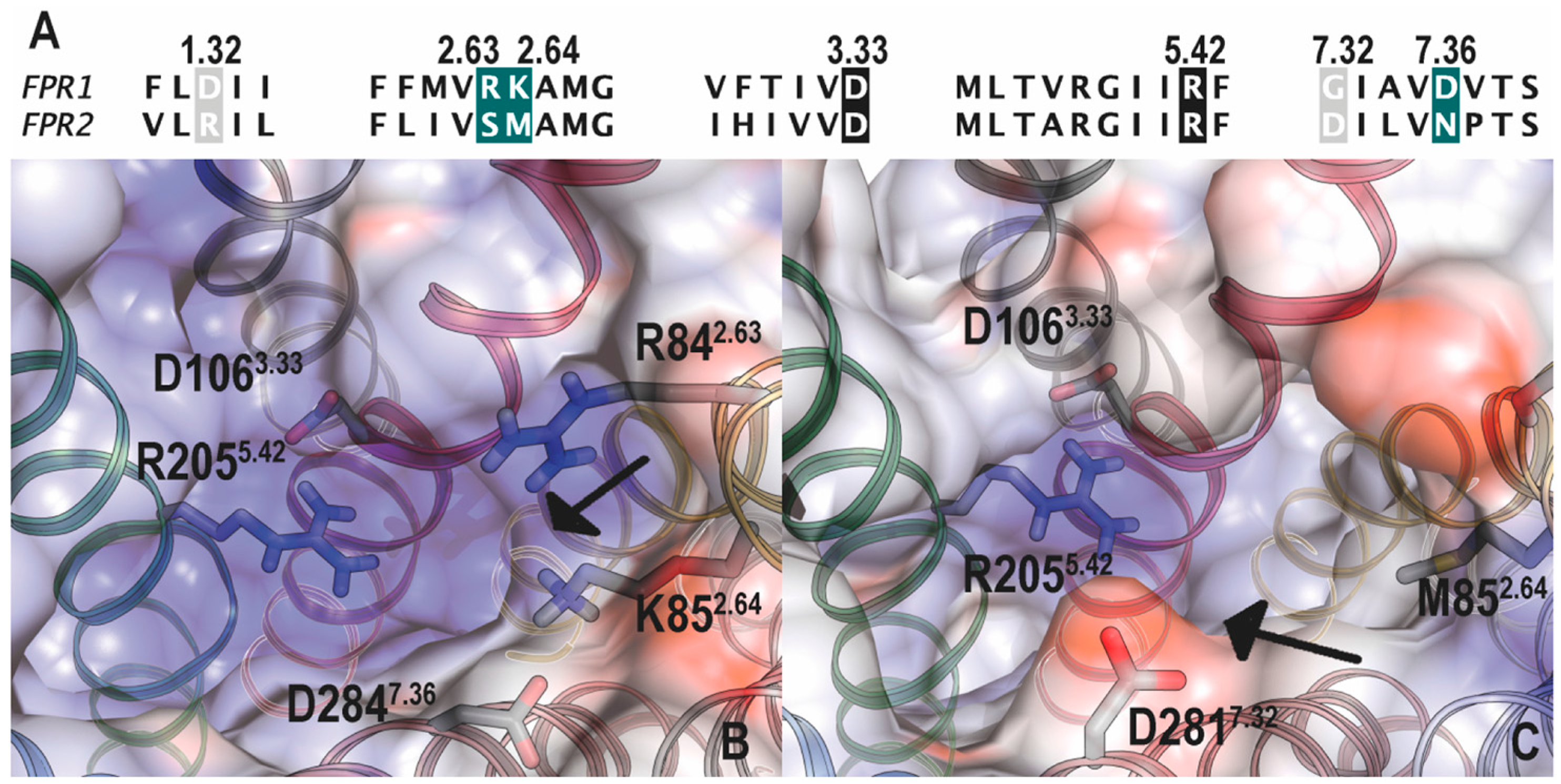
5.1. Binding Sites for Formyl Peptides
5.2. Binding Sites for Other FPR Ligands
5.3. Docking Studies
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| AhR | aryl hydrocarbon receptor |
| Aβ | β-amyloid |
| BLT1 | Leukotriene B4 receptor 1 |
| CB1 | cannabinoid receptor type 1 |
| C5aR | complement component 5a receptor |
| CDCA | chenodeoxycholic acid |
| CHIPS | Chemotaxis inhibitory protein of S. aureus |
| CHO | Chinese hamster ovary |
| CsA | Cyclosporin A |
| CsH | Cyclosporin H |
| CXCR1 | C-X-C motif chemokine receptor 1 |
| CXCR4 | C-X-C chemokine receptor type 4 |
| DAMP | damage-associated molecular pattern |
| DCA | deoxycholic acid |
| EC50 | half maximal effective concentration |
| E. coli | Escherichia coli |
| FAM3D | family with sequence similarity 3 member D |
| FPR | Formyl peptide receptor |
| GPCR | G protein-coupled receptor |
| H. pylori | Helicobacter pylori |
| HTS | high throughput screening |
| i-BOC | iso-butyloxycarbonyl |
| IC50 | half maximal inhibitory concentration |
| Kd | dissociation constant |
| Ki | inhibitor constant |
| L. monocytogenes | Listeria monocytogenes |
| LXA4 | lipoxin A4 |
| μOR | μ-opioid receptor |
| oxLDL | oxidized low-density lipoprotein |
| PAMP | pathogen-associated molecular pattern |
| P2Y2R | P2Y purinoceptor 2 |
| ROS | reactive oxygen species |
| SAA | serum amyloid A |
| SAR | structure-activity relationships |
| S. aureus | Staphylococcus aureus |
| t-Boc | tert-butyloxycarbonyl |
| uPA | Urokinase plasminogen activator |
| UG | Uteroglobin |
References
- Migeotte, I.; Communi, D.; Parmentier, M. Formyl peptide receptors: A promiscuous subfamily of g protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev. 2006, 17, 501–519. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.D.; Boulay, F.; Wang, J.M.; Dahlgren, C.; Gerard, C.; Parmentier, M.; Serhan, C.N.; Murphy, P.M. International union of basic and clinical pharmacology. Lxxiii. Nomenclature for the formyl peptide receptor (fpr) family. Pharmacol. Rev. 2009, 61, 119–161. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, E.; Corcoran, B.A.; Wahl, S.M. N-formylmethionyl peptides as chemoattractants for leucocytes. Proc. Natl. Acad. Sci. USA 1975, 72, 1059–1062. [Google Scholar] [CrossRef] [PubMed]
- Carp, H. Mitochondrial N-formylmethionyl proteins as chemoattractants for neutrophils. J. Exp. Med. 1982, 155, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, C.; Gabl, M.; Holdfeldt, A.; Winther, M.; Forsman, H. Basic characteristics of the neutrophil receptors that recognize formylated peptides, a danger-associated molecular pattern generated by bacteria and mitochondria. Biochem. Pharmacol. 2016, 114, 22–39. [Google Scholar] [CrossRef] [PubMed]
- Schepetkin, I.A.; Khlebnikov, A.I.; Giovannoni, M.P.; Kirpotina, L.N.; Cilibrizzi, A.; Quinn, M.T. Development of small molecule non-peptide formyl peptide receptor (fpr) ligands and molecular modeling of their recognition. Curr. Med. Chem. 2014, 21, 1478–1504. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, K.; Xiang, Y.; Yoshimura, T.; Su, S.; Zhu, J.; Bian, X.W.; Wang, J.M. New development in studies of formyl-peptide receptors: Critical roles in host defense. J. Leukoc. Biol. 2016, 99, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Boulay, F.; Tardif, M.; Brouchon, L.; Vignais, P. Synthesis and use of a novel n-formyl peptide derivative to isolate a human n-formyl peptide receptor cdna. Biochem. Biophys. Res. Commun. 1990, 168, 1103–1109. [Google Scholar] [CrossRef]
- Boulay, F.; Tardif, M.; Brouchon, L.; Vignais, P. The human N-formylpeptide receptor. Characterization of two cdna isolates and evidence for a new subfamily of g-protein-coupled receptors. Biochemistry 1990, 29, 11123–11133. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.D.; Cavanagh, S.L.; Quehenberger, O.; Prossnitz, E.R.; Cochrane, C.G. Isolation of a cdna that encodes a novel granulocyte N-formyl peptide receptor. Biochem. Biophys. Res. Commun. 1992, 184, 582–589. [Google Scholar] [CrossRef]
- Murphy, P.M.; Ozcelik, T.; Kenney, R.T.; Tiffany, H.L.; McDermott, D.; Francke, U. A structural homologue of the n-formyl peptide receptor. Characterization and chromosome mapping of a peptide chemoattractant receptor family. J. Biol. Chem. 1992, 267, 7637–7643. [Google Scholar] [PubMed]
- Bao, L.; Gerard, N.P.; Eddy, R.L., Jr.; Shows, T.B.; Gerard, C. Mapping of genes for the human c5a receptor (c5ar), human fmlp receptor (fpr), and two fmlp receptor homologue orphan receptors (fprh1, fprh2) to chromosome 19. Genomics 1992, 13, 437–440. [Google Scholar] [CrossRef]
- Perez, H.D.; Holmes, R.; Kelly, E.; McClary, J.; Andrews, W.H. Cloning of a cdna encoding a receptor related to the formyl peptide receptor of human neutrophils. Gene 1992, 118, 303–304. [Google Scholar] [CrossRef]
- Muto, Y.; Guindon, S.; Umemura, T.; Kohidai, L.; Ueda, H. Adaptive evolution of formyl peptide receptors in mammals. J. Mol. Evol. 2015, 80, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Le, Y.; Yazawa, H.; Gong, W.; Wang, J.M. Potential role of the formyl peptide receptor-like 1 (fprl1) in inflammatory aspects of Alzheimer’s disease. J. Leukoc. Biol. 2002, 72, 628–635. [Google Scholar] [PubMed]
- Cui, Y.H.; Le, Y.; Gong, W.; Proost, P.; Van Damme, J.; Murphy, W.J.; Wang, J.M. Bacterial lipopolysaccharide selectively up-regulates the function of the chemotactic peptide receptor formyl peptide receptor 2 in murine microglial cells. J. Immunol. 2002, 168, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Bennett, L.; Palucka, A.K.; Arce, E.; Cantrell, V.; Borvak, J.; Banchereau, J.; Pascual, V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 2003, 197, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Iribarren, P.; Zhou, Y.; Hu, J.; Le, Y.; Wang, J.M. Role of formyl peptide receptor-like 1 (fprl1/fpr2) in mononuclear phagocyte responses in Alzheimer disease. Immunol. Res. 2005, 31, 165–176. [Google Scholar] [CrossRef]
- Wan, W.; Gao, J.L. Leukocyte chemoattractant receptor fpr2 may accelerate atherogenesis. Med. Hypotheses 2012, 79, 101–103. [Google Scholar] [CrossRef] [PubMed]
- Prevete, N.; Liotti, F.; Marone, G.; Melillo, R.M.; de Paulis, A. Formyl peptide receptors at the interface of inflammation, angiogenesis and tumor growth. Pharmacol. Res. 2015, 102, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.L.; Lee, E.J.; Murphy, P.M. Impaired antibacterial host defense in mice lacking the n-formylpeptide receptor. J. Exp. Med. 1999, 189, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Chen, K.; Yoshimura, T.; Liu, Y.; Gong, W.; Wang, A.; Gao, J.L.; Murphy, P.M.; Wang, J.M. Formylpeptide receptors are critical for rapid neutrophil mobilization in host defense against Listeria monocytogenes. Sci. Rep. 2012, 2, 786. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.L.; Schneider, E.H.; Dimitrov, E.L.; Haun, F.; Pham, T.M.; Mohammed, A.H.; Usdin, T.B.; Murphy, P.M. Reduced fear memory and anxiety-like behavior in mice lacking formylpeptide receptor 1. Behav. Genet. 2011, 41, 724–733. [Google Scholar] [CrossRef] [PubMed]
- Hartt, J.K.; Barish, G.; Murphy, P.M.; Gao, J.L. N-formylpeptides induce two distinct concentration optima for mouse neutrophil chemotaxis by differential interaction with two n-formylpeptide receptor (fpr) subtypes. Molecular characterization of fpr2, a second mouse neutrophil fpr. J. Exp. Med. 1999, 190, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Southgate, E.L.; He, R.L.; Gao, J.L.; Murphy, P.M.; Nanamori, M.; Ye, R.D. Identification of formyl peptides from Listeria monocytogenes and Staphylococcus aureus as potent chemoattractants for mouse neutrophils. J. Immunol. 2008, 181, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- He, H.Q.; Liao, D.; Wang, Z.G.; Wang, Z.L.; Zhou, H.C.; Wang, M.W.; Ye, R.D. Functional characterization of three mouse formyl peptide receptors. Mol. Pharmacol. 2013, 83, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.S.; Wang, J.M.; Murphy, P.M.; Gao, J.L. Serum amyloid a is a chemotactic agonist at fpr2, a low-affinity n-formylpeptide receptor on mouse neutrophils. Biochem. Biophys. Res. Commun. 2000, 270, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Tiffany, H.L.; Lavigne, M.C.; Cui, Y.H.; Wang, J.M.; Leto, T.L.; Gao, J.L.; Murphy, P.M. Amyloid-beta induces chemotaxis and oxidant stress by acting at formylpeptide receptor 2, a g protein-coupled receptor expressed in phagocytes and brain. J. Biol. Chem. 2001, 276, 23645–23652. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.L.; Guillabert, A.; Hu, J.; Le, Y.; Urizar, E.; Seligman, E.; Fang, K.J.; Yuan, X.; Imbault, V.; Communi, D.; et al. F2l, a peptide derived from heme-binding protein, chemoattracts mouse neutrophils by specifically activating fpr2, the low-affinity n-formylpeptide receptor. J. Immunol. 2007, 178, 1450–1456. [Google Scholar] [CrossRef] [PubMed]
- Migeotte, I.; Riboldi, E.; Franssen, J.D.; Gregoire, F.; Loison, C.; Wittamer, V.; Detheux, M.; Robberecht, P.; Costagliola, S.; Vassart, G.; et al. Identification and characterization of an endogenous chemotactic ligand specific for fprl2. J. Exp. Med. 2005, 201, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Tiffany, H.L.; Gao, J.L.; Roffe, E.; Sechler, J.M.; Murphy, P.M. Characterization of fpr-rs8, an atypical member of the mouse formyl peptide receptor gene family. J. Innate Immun. 2011, 3, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Riviere, S.; Challet, L.; Fluegge, D.; Spehr, M.; Rodriguez, I. Formyl peptide receptor-like proteins are a novel family of vomeronasal chemosensors. Nature 2009, 459, 574–577. [Google Scholar] [CrossRef] [PubMed]
- Liberles, S.D.; Horowitz, L.F.; Kuang, D.; Contos, J.J.; Wilson, K.L.; Siltberg-Liberles, J.; Liberles, D.A.; Buck, L.B. Formyl peptide receptors are candidate chemosensory receptors in the vomeronasal organ. Proc. Natl. Acad. Sci. USA 2009, 106, 9842–9847. [Google Scholar] [CrossRef] [PubMed]
- Takano, T.; Fiore, S.; Maddox, J.F.; Brady, H.R.; Petasis, N.A.; Serhan, C.N. Aspirin-triggered 15-epi-lipoxin a4 (lxa4) and lxa4 stable analogues are potent inhibitors of acute inflammation: Evidence for anti-inflammatory receptors. J. Exp. Med. 1997, 185, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.G.; Ye, R.D. Characterization of two new members of the formyl peptide receptor gene family from 129s6 mice. Gene 2002, 299, 57–63. [Google Scholar] [CrossRef]
- McDonald, B.; Pittman, K.; Menezes, G.B.; Hirota, S.A.; Slaba, I.; Waterhouse, C.C.; Beck, P.L.; Muruve, D.A.; Kubes, P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 2010, 330, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial damps cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Honda, M.; Takeichi, T.; Hashimoto, S.; Yoshii, D.; Isono, K.; Hayashida, S.; Ohya, Y.; Yamamoto, H.; Sugawara, Y.; Inomata, Y. Intravital imaging of neutrophil recruitment reveals the efficacy of fpr1 blockade in hepatic ischemia-reperfusion injury. J. Immunol. 2017, 198, 1718–1728. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, E.; Showell, H.V.; Corcoran, B.A.; Ward, P.A.; Smith, E.; Becker, E.L. The isolation and partial characterization of neutrophil chemotactic factors from escherichia coli. J. Immunol. 1975, 114, 1831–1837. [Google Scholar] [PubMed]
- Freer, R.J.; Day, A.R.; Radding, J.A.; Schiffmann, E.; Aswanikumar, S.; Showell, H.J.; Becker, E.L. Further studies on the structural requirements for synthetic peptide chemoattractants. Biochemistry 1980, 19, 2404–2410. [Google Scholar] [CrossRef] [PubMed]
- Marasco, W.A.; Phan, S.H.; Krutzsch, H.; Showell, H.J.; Feltner, D.E.; Nairn, R.; Becker, E.L.; Ward, P.A. Purification and identification of formyl-methionyl-leucyl-phenylalanine as the major peptide neutrophil chemotactic factor produced by Escherichia coli. J. Biol. Chem. 1984, 259, 5430–5439. [Google Scholar] [PubMed]
- Chester, J.F.; Ross, J.S.; Malt, R.A.; Weitzman, S.A. Acute colitis produced by chemotactic peptides in rats and mice. Am. J. Pathol. 1985, 121, 284–290. [Google Scholar] [PubMed]
- LeDuc, L.E.; Nast, C.C. Chemotactic peptide-induced acute colitis in rabbits. Gastroenterology 1990, 98, 929–935. [Google Scholar] [CrossRef]
- Colucci, M.; Mastriota, M.; Maione, F.; Di Giannuario, A.; Mascolo, N.; Palmery, M.; Severini, C.; Perretti, M.; Pieretti, S. Guinea pig ileum motility stimulation elicited by n-formyl-met-leu-phe (fmlf) involves neurotransmitters and prostanoids. Peptides 2011, 32, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Nast, C.C.; LeDuc, L.E. Chemotactic peptides. Mechanisms, functions, and possible role in inflammatory bowel disease. Dig. Dis. Sci. 1988, 33, 50S–57S. [Google Scholar] [CrossRef] [PubMed]
- Anton, P.A.; Targan, S.R.; Shanahan, F. Increased neutrophil receptors for and response to the proinflammatory bacterial peptide formyl-methionyl-leucyl-phenylalanine in crohn’s disease. Gastroenterology 1989, 97, 20–28. [Google Scholar] [CrossRef]
- Perez, H.D.; Kelly, E.; Elfman, F.; Armitage, G.; Winkler, J. Defective polymorphonuclear leukocyte formyl peptide receptor(s) in juvenile periodontitis. J. Clin. Investig. 1991, 87, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Berend, N.; Armour, C.L.; Black, J.L. Formyl-methionyl-leucyl-phenylalanine causes bronchoconstriction in rabbits. Agents Actions 1986, 17, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.J.; Breslin, A.B.; Kemp, A.S.; Chu, J.; Berend, N. Haematological effects of inhalation of n-formyl-methionyl-leucyl-phenylalanine in man. Thorax 1992, 47, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, M.; Tzanela, M.; Kolbeck, R.C.; McCormick, J.R. Hemodynamic and metabolic effects of intravenous formyl-methionyl-leucyl-phenylalanine (fmlp) in rabbits. In Vivo 1997, 11, 133–139. [Google Scholar] [PubMed]
- Tzanela, M.; Orfanos, S.; McCormick, J.R. Endotoxin augments hemodynamic and metabolic effects of formyl-methionyl-leucyl-phenylalanine (fmlp) in rabbits. In Vivo 2007, 21, 81–87. [Google Scholar] [PubMed]
- Forsman, H.; Winther, M.; Gabl, M.; Skovbakke, S.L.; Boulay, F.; Rabiet, M.J.; Dahlgren, C. Structural changes of the ligand and of the receptor alters the receptor preference for neutrophil activating peptides starting with a formylmethionyl group. Biochim. Biophys. Acta 2015, 1853, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Rabiet, M.J.; Huet, E.; Boulay, F. Human mitochondria-derived n-formylated peptides are novel agonists equally active on fpr and fprl1, while Listeria monocytogenes-derived peptides preferentially activate fpr. Eur. J. Immunol. 2005, 35, 2486–2495. [Google Scholar] [CrossRef] [PubMed]
- Seki, T.; Fukamizu, A.; Kiso, Y.; Mukai, H. Mitocryptide-2, a neutrophil-activating cryptide, is a specific endogenous agonist for formyl-peptide receptor-like 1. Biochem. Biophys. Res. Commun. 2011, 404, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Betten, A.; Bylund, J.; Christophe, T.; Boulay, F.; Romero, A.; Hellstrand, K.; Dahlgren, C. A proinflammatory peptide from Helicobacter pylori activates monocytes to induce lymphocyte dysfunction and apoptosis. J. Clin. Investig. 2001, 108, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Su, S.B.; Gong, W.; Gao, J.L.; Shen, W.; Murphy, P.M.; Oppenheim, J.J.; Wang, J.M. A seven-transmembrane, g protein-coupled receptor, fprl1, mediates the chemotactic activity of serum amyloid a for human phagocytic cells. J. Exp. Med. 1999, 189, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Yu, Y.; Zhu, I.; Cheng, Y.; Sun, P.D. Structural mechanism of serum amyloid a-mediated inflammatory amyloidosis. Proc. Natl. Acad. Sci. USA 2014, 111, 5189–5194. [Google Scholar] [CrossRef] [PubMed]
- Le, Y.; Gong, W.; Tiffany, H.L.; Tumanov, A.; Nedospasov, S.; Shen, W.; Dunlop, N.M.; Gao, J.L.; Murphy, P.M.; Oppenheim, J.J.; et al. Amyloid (beta)42 activates a g-protein-coupled chemoattractant receptor, fpr-like-1. J. Neurosci. 2001, 21, RC123. [Google Scholar] [PubMed]
- Perretti, M.; Chiang, N.; La, M.; Fierro, I.M.; Marullo, S.; Getting, S.J.; Solito, E.; Serhan, C.N. Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin a4 receptor. Nat. Med. 2002, 8, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Hayhoe, R.P.; Kamal, A.M.; Solito, E.; Flower, R.J.; Cooper, D.; Perretti, M. Annexin 1 and its bioactive peptide inhibit neutrophil-endothelium interactions under flow: Indication of distinct receptor involvement. Blood 2006, 107, 2123–2130. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Chen, Q.; Schmidt, A.P.; Anderson, G.M.; Wang, J.M.; Wooters, J.; Oppenheim, J.J.; Chertov, O. Ll-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (fprl1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and t cells. J. Exp. Med. 2000, 192, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Gargiulo, L.; Longanesi-Cattani, I.; Bifulco, K.; Franco, P.; Raiola, R.; Campiglia, P.; Grieco, P.; Peluso, G.; Stoppelli, M.P.; Carriero, M.V. Cross-talk between fmlp and vitronectin receptors triggered by urokinase receptor-derived srsry peptide. J. Biol. Chem. 2005, 280, 25225–25232. [Google Scholar] [CrossRef] [PubMed]
- De Paulis, A.; Montuori, N.; Prevete, N.; Fiorentino, I.; Rossi, F.W.; Visconte, V.; Rossi, G.; Marone, G.; Ragno, P. Urokinase induces basophil chemotaxis through a urokinase receptor epitope that is an endogenous ligand for formyl peptide receptor-like 1 and -like 2. J. Immunol. 2004, 173, 5739–5748. [Google Scholar] [CrossRef] [PubMed]
- Le, Y.; Yazawa, H.; Gong, W.; Yu, Z.; Ferrans, V.J.; Murphy, P.M.; Wang, J.M. The neurotoxic prion peptide fragment prp(106–126) is a chemotactic agonist for the g protein-coupled receptor formyl peptide receptor-like 1. J. Immunol. 2001, 166, 1448–1451. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Premack, B.A.; Wei, Z.; Wang, Y.; Gerard, C.; Showell, H.; Howard, M.; Schall, T.J.; Berahovich, R. Proinflammatory proteases liberate a discrete high-affinity functional fprl1 (ccr12) ligand from ccl23. J. Immunol. 2007, 178, 7395–7404. [Google Scholar] [CrossRef] [PubMed]
- El Zein, N.; Badran, B.; Sariban, E. Vip differentially activates beta2 integrins, cr1, and matrix metalloproteinase-9 in human monocytes through camp/pka, epac, and pi-3k signaling pathways via vip receptor type 1 and fprl1. J. Leukoc. Biol. 2008, 83, 972–981. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.K.; Choi, S.Y.; Kim, Y.; Baek, S.H.; Kim, K.T.; Chae, C.B.; Lambeth, J.D.; Suh, P.G.; Ryu, S.H. A peptide with unique receptor specificity: Stimulation of phosphoinositide hydrolysis and induction of superoxide generation in human neutrophils. J. Immunol. 1997, 158, 1895–1901. [Google Scholar] [PubMed]
- Le, Y.; Gong, W.; Li, B.; Dunlop, N.M.; Shen, W.; Su, S.B.; Ye, R.D.; Wang, J.M. Utilization of two seven-transmembrane, g protein-coupled receptors, formyl peptide receptor-like 1 and formyl peptide receptor, by the synthetic hexapeptide wkymvm for human phagocyte activation. J. Immunol. 1999, 163, 6777–6784. [Google Scholar] [PubMed]
- Christophe, T.; Karlsson, A.; Dugave, C.; Rabiet, M.J.; Boulay, F.; Dahlgren, C. The synthetic peptide trp-lys-tyr-met-val-met-nh2 specifically activates neutrophils through fprl1/lipoxin a4 receptors and is an agonist for the orphan monocyte-expressed chemoattractant receptor fprl2. J. Biol. Chem. 2001, 276, 21585–21593. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Paul, J.I.; Sauve, K.; Schmidt, M.M.; Arcangeli, L.; Ransom, J.; Trueheart, J.; Manfredi, J.P.; Broach, J.R.; Murphy, A.J. Identification of surrogate agonists for the human fprl-1 receptor by autocrine selection in yeast. Nat. Biotechnol. 1998, 16, 1334–1337. [Google Scholar] [CrossRef] [PubMed]
- Madenspacher, J.H.; Azzam, K.M.; Gong, W.; Gowdy, K.M.; Vitek, M.P.; Laskowitz, D.T.; Remaley, A.T.; Wang, J.M.; Fessler, M.B. Apolipoproteins and apolipoprotein mimetic peptides modulate phagocyte trafficking through chemotactic activity. J. Biol. Chem. 2012, 287, 43730–43740. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Kim, S.D.; Shim, J.W.; Kim, H.J.; Kwon, J.Y.; Kim, J.M.; Baek, S.H.; Park, J.S.; Bae, Y.S. Activation of human monocytes by a formyl peptide receptor 2-derived pepducin. FEBS Lett. 2010, 584, 4102–4108. [Google Scholar] [CrossRef] [PubMed]
- Forsman, H.; Bylund, J.; Oprea, T.I.; Karlsson, A.; Boulay, F.; Rabiet, M.J.; Dahlgren, C. The leukocyte chemotactic receptor fpr2, but not the closely related fpr1, is sensitive to cell-penetrating pepducins with amino acid sequences descending from the third intracellular receptor loop. Biochim. Biophys. Acta 2013, 1833, 1914–1923. [Google Scholar] [CrossRef] [PubMed]
- Fiore, S.; Maddox, J.F.; Perez, H.D.; Serhan, C.N. Identification of a human cdna encoding a functional high affinity lipoxin a4 receptor. J. Exp. Med. 1994, 180, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Nanamori, M.; Cheng, X.; Mei, J.; Sang, H.; Xuan, Y.; Zhou, C.; Wang, M.W.; Ye, R.D. A novel nonpeptide ligand for formyl peptide receptor-like 1. Mol. Pharmacol. 2004, 66, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- Kirpotina, L.N.; Khlebnikov, A.I.; Schepetkin, I.A.; Ye, R.D.; Rabiet, M.J.; Jutila, M.A.; Quinn, M.T. Identification of novel small-molecule agonists for human formyl peptide receptors and pharmacophore models of their recognition. Mol. Pharmacol. 2010, 77, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Frohn, M.; Xu, H.; Zou, X.; Chang, C.; McElvaine, M.; Plant, M.H.; Wong, M.; Tagari, P.; Hungate, R.; Burli, R.W. New ‘chemical probes’ to examine the role of the hfprl1 (or alxr) receptor in inflammation. Bioorg. Med. Chem. Lett. 2007, 17, 6633–6637. [Google Scholar] [CrossRef] [PubMed]
- Burli, R.W.; Xu, H.; Zou, X.; Muller, K.; Golden, J.; Frohn, M.; Adlam, M.; Plant, M.H.; Wong, M.; McElvain, M.; et al. Potent hfprl1 (alxr) agonists as potential anti-inflammatory agents. Bioorg. Med. Chem. Lett. 2006, 16, 3713–3718. [Google Scholar] [CrossRef] [PubMed]
- Sogawa, Y.; Ohyama, T.; Maeda, H.; Hirahara, K. Formyl peptide receptor 1 and 2 dual agonist inhibits human neutrophil chemotaxis by the induction of chemoattractant receptor cross-desensitization. J. Pharmacol. Sci. 2011, 115, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Crocetti, L.; Claudia, V.; Cilibrizzi, A.; Graziano, A.; Khlebnikov, A.I.; Kirpotina, L.N.; Schepetkin, I.A.; Quinn, M.T.; Giovannoni, M.P. Synthesis and pharmacological evaluation of new pyridazin-based thioderivatives as formyl peptide receptor (fpr) agonists. Drug Dev. Res. 2013, 74, 12. [Google Scholar] [CrossRef]
- Cilibrizzi, A.; Schepetkin, I.A.; Bartolucci, G.; Crocetti, L.; Dal Piaz, V.; Giovannoni, M.P.; Graziano, A.; Kirpotina, L.N.; Quinn, M.T.; Vergelli, C. Synthesis, enantioresolution, and activity profile of chiral 6-methyl-2,4-disubstituted pyridazin-3(2h)-ones as potent n-formyl peptide receptor agonists. Bioorg. Med. Chem. 2012, 20, 3781–3792. [Google Scholar] [CrossRef] [PubMed]
- Schepetkin, I.A.; Kirpotina, L.N.; Khlebnikov, A.I.; Leopoldo, M.; Lucente, E.; Lacivita, E.; De Giorgio, P.; Quinn, M.T. 3-(1h-indol-3-yl)-2-[3-(4-nitrophenyl)ureido]propanamide enantiomers with human formyl-peptide receptor agonist activity: Molecular modeling of chiral recognition by fpr2. Biochem. Pharmacol. 2013, 85, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Lacivita, E.; Schepetkin, I.A.; Stama, M.L.; Kirpotina, L.N.; Colabufo, N.A.; Perrone, R.; Khlebnikov, A.I.; Quinn, M.T.; Leopoldo, M. Novel 3-(1h-indol-3-yl)-2-[3-(4-methoxyphenyl)ureido]propanamides as selective agonists of human formyl-peptide receptor 2. Bioorg. Med. Chem. 2015, 23, 3913–3924. [Google Scholar] [CrossRef] [PubMed]
- Pinilla, C.; Edwards, B.S.; Appel, J.R.; Yates-Gibbins, T.; Giulianotti, M.A.; Medina-Franco, J.L.; Young, S.M.; Santos, R.G.; Sklar, L.A.; Houghten, R.A. Selective agonists and antagonists of formylpeptide receptors: Duplex flow cytometry and mixture-based positional scanning libraries. Mol. Pharmacol. 2013, 84, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Haas, P.J.; de Haas, C.J.; Kleibeuker, W.; Poppelier, M.J.; van Kessel, K.P.; Kruijtzer, J.A.; Liskamp, R.M.; van Strijp, J.A. N-terminal residues of the chemotaxis inhibitory protein of staphylococcus aureus are essential for blocking formylated peptide receptor but not c5a receptor. J. Immunol. 2004, 173, 5704–5711. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.; Zhang, Z.; Lee, Y.C.; Gao, J.L.; Mukherjee, A.B. Uteroglobin suppresses allergen-induced th2 differentiation by down-regulating the expression of serum amyloid a and socs-3 genes. FEBS Lett. 2006, 580, 6022–6026. [Google Scholar] [CrossRef] [PubMed]
- Wenzel-Seifert, K.; Seifert, R. Cyclosporin h is a potent and selective formyl peptide receptor antagonist. Comparison with n-t-butoxycarbonyl-l-phenylalanyl-l-leucyl-l-phenylalanyl-l- leucyl-l-phenylalanine and cyclosporins a, b, c, d, and e. J. Immunol. 1993, 150, 4591–4599. [Google Scholar] [PubMed]
- Yousif, A.M.; Minopoli, M.; Bifulco, K.; Ingangi, V.; Di Carluccio, G.; Merlino, F.; Motti, M.L.; Grieco, P.; Carriero, M.V. Cyclization of the urokinase receptor-derived ser-arg-ser-arg-tyr peptide generates a potent inhibitor of trans-endothelial migration of monocytes. PLoS ONE 2015, 10, e0126172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, Y.S.; Lee, H.Y.; Jo, E.J.; Kim, J.I.; Kang, H.K.; Ye, R.D.; Kwak, J.Y.; Ryu, S.H. Identification of peptides that antagonize formyl peptide receptor-like 1-mediated signaling. J. Immunol. 2004, 173, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Forsman, H.; Andreasson, E.; Karlsson, J.; Boulay, F.; Rabiet, M.J.; Dahlgren, C. Structural characterization and inhibitory profile of formyl peptide receptor 2 selective peptides descending from a pip2-binding domain of gelsolin. J. Immunol. 2012, 189, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Winther, M.; Gabl, M.; Welin, A.; Dahlgren, C.; Forsman, H. A neutrophil inhibitory pepducin derived from fpr1 expected to target fpr1 signaling hijacks the closely related fpr2 instead. FEBS Lett. 2015, 589, 1832–1839. [Google Scholar] [CrossRef] [PubMed]
- Skovbakke, S.L.; Heegaard, P.M.; Larsen, C.J.; Franzyk, H.; Forsman, H.; Dahlgren, C. The proteolytically stable peptidomimetic pam-(lys-betanspe)6-nh2 selectively inhibits human neutrophil activation via formyl peptide receptor 2. Biochem. Pharmacol. 2015, 93, 182–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skovbakke, S.L.; Winther, M.; Gabl, M.; Holdfeldt, A.; Linden, S.; Wang, J.M.; Dahlgren, C.; Franzyk, H.; Forsman, H. The peptidomimetic lau-(lys-betanspe)6-nh2 antagonizes formyl peptide receptor 2 expressed in mouse neutrophils. Biochem. Pharmacol. 2016, 119, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Schepetkin, I.A.; Kirpotina, L.N.; Khlebnikov, A.I.; Cheng, N.; Ye, R.D.; Quinn, M.T. Antagonism of human formyl peptide receptor 1 (fpr1) by chromones and related isoflavones. Biochem. Pharmacol. 2014, 92, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Zhang, S.; Nanamori, M.; Zhang, Y.; Liu, Q.; Li, N.; Sun, M.; Tian, J.; Ye, P.P.; Cheng, N.; et al. Pharmacological characterization of a novel nonpeptide antagonist for formyl peptide receptor-like 1. Mol. Pharmacol. 2007, 72, 976–983. [Google Scholar] [CrossRef] [PubMed]
- He, H.Q.; Troksa, E.L.; Caltabiano, G.; Pardo, L.; Ye, R.D. Structural determinants for the interaction of formyl peptide receptor 2 with peptide ligands. J. Biol. Chem. 2014, 289, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Otto, M. Community-associated mrsa: What makes them special? Int. J. Med. Microbiol. 2013, 303, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Kretschmer, D.; Gleske, A.K.; Rautenberg, M.; Wang, R.; Koberle, M.; Bohn, E.; Schoneberg, T.; Rabiet, M.J.; Boulay, F.; Klebanoff, S.J.; et al. Human formyl peptide receptor 2 senses highly pathogenic Staphylococcus aureus. Cell Host Microbe 2010, 7, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Mader, D.; Rabiet, M.J.; Boulay, F.; Peschel, A. Formyl peptide receptor-mediated proinflammatory consequences of peptide deformylase inhibition in Staphylococcus aureus. Microbes Infect. 2010, 12, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Forsman, H.; Christenson, K.; Bylund, J.; Dahlgren, C. Receptor-dependent and -independent immunomodulatory effects of phenol-soluble modulin peptides from Staphylococcus aureus on human neutrophils are abrogated through peptide inactivation by reactive oxygen species. Infect. Immun. 2012, 80, 1987–1995. [Google Scholar] [CrossRef] [PubMed]
- Su, S.B.; Gao, J.; Gong, W.; Dunlop, N.M.; Murphy, P.M.; Oppenheim, J.J.; Wang, J.M. T21/dp107, a synthetic leucine zipper-like domain of the hiv-1 envelope gp41, attracts and activates human phagocytes by using g-protein-coupled formyl peptide receptors. J. Immunol. 1999, 162, 5924–5930. [Google Scholar] [PubMed]
- Su, S.B.; Gong, W.H.; Gao, J.L.; Shen, W.P.; Grimm, M.C.; Deng, X.; Murphy, P.M.; Oppenheim, J.J.; Wang, J.M. T20/dp178, an ectodomain peptide of human immunodeficiency virus type 1 gp41, is an activator of human phagocyte n-formyl peptide receptor. Blood 1999, 93, 3885–3892. [Google Scholar] [PubMed]
- Deng, X.; Ueda, H.; Su, S.B.; Gong, W.; Dunlop, N.M.; Gao, J.L.; Murphy, P.M.; Wang, J.M. A synthetic peptide derived from human immunodeficiency virus type 1 gp120 downregulates the expression and function of chemokine receptors ccr5 and cxcr4 in monocytes by activating the 7-transmembrane g-protein-coupled receptor fprl1/lxa4r. Blood 1999, 94, 1165–1173. [Google Scholar] [PubMed]
- Le, Y.; Jiang, S.; Hu, J.; Gong, W.; Su, S.; Dunlop, N.M.; Shen, W.; Li, B.; Wang, J.M. N36, a synthetic n-terminal heptad repeat domain of the hiv-1 envelope protein gp41, is an activator of human phagocytes. Clin. Immunol. 2000, 96, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Proost, P.; Li, B.; Gong, W.; Le, Y.; Sargeant, R.; Murphy, P.M.; Van Damme, J.; Wang, J.M. Activation of the chemotactic peptide receptor fprl1 in monocytes phosphorylates the chemokine receptor ccr5 and attenuates cell responses to selected chemokines. Biochem. Biophys. Res. Commun. 2000, 272, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Hartt, J.K.; Liang, T.; Sahagun-Ruiz, A.; Wang, J.M.; Gao, J.L.; Murphy, P.M. The hiv-1 cell entry inhibitor t-20 potently chemoattracts neutrophils by specifically activating the n-formylpeptide receptor. Biochem. Biophys. Res. Commun. 2000, 272, 699–704. [Google Scholar] [CrossRef] [PubMed]
- De Paulis, A.; Prevete, N.; Fiorentino, I.; Walls, A.F.; Curto, M.; Petraroli, A.; Castaldo, V.; Ceppa, P.; Fiocca, R.; Marone, G. Basophils infiltrate human gastric mucosa at sites of Helicobacter pylori infection, and exhibit chemotaxis in response to h. Pylori-derived peptide hp(2–20). J. Immunol. 2004, 172, 7734–7743. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.J.; Lee, S.K.; Jung, Y.S.; Lee, M.; Lee, H.Y.; Kim, S.D.; Park, J.S.; Koo, J.; Hwang, J.S.; Bae, Y.S. Promotion of formyl peptide receptor 1-mediated neutrophil chemotactic migration by antimicrobial peptides isolated from the centipede scolopendra subspinipes mutilans. BMB Rep. 2016, 49, 520–525. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Sang, H.; Ye, R.D. Serum amyloid a induces il-8 secretion through a g protein-coupled receptor, fprl1/lxa4r. Blood 2003, 101, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, R.; Murphy, E.P.; Whitehead, A.S.; FitzGerald, O.; Bresnihan, B. Local expression of the serum amyloid a and formyl peptide receptor-like 1 genes in synovial tissue is associated with matrix metalloproteinase production in patients with inflammatory arthritis. Arthritis Rheum. 2004, 50, 1788–1799. [Google Scholar] [CrossRef] [PubMed]
- Sodin-Semrl, S.; Spagnolo, A.; Mikus, R.; Barbaro, B.; Varga, J.; Fiore, S. Opposing regulation of interleukin-8 and nf-kappab responses by lipoxin a4 and serum amyloid a via the common lipoxin a receptor. Int. J. Immunopathol. Pharmacol. 2004, 17, 145–156. [Google Scholar] [PubMed]
- Lee, H.Y.; Jo, S.H.; Lee, C.; Baek, S.H.; Bae, Y.S. Differential production of leukotriene b4 or prostaglandin e2 by wkymvm or serum amyloid a via formyl peptide receptor-like 1. Biochem. Pharmacol. 2006, 72, 860–868. [Google Scholar] [CrossRef] [PubMed]
- Bozinovski, S.; Uddin, M.; Vlahos, R.; Thompson, M.; McQualter, J.L.; Merritt, A.S.; Wark, P.A.; Hutchinson, A.; Irving, L.B.; Levy, B.D.; et al. Serum amyloid a opposes lipoxin a(4) to mediate glucocorticoid refractory lung inflammation in chronic obstructive pulmonary disease. Proc. Natl. Acad. Sci. USA 2012, 109, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Bjorkman, L.; Karlsson, J.; Karlsson, A.; Rabiet, M.J.; Boulay, F.; Fu, H.; Bylund, J.; Dahlgren, C. Serum amyloid a mediates human neutrophil production of reactive oxygen species through a receptor independent of formyl peptide receptor like-1. J. Leukoc. Biol. 2008, 83, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Christenson, K.; Bjorkman, L.; Ahlin, S.; Olsson, M.; Sjoholm, K.; Karlsson, A.; Bylund, J. Endogenous acute phase serum amyloid a lacks pro-inflammatory activity, contrasting the two recombinant variants that activate human neutrophils through different receptors. Front. Immunol. 2013, 4, 92. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhou, H.; Cheng, N.; Qian, F.; Ye, R.D. Serum amyloid a1 isoforms display different efficacy at toll-like receptor 2 and formyl peptide receptor 2. Immunobiology 2014, 219, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Sano, T.; Huang, W.; Hall, J.A.; Yang, Y.; Chen, A.; Gavzy, S.J.; Lee, J.Y.; Ziel, J.W.; Miraldi, E.R.; Domingos, A.I.; et al. An il-23r/il-22 circuit regulates epithelial serum amyloid a to promote local effector th17 responses. Cell 2015, 163, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Ando, M.; Kamada, N.; Nagano, Y.; Narushima, S.; Suda, W.; Imaoka, A.; Setoyama, H.; Nagamori, T.; et al. Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell 2015, 163, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Le, Y.; Oppenheim, J.J.; Wang, J.M. Pleiotropic roles of formyl peptide receptors. Cytokine Growth Factor Rev. 2001, 12, 91–105. [Google Scholar] [CrossRef]
- Yazawa, H.; Yu, Z.X.; Takeda; Le, Y.; Gong, W.; Ferrans, V.J.; Oppenheim, J.J.; Li, C.C.; Wang, J.M. Beta amyloid peptide (abeta42) is internalized via the g-protein-coupled receptor fprl1 and forms fibrillar aggregates in macrophages. FASEB J. 2001, 15, 2454–2462. [Google Scholar] [CrossRef] [PubMed]
- Ying, G.; Iribarren, P.; Zhou, Y.; Gong, W.; Zhang, N.; Yu, Z.X.; Le, Y.; Cui, Y.; Wang, J.M. Humanin, a newly identified neuroprotective factor, uses the g protein-coupled formylpeptide receptor-like-1 as a functional receptor. J. Immunol. 2004, 172, 7078–7085. [Google Scholar] [CrossRef] [PubMed]
- Harada, M.; Habata, Y.; Hosoya, M.; Nishi, K.; Fujii, R.; Kobayashi, M.; Hinuma, S. N-formylated humanin activates both formyl peptide receptor-like 1 and 2. Biochem. Biophys. Res. Commun. 2004, 324, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Ernst, S.; Lange, C.; Wilbers, A.; Goebeler, V.; Gerke, V.; Rescher, U. An annexin 1 n-terminal peptide activates leukocytes by triggering different members of the formyl peptide receptor family. J. Immunol. 2004, 172, 7669–7676. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, J.; Fu, H.; Boulay, F.; Dahlgren, C.; Hellstrand, K.; Movitz, C. Neutrophil nadph-oxidase activation by an annexin ai peptide is transduced by the formyl peptide receptor (fpr), whereas an inhibitory signal is generated independently of the fpr family receptors. J. Leukoc. Biol. 2005, 78, 762–771. [Google Scholar] [CrossRef] [PubMed]
- Kurosaka, K.; Chen, Q.; Yarovinsky, F.; Oppenheim, J.J.; Yang, D. Mouse cathelin-related antimicrobial peptide chemoattracts leukocytes using formyl peptide receptor-like 1/mouse formyl peptide receptor-like 2 as the receptor and acts as an immune adjuvant. J. Immunol. 2005, 174, 6257–6265. [Google Scholar] [CrossRef] [PubMed]
- Iaccio, A.; Cattaneo, F.; Mauro, M.; Ammendola, R. Fprl1-mediated induction of superoxide in ll-37-stimulated imr90 human fibroblast. Arch. Biochem. Biophys. 2009, 481, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Kim, S.D.; Shim, J.W.; Lee, S.Y.; Yun, J.; Bae, Y.S. Ll-37 inhibits serum amyloid a-induced il-8 production in human neutrophils. Exp. Mol. Med. 2009, 41, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Shaykhiev, R.; Beisswenger, C.; Kandler, K.; Senske, J.; Puchner, A.; Damm, T.; Behr, J.; Bals, R. Human endogenous antibiotic ll-37 stimulates airway epithelial cell proliferation and wound closure. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 289, L842–L848. [Google Scholar] [CrossRef] [PubMed]
- Koczulla, R.; von Degenfeld, G.; Kupatt, C.; Krotz, F.; Zahler, S.; Gloe, T.; Issbrucker, K.; Unterberger, P.; Zaiou, M.; Lebherz, C.; et al. An angiogenic role for the human peptide antibiotic ll-37/hcap-18. J. Clin. Investig. 2003, 111, 1665–1672. [Google Scholar] [CrossRef] [PubMed]
- Coffelt, S.B.; Marini, F.C.; Watson, K.; Zwezdaryk, K.J.; Dembinski, J.L.; LaMarca, H.L.; Tomchuck, S.L.; Honer zu Bentrup, K.; Danka, E.S.; Henkle, S.L.; et al. The pro-inflammatory peptide ll-37 promotes ovarian tumor progression through recruitment of multipotent mesenchymal stromal cells. Proc. Natl. Acad. Sci. USA 2009, 106, 3806–3811. [Google Scholar] [CrossRef] [PubMed]
- Coffelt, S.B.; Tomchuck, S.L.; Zwezdaryk, K.J.; Danka, E.S.; Scandurro, A.B. Leucine leucine-37 uses formyl peptide receptor-like 1 to activate signal transduction pathways, stimulate oncogenic gene expression, and enhance the invasiveness of ovarian cancer cells. Mol. Cancer Res. 2009, 7, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, Q.; Tang, R.; Shen, Y.; Liu, W.D. The expression of beta-defensin-2, 3 and ll-37 induced by candida albicans phospholipomannan in human keratinocytes. J. Dermatol. Sci. 2011, 61, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Wan, M.; Godson, C.; Guiry, P.J.; Agerberth, B.; Haeggstrom, J.Z. Leukotriene b4/antimicrobial peptide ll-37 proinflammatory circuits are mediated by blt1 and fpr2/alx and are counterregulated by lipoxin a4 and resolvin e1. FASEB J. 2011, 25, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Resnati, M.; Pallavicini, I.; Wang, J.M.; Oppenheim, J.; Serhan, C.N.; Romano, M.; Blasi, F. The fibrinolytic receptor for urokinase activates the g protein-coupled chemotactic receptor fprl1/lxa4r. Proc. Natl. Acad. Sci. USA 2002, 99, 1359–1364. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Xu, E.; Liang, W.; Pei, X.; Chen, D.; Zheng, D.; Zhang, Y.; Zheng, C.; Wang, P.; She, S.; et al. Identification of fam3d as a new endogenous chemotaxis agonist for the formyl peptide receptors. J. Cell Sci. 2016, 129, 1831–1842. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, F.; Parisi, M.; Ammendola, R. Distinct signaling cascades elicited by different formyl peptide receptor 2 (fpr2) agonists. Int. J. Mol. Sci. 2013, 14, 7193–7230. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.K.; Lee, H.Y.; Kim, M.K.; Park, K.S.; Park, Y.M.; Kwak, J.Y.; Bae, Y.S. The synthetic peptide trp-lys-tyr-met-val-d-met inhibits human monocyte-derived dendritic cell maturation via formyl peptide receptor and formyl peptide receptor-like 2. J. Immunol. 2005, 175, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.Y.; Le, Y.; Gong, W.; Dunlop, N.M.; Gao, J.L.; Murphy, P.M.; Wang, J.M. Synthetic peptide mmk-1 is a highly specific chemotactic agonist for leukocyte fprl1. J. Leukoc. Biol. 2001, 70, 155–161. [Google Scholar] [PubMed]
- Hecht, I.; Rong, J.; Sampaio, A.L.; Hermesh, C.; Rutledge, C.; Shemesh, R.; Toporik, A.; Beiman, M.; Dassa, L.; Niv, H.; et al. A novel peptide agonist of formyl-peptide receptor-like 1 (alx) displays anti-inflammatory and cardioprotective effects. J. Pharmacol. Exp. Ther. 2009, 328, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Bernstein, H.S.; Chen, M.; Wang, L.; Ishii, M.; Turck, C.W.; Coughlin, S.R. Tethered ligand library for discovery of peptide agonists. J. Biol. Chem. 1995, 270, 23398–23401. [Google Scholar] [CrossRef]
- Hu, Y.; Cheng, N.; Wu, H.; Kang, S.; Ye, R.D.; Cai, J. Design, synthesis and characterization of fmlf-mimicking aapeptides. ChemBioChem 2014, 15, 2420–2426. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Cheng, N.; Gao, W.W.; Zhang, M.; Zhang, Y.Y.; Ye, R.D.; Wang, M.W. Characterization of quin-c1 for its anti-inflammatory property in a mouse model of bleomycin-induced lung injury. Acta Pharmacol. Sin. 2011, 32, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Forsman, H.; Kalderen, C.; Nordin, A.; Nordling, E.; Jensen, A.J.; Dahlgren, C. Stable formyl peptide receptor agonists that activate the neutrophil nadph-oxidase identified through screening of a compound library. Biochem. Pharmacol. 2011, 81, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Cevik-Aras, H.; Kalderen, C.; Jenmalm Jensen, A.; Oprea, T.; Dahlgren, C.; Forsman, H. A non-peptide receptor inhibitor with selectivity for one of the neutrophil formyl peptide receptors, fpr 1. Biochem. Pharmacol. 2012, 83, 1655–1662. [Google Scholar] [CrossRef] [PubMed]
- Khlebnikov, A.I.; Schepetkin, I.A.; Kirpotina, L.N.; Brive, L.; Dahlgren, C.; Jutila, M.A.; Quinn, M.T. Molecular docking of 2-(benzimidazol-2-ylthio)-n-phenylacetamide-derived small-molecule agonists of human formyl peptide receptor 1. J. Mol. Model. 2012, 18, 2831–2843. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, M.P.; Schepetkin, I.A.; Cilibrizzi, A.; Crocetti, L.; Khlebnikov, A.I.; Dahlgren, C.; Graziano, A.; Dal Piaz, V.; Kirpotina, L.N.; Zerbinati, S.; et al. Further studies on 2-arylacetamide pyridazin-3(2H)-ones: Design, synthesis and evaluation of 4,6-disubstituted analogs as formyl peptide receptors (fprs) agonists. Eur. J. Med. Chem. 2013, 64, 512–528. [Google Scholar] [CrossRef] [PubMed]
- Schepetkin, I.A.; Kirpotina, L.N.; Khlebnikov, A.I.; Quinn, M.T. High-throughput screening for small-molecule activators of neutrophils: Identification of novel n-formyl peptide receptor agonists. Mol. Pharmacol. 2007, 71, 1061–1074. [Google Scholar] [CrossRef] [PubMed]
- Schepetkin, I.A.; Kirpotina, L.N.; Khlebnikov, A.I.; Jutila, M.A.; Quinn, M.T. Gastrin-releasing peptide/neuromedin b receptor antagonists pd176252, pd168368, and related analogs are potent agonists of human formyl-peptide receptors. Mol. Pharmacol. 2011, 79, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot. Essent. Fat. Acids 2005, 73, 141–162. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.; Serhan, C.N.; Dahlen, S.E.; Drazen, J.M.; Hay, D.W.; Rovati, G.E.; Shimizu, T.; Yokomizo, T.; Brink, C. The lipoxin receptor alx: Potent ligand-specific and stereoselective actions in vivo. Pharmacol. Rev. 2006, 58, 463–487. [Google Scholar] [CrossRef] [PubMed]
- O’Meara, S.J.; Rodgers, K.; Godson, C. Lipoxins: Update and impact of endogenous pro-resolution lipid mediators. Rev. Physiol. Biochem. Pharmacol. 2008, 160, 47–70. [Google Scholar] [PubMed]
- Serhan, C.N.; Brain, S.D.; Buckley, C.D.; Gilroy, D.W.; Haslett, C.; O’Neill, L.A.; Perretti, M.; Rossi, A.G.; Wallace, J.L. Resolution of inflammation: State of the art, definitions and terms. FASEB J. 2007, 21, 325–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughn, M.W.; Proske, R.J.; Haviland, D.L. Identification, cloning, and functional characterization of a murine lipoxin a4 receptor homologue gene. J. Immunol. 2002, 169, 3363–3369. [Google Scholar] [CrossRef] [PubMed]
- Forsman, H.; Dahlgren, C. Lipoxin a(4) metabolites/analogues from two commercial sources have no effects on tnf-alpha-mediated priming or activation through the neutrophil formyl peptide receptors. Scand. J. Immunol. 2009, 70, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Forsman, H.; Onnheim, K.; Andreasson, E.; Dahlgren, C. What formyl peptide receptors, if any, are triggered by compound 43 and lipoxin a4? Scand. J. Immunol. 2011, 74, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Hanson, J.; Ferreiros, N.; Pirotte, B.; Geisslinger, G.; Offermanns, S. Heterologously expressed formyl peptide receptor 2 (fpr2/alx) does not respond to lipoxin a(4). Biochem. Pharmacol. 2013, 85, 1795–1802. [Google Scholar] [CrossRef] [PubMed]
- Pamplona, F.A.; Ferreira, J.; Menezes de Lima, O., Jr.; Duarte, F.S.; Bento, A.F.; Forner, S.; Villarinho, J.G.; Bellocchio, L.; Wotjak, C.T.; Lerner, R.; et al. Anti-inflammatory lipoxin a4 is an endogenous allosteric enhancer of cb1 cannabinoid receptor. Proc. Natl. Acad. Sci. USA 2012, 109, 21134–21139. [Google Scholar] [CrossRef] [PubMed]
- Schaldach, C.M.; Riby, J.; Bjeldanes, L.F. Lipoxin a4: A new class of ligand for the ah receptor. Biochemistry 1999, 38, 7594–7600. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.; Gori, I.; Pellegrini, C.; Kumar, R.; Achtari, C.; Canny, G.O. Lipoxin a4 is a novel estrogen receptor modulator. FASEB J. 2011, 25, 4326–4337. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, S.; Recchiuti, A.; Chiang, N.; Yacoubian, S.; Lee, C.H.; Yang, R.; Petasis, N.A.; Serhan, C.N. Resolvin d1 binds human phagocytes with evidence for proresolving receptors. Proc. Natl. Acad. Sci. USA 2010, 107, 1660–1665. [Google Scholar] [CrossRef] [PubMed]
- Odusanwo, O.; Chinthamani, S.; McCall, A.; Duffey, M.E.; Baker, O.J. Resolvin d1 prevents tnf-alpha-mediated disruption of salivary epithelial formation. Am. J. Physiol. Cell Physiol. 2012, 302, C1331–C1345. [Google Scholar] [CrossRef] [PubMed]
- Eickmeier, O.; Seki, H.; Haworth, O.; Hilberath, J.N.; Gao, F.; Uddin, M.; Croze, R.H.; Carlo, T.; Pfeffer, M.A.; Levy, B.D. Aspirin-triggered resolvin d1 reduces mucosal inflammation and promotes resolution in a murine model of acute lung injury. Mucosal Immunol. 2013, 6, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Claria, J.; Dalli, J.; Yacoubian, S.; Gao, F.; Serhan, C.N. Resolvin d1 and resolvin d2 govern local inflammatory tone in obese fat. J. Immunol. 2012, 189, 2597–2605. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Oh, E.; Kim, S.D.; Seo, J.K.; Bae, Y.S. Oxidized low-density lipoprotein-induced foam cell formation is mediated by formyl peptide receptor 2. Biochem. Biophys. Res. Commun. 2014, 443, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.; Agarwal, A.; Devi, L.A.; Fontanini, K.; Hamilton, J.A.; Pin, J.P.; Shields, D.C.; Spek, C.A.; Sakmar, T.P.; Kuliopulos, A.; et al. Insider access: Pepducin symposium explores a new approach to gpcr modulation. Ann. N. Y. Acad. Sci. 2009, 1180 (Suppl. S1), E1–E12. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, K.; Kuliopulos, A.; Covic, L. Turning receptors on and off with intracellular pepducins: New insights into g-protein-coupled receptor drug development. J. Biol. Chem. 2012, 287, 12787–12796. [Google Scholar] [CrossRef] [PubMed]
- Gabl, M.; Winther, M.; Skovbakke, S.L.; Bylund, J.; Dahlgren, C.; Forsman, H. A pepducin derived from the third intracellular loop of fpr2 is a partial agonist for direct activation of this receptor in neutrophils but a full agonist for cross-talk triggered reactivation of fpr2. PLoS ONE 2014, 9, e109516. [Google Scholar] [CrossRef] [PubMed]
- Gabl, M.; Holdfeldt, A.; Winther, M.; Oprea, T.; Bylund, J.; Dahlgren, C.; Forsman, H. A pepducin designed to modulate p2y2r function interacts with fpr2 in human neutrophils and transfers atp to an nadph-oxidase-activating ligand through a receptor cross-talk mechanism. Biochim. Biophys. Acta 2016, 1863, 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Holdfeldt, A.; Winther, M.; Gabl, M.; Dahlgren, C.; Forsman, H. Data on human neutrophil activation induced by pepducins with amino acid sequences derived from β2ar and cxcr4. Data Brief 2016, 8, 411–414. [Google Scholar] [CrossRef] [PubMed]
- Babich, J.W.; Tompkins, R.G.; Graham, W.; Barrow, S.A.; Fischman, A.J. Localization of radiolabeled chemotactic peptide at focal sites of escherichia coli infection in rabbits: Evidence for a receptor-specific mechanism. J. Nucl. Med. 1997, 38, 1316–1322. [Google Scholar] [PubMed]
- Locke, L.W.; Chordia, M.D.; Zhang, Y.; Kundu, B.; Kennedy, D.; Landseadel, J.; Xiao, L.; Fairchild, K.D.; Berr, S.S.; Linden, J.; et al. A novel neutrophil-specific pet imaging agent: Cflflfk-peg-64cu. J. Nucl. Med. 2009, 50, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Derian, C.K.; Solomon, H.F.; Higgins, J.D., 3rd; Beblavy, M.J.; Santulli, R.J.; Bridger, G.J.; Pike, M.C.; Kroon, D.J.; Fischman, A.J. Selective inhibition of n-formylpeptide-induced neutrophil activation by carbamate-modified peptide analogues. Biochemistry 1996, 35, 1265–1269. [Google Scholar] [CrossRef] [PubMed]
- Aswanikumar, S.; Schiffmann, E.; Corcoran, B.A.; Pert, C.B.; Morell, J.L.; Gross, E. Antibiotics and peptides with agonist and antagonist chemotactic activity. Biochem. Biophys. Res. Commun. 1978, 80, 464–471. [Google Scholar] [CrossRef]
- Stenfeldt, A.L.; Karlsson, J.; Wenneras, C.; Bylund, J.; Fu, H.; Dahlgren, C. Cyclosporin h, boc-mlf and boc-flflf are antagonists that preferentially inhibit activity triggered through the formyl peptide receptor. Inflammation 2007, 30, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, R.; Kitajima, T.; Mizuguchi, H.; Fujimoto, M.; Yamaguchi, A.; Koga, S.; Koga, Y.; Osada, S.; Kodama, H. Development of potent antagonists for formyl peptide receptor 1 based on boc-phe-d-leu-phe-d-leu-phe-oh. Bioorg. Med. Chem. 2014, 22, 3824–3828. [Google Scholar] [CrossRef] [PubMed]
- Postma, B.; Poppelier, M.J.; van Galen, J.C.; Prossnitz, E.R.; van Strijp, J.A.; de Haas, C.J.; van Kessel, K.P. Chemotaxis inhibitory protein of Staphylococcus aureus binds specifically to the c5a and formylated peptide receptor. J. Immunol. 2004, 172, 6994–7001. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.J.; Higginbottom, A.; Philippe, D.; Upadhyay, A.; Bagby, S.; Read, R.C.; Monk, P.N.; Partridge, L.J. Characterisation of receptor binding by the chemotaxis inhibitory protein of Staphylococcus aureus and the effects of the host immune response. Mol. Immunol. 2007, 44, 2507–2517. [Google Scholar] [CrossRef] [PubMed]
- Prat, C.; Bestebroer, J.; de Haas, C.J.; van Strijp, J.A.; van Kessel, K.P. A new staphylococcal anti-inflammatory protein that antagonizes the formyl peptide receptor-like 1. J. Immunol. 2006, 177, 8017–8026. [Google Scholar] [CrossRef] [PubMed]
- Oostendorp, R.A.; Knol, E.F.; Verhoeven, A.J.; Scheper, R.J. An immunosuppressive retrovirus-derived hexapeptide interferes with intracellular signaling in monocytes and granulocytes through n-formylpeptide receptors. J. Immunol. 1992, 149, 1010–1015. [Google Scholar] [PubMed]
- Mills, J.S. Peptides derived from HIV-1, HIV-2, Ebola virus, SARS coronavirus and coronavirus 229E exhibit high affinity binding to the formyl peptide receptor. Biochim Biophys Acta 2006, 1762, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Loor, F.; Tiberghien, F.; Wenandy, T.; Didier, A.; Traber, R. Cyclosporins: Structure-activity relationships for the inhibition of the human fpr1 formylpeptide receptor. J. Med. Chem. 2002, 45, 4613–4628. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Nanamori, M.; Sun, M.; Zhou, C.; Cheng, N.; Li, N.; Zheng, W.; Xiao, L.; Xie, X.; Ye, R.D.; et al. The immunosuppressant cyclosporin a antagonizes human formyl peptide receptor through inhibition of cognate ligand binding. J. Immunol. 2006, 177, 7050–7058. [Google Scholar] [CrossRef] [PubMed]
- Cardini, S.; Dalli, J.; Fineschi, S.; Perretti, M.; Lungarella, G.; Lucattelli, M. Genetic ablation of the fpr1 gene confers protection from smoking-induced lung emphysema in mice. Am. J. Respir. Cell Mol. Biol. 2012, 47, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Rittner, H.L.; Hackel, D.; Voigt, P.; Mousa, S.; Stolz, A.; Labuz, D.; Schafer, M.; Schaefer, M.; Stein, C.; Brack, A. Mycobacteria attenuate nociceptive responses by formyl peptide receptor triggered opioid peptide release from neutrophils. PLoS Pathog. 2009, 5, e1000362. [Google Scholar] [CrossRef] [PubMed]
- Vasanthakumar, G.; Manjunath, R.; Mukherjee, A.B.; Warabi, H.; Schiffmann, E. Inhibition of phagocyte chemotaxis by uteroglobin, an inhibitor of blastocyst rejection. Biochem. Pharmacol. 1988, 37, 389–394. [Google Scholar] [CrossRef]
- Antico, G.; Aloman, M.; Lakota, K.; Miele, L.; Fiore, S.; Sodin-Semrl, S. Uteroglobin, a possible ligand of the lipoxin receptor inhibits serum amyloid a-driven inflammation. Mediators Inflamm. 2014, 2014, 876395. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.C.; Lin, C.F.; Chang, W.Y.; Kuo, J.; Huang, Y.T.; Chung, P.J.; Hwang, T.L. Bioactive secondary metabolites of a marine bacillus sp. Inhibit superoxide generation and elastase release in human neutrophils by blocking formyl peptide receptor 1. Molecules 2013, 18, 6455–6468. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.S.; Gao, J.L.; Fatemi, O.; Lavigne, M.; Leto, T.L.; Murphy, P.M. The endogenous opioid spinorphin blocks fmet-leu-phe-induced neutrophil chemotaxis by acting as a specific antagonist at the n-formylpeptide receptor subtype fpr. J. Immunol. 2001, 167, 6609–6614. [Google Scholar] [CrossRef] [PubMed]
- Nwodo, N.J.; Okoye, F.B.; Lai, D.; Debbab, A.; Brun, R.; Proksch, P. Two trypanocidal dipeptides from the roots of zapoteca portoricensis (fabaceae). Molecules 2014, 19, 5470–5477. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Kanazawa, T.; Shimamura, M.; Ueki, M.; Hazato, T. Inhibitory effects of spinorphin, a novel endogenous regulator, on chemotaxis, o2- generation, and exocytosis by n-formylmethionyl-leucyl-phenylalanine (fmlp)-stimulated neutrophils. Biochem. Pharmacol. 1997, 54, 695–701. [Google Scholar] [CrossRef]
- Tsai, P.L.; Wang, J.P.; Chang, C.W.; Kuo, S.C.; Chao, P.D. Constituents and bioactive principles of polygonum chinensis. Phytochemistry 1998, 49, 1663–1666. [Google Scholar] [CrossRef]
- Yen, C.T.; Hwang, T.L.; Wu, Y.C.; Hsieh, P.W. Design and synthesis of new n-(fluorenyl-9-methoxycarbonyl) (fmoc)-dipeptides as anti-inflammatory agents. Eur. J. Med. Chem. 2009, 44, 1933–1940. [Google Scholar] [CrossRef] [PubMed]
- Hwang, T.L.; Hung, C.H.; Hsu, C.Y.; Huang, Y.T.; Tsai, Y.C.; Hsieh, P.W. Design and synthesis of tryptophan containing dipeptide derivatives as formyl peptide receptor 1 antagonist. Org. Biomol. Chem. 2013, 11, 3742–3755. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.H.; Lee, H.Y.; Kim, S.D.; Jo, S.H.; Kim, M.K.; Park, K.S.; Lee, H.; Bae, Y.S. Trp-arg-trp-trp-trp-trp antagonizes formyl peptide receptor like 2-mediated signaling. Biochem. Biophys. Res. Commun. 2006, 341, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Dahinden, C.; Fehr, J. Receptor-directed inhibition of chemotactic factor-induced neutrophil hyperactivity by pyrazolon derivatives. Definition of a chemotactic peptide antagonist. J. Clin. Investig. 1980, 66, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Levesque, L.; Gaudreault, R.C.; Marceau, F. The interaction of 3,5-pyrazolidinedione drugs with receptors for f-met-leu-phe on human neutrophil leukocytes: A study of the structure-activity relationship. Can. J. Physiol. Pharmacol. 1991, 69, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Del Maschio, A.; Livio, M.; Cerletti, C.; De Gaetano, G. Inhibition of human platelet cyclo-oxygenase activity by sulfinpyrazone and three of its metabolites. Eur. J. Pharmacol. 1984, 101, 209–214. [Google Scholar] [CrossRef]
- Young, S.M.; Bologa, C.; Prossnitz, E.R.; Oprea, T.I.; Sklar, L.A.; Edwards, B.S. High-throughput screening with hypercyt flow cytometry to detect small molecule formylpeptide receptor ligands. J. Biomol. Screen. 2005, 10, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Hwang, T.L.; Wang, C.C.; Kuo, Y.H.; Huang, H.C.; Wu, Y.C.; Kuo, L.M.; Wu, Y.H. The hederagenin saponin smg-1 is a natural fmlp receptor inhibitor that suppresses human neutrophil activation. Biochem. Pharmacol. 2010, 80, 1190–1200. [Google Scholar] [CrossRef] [PubMed]
- Schepetkin, I.A.; Khlebnikov, A.I.; Kirpotina, L.N.; Quinn, M.T. Antagonism of human formyl peptide receptor 1 with natural compounds and their synthetic derivatives. Int. Immunopharmacol. 2016, 37, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.J.; Chen, I.S.; Tseng, C.P.; Day, Y.J.; Lin, Y.C.; Liao, C.H. (2r,3r)-2-(3′,4′-dihydroxybenzyl)-3-(3′′,4′′-dimethoxybenzyl)butyrolactone suppresses fmlp-induced superoxide production by inhibiting fmlp-receptor binding in human neutrophils. Biochem. Pharmacol. 2008, 75, 688–697. [Google Scholar] [CrossRef] [PubMed]
- Edwards, B.S.; Young, S.M.; Ivnitsky-Steele, I.; Ye, R.D.; Prossnitz, E.R.; Sklar, L.A. High-content screening: Flow cytometry analysis. Methods Mol. Biol. 2009, 486, 151–165. [Google Scholar] [PubMed]
- Edwards, B.S.; Young, S.M.; Oprea, T.I.; Bologa, C.G.; Prossnitz, E.R.; Sklar, L.A. Biomolecular screening of formylpeptide receptor ligands with a sensitive, quantitative, high-throughput flow cytometry platform. Nat. Protoc. 2006, 1, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Strouse, J.J.; Young, S.M.; Mitchell, H.D.; Ye, R.D.; Prossnitz, E.R.; Sklar, L.A.; Edwards, B.S. A novel fluorescent cross-reactive formylpeptide receptor/formylpeptide receptor-like 1 hexapeptide ligand. Cytom. A 2009, 75, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Young, S.M.; Bologa, C.M.; Fara, D.; Bryant, B.K.; Strouse, J.J.; Arterburn, J.B.; Ye, R.D.; Oprea, T.I.; Prossnitz, E.R.; Sklar, L.A.; et al. Duplex high-throughput flow cytometry screen identifies two novel formylpeptide receptor family probes. Cytom. A 2009, 75, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Quehenberger, O.; Prossnitz, E.R.; Cavanagh, S.L.; Cochrane, C.G.; Ye, R.D. Multiple domains of the n-formyl peptide receptor are required for high-affinity ligand binding. Construction and analysis of chimeric n-formyl peptide receptors. J. Biol. Chem. 1993, 268, 18167–18175. [Google Scholar] [PubMed]
- Gao, J.L.; Murphy, P.M. Species and subtype variants of the n-formyl peptide chemotactic receptor reveal multiple important functional domains. J. Biol. Chem. 1993, 268, 25395–25401. [Google Scholar] [PubMed]
- Le, Y.; Ye, R.D.; Gong, W.; Li, J.; Iribarren, P.; Wang, J.M. Identification of functional domains in the formyl peptide receptor-like 1 for agonist-induced cell chemotaxis. FEBS J. 2005, 272, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, H.M.; Mills, J.S.; Gripentrog, J.M.; Dratz, E.A.; Granger, B.L.; Jesaitis, A.J. The ligand binding site of the formyl peptide receptor maps in the transmembrane region. J. Immunol. 1997, 159, 4045–4054. [Google Scholar] [PubMed]
- Quehenberger, O.; Pan, Z.K.; Prossnitz, E.R.; Cavanagh, S.L.; Cochrane, C.G.; Ye, R.D. Identification of an n-formyl peptide receptor ligand binding domain by a gain-of-function approach. Biochem. Biophys. Res. Commun. 1997, 238, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Lala, A.; Gwinn, M.; De Nardin, E. Human formyl peptide receptor function role of conserved and nonconserved charged residues. Eur. J. Biochem. 1999, 264, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.S.; Miettinen, H.M.; Cummings, D.; Jesaitis, A.J. Characterization of the binding site on the formyl peptide receptor using three receptor mutants and analogs of met-leu-phe and met-met-trp-leu-leu. J. Biol. Chem. 2000, 275, 39012–39017. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.S.; Miettinen, H.M.; Barnidge, D.; Vlases, M.J.; Wimer-Mackin, S.; Dratz, E.A.; Sunner, J.; Jesaitis, A.J. Identification of a ligand binding site in the human neutrophil formyl peptide receptor using a site-specific fluorescent photoaffinity label and mass spectrometry. J. Biol. Chem. 1998, 273, 10428–10435. [Google Scholar] [CrossRef] [PubMed]
- Bokoch, M.P.; Zou, Y.; Rasmussen, S.G.; Liu, C.W.; Nygaard, R.; Rosenbaum, D.M.; Fung, J.J.; Choi, H.J.; Thian, F.S.; Kobilka, T.S.; et al. Ligand-specific regulation of the extracellular surface of a g-protein-coupled receptor. Nature 2010, 463, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Jongejan, A.; Bruysters, M.; Ballesteros, J.A.; Haaksma, E.; Bakker, R.A.; Pardo, L.; Leurs, R. Linking agonist binding to histamine h1 receptor activation. Nat. Chem. Biol. 2005, 1, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Cheung, G.Y.; Kretschmer, D.; Queck, S.Y.; Joo, H.S.; Wang, R.; Duong, A.C.; Nguyen, T.H.; Bach, T.H.; Porter, A.R.; DeLeo, F.R.; et al. Insight into structure-function relationship in phenol-soluble modulins using an alanine screen of the phenol-soluble modulin (psm) alpha3 peptide. FASEB J. 2014, 28, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Kretschmer, D.; Rautenberg, M.; Linke, D.; Peschel, A. Peptide length and folding state govern the capacity of staphylococcal beta-type phenol-soluble modulins to activate human formyl-peptide receptors 1 or 2. J. Leukoc. Biol. 2015, 97, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.; Fierro, I.M.; Gronert, K.; Serhan, C.N. Activation of lipoxin a(4) receptors by aspirin-triggered lipoxins and select peptides evokes ligand-specific responses in inflammation. J. Exp. Med. 2000, 191, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Movitz, C.; Brive, L.; Hellstrand, K.; Rabiet, M.J.; Dahlgren, C. The annexin i sequence gln(9)-ala(10)-trp(11)-phe(12) is a core structure for interaction with the formyl peptide receptor 1. J. Biol. Chem. 2010, 285, 14338–14345. [Google Scholar] [CrossRef] [PubMed]
- Bena, S.; Brancaleone, V.; Wang, J.M.; Perretti, M.; Flower, R.J. Annexin a1 interaction with the fpr2/alx receptor: Identification of distinct domains and downstream associated signaling. J. Biol. Chem. 2012, 287, 24690–24697. [Google Scholar] [CrossRef] [PubMed]
- Palczewski, K.; Kumasaka, T.; Hori, T.; Behnke, C.A.; Motoshima, H.; Fox, B.A.; Le Trong, I.; Teller, D.C.; Okada, T.; Stenkamp, R.E.; et al. Crystal structure of rhodopsin: A g protein-coupled receptor. Science 2000, 289, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Chien, E.Y.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F.C.; et al. Structures of the cxcr4 chemokine gpcr with small-molecule and cyclic peptide antagonists. Science 2010, 330, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Stepniewski, T.M.; Filipek, S. Non-peptide ligand binding to the formyl peptide receptor fpr2—A comparison to peptide ligand binding modes. Bioorg. Med. Chem. 2015, 23, 4072–4081. [Google Scholar] [CrossRef] [PubMed]
- Edwards, B.S.; Bologa, C.; Young, S.M.; Balakin, K.V.; Prossnitz, E.R.; Savchuck, N.P.; Sklar, L.A.; Oprea, T.I. Integration of virtual screening with high-throughput flow cytometry to identify novel small molecule formylpeptide receptor antagonists. Mol. Pharmacol. 2005, 68, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, C.; Macchiarulo, A.; Costantino, G.; Pellicciari, R. Pharmacophore model for bile acids recognition by the fpr receptor. J. Comput. Aided Mol. Des. 2006, 20, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Dalpiaz, A.; Ferretti, M.E.; Vertuani, G.; Traniello, S.; Scatturin, A.; Spisani, S. C- and n-terminal residue effect on peptide derivatives’ antagonism toward the formyl-peptide receptor. Eur. J. Pharmacol. 2002, 436, 187–196. [Google Scholar] [CrossRef]
- Schepetkin, I.A.; Kirpotina, L.N.; Tian, J.; Khlebnikov, A.I.; Ye, R.D.; Quinn, M.T. Identification of novel formyl peptide receptor-like 1 agonists that induce macrophage tumor necrosis factor alpha production. Mol. Pharmacol. 2008, 74, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Ghoshdastider, U.; Trzaskowski, B.; Latek, D.; Debinski, A.; Pulawski, W.; Wu, R.; Gerke, V.; Filipek, S. The role of water in activation mechanism of human n-formyl peptide receptor 1 (fpr1) based on molecular dynamics simulations. PLoS ONE 2012, 7, e47114. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Kato, T.; Watanabe, N.; Takahashi, T.; Kitagawa, S. Stimulation of human formyl peptide receptors by calpain inhibitors: Homology modeling of receptors and ligand docking simulation. Arch. Biochem. Biophys. 2011, 516, 121–127. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Ligands Representatives | Sequence/Structure | Selectivity | Reference |
|---|---|---|---|
| Agonists | |||
| N-Formyl peptides | |||
| |||
| fMLF | formyl-Met-Leu-Phe | FPR1 > FPR2 | [40] |
| PSMα peptide | formyl-MGIIAGIIKFI KGLIEKFTGK | FPR2 > FPR1 | [52] |
| |||
| fMMYALF | formyl-Met-Met-Tyr-Ala-Leu-Phe | FPR1, FPR2 | [53] |
| Mitocryptide-2 | formyl-MTNIRKSHPLMKIIN | FPR2 | [54] |
| Non-formyl peptides | |||
| |||
| Hp2-20 | AKKVFKRLEKLFSKIQNDK | FPR2 >> FPR3 | [55] |
| |||
| SAA1.1 |  | FPR2, others | [56,57] |
| Aβ42 | DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA | FPR2 | [28,58] |
| Ac2–26 | Ac-AMVSEFLKQAWFIENEEQEYVQTVK | FPR1, FPR2 | [59,60] |
| LL-37 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | FPR2 | [61] |
| uPAR88-92 | 88Ser-Arg-Ser-Arg-Tyr92 (SRSRY) | FPR1 | [62] |
| uPAR84-95 | AVTYSRSRYLEC | FPR2, FPR3 | [63] |
| PrP(106-126) | KTNMKHMAGAAAAGAVVGGLG | FPR2 | [64] |
| SHAAGtide | MLWRRKIGPQMTLSHAAG | FPR2 > CCR1 | [65] |
| VIP | HSDAVFTDNYTRLRKQMAVKKYLNSILN | FPR2, VPAC1 | [66] |
| |||
| W peptides | WKYMVm(Trp-Lys-Tyr-Met-Val-D-Met-NH2) | FPR2 > FPR1 >> FPR3 | [67,68,69] |
| WKYMVM(Trp-Lys-Tyr-Met-Val-Met-NH2) | FPR2 >> FPR3 | [69] | |
| MMK1 | LESIFRSLLFRVM | FPR2 | [70] |
| L-37pA | DWLKAFYDKVAEKLKEAFPDWLKAFYDKVAEKLKEAF | FPR2 | [71] |
| CGEN-855A | TIPMFVPESTSKLQKFTSWFM | FPR2, FPR3 | [72] |
| |||
| F2Pal16 | Pam-KIHKKGMIKSSRPLRV | FPR2 | [72,73] |
| Eicosanoids | |||
| Lipoxin A4 |  | [74] | |
| Small molecules | |||
|  | FPR2 >> FPR1 | [75] |
|  | FPR1 | [76] |
|  | FPR2 | [77] |
|  | FPR2 > FPR1 | [78,79] |
|  | FPR1 | [80] |
| Pyridazin-3(2H)-one derivative 2 |  | FPR2 | [80] |
|  | FPR2 | [76] |
|  R = n-C3H7, i-C3H7, n-C4H9 | FPR1, FPR2 | [81] |
|  | FPR2 | [76] |
|  | FPR2 | [82,83] |
|  | FPR1 | [84] |
| Antagonists | |||
| Peptides | |||
| |||
| Boc-1 | N-tert-butoxycarbonyl-MLF | FPR1 >> FPR2 | [40] |
| Boc-2 | N-tert-butoxycarbonyl-FLFLF | FPR1 >> FPR2 | [40] |
| CHIP peptide Uteroglobin (UG) | FTFEPF MKLAVTLTLVTLALCCSSASAEICPSFQRVIETLLMDTPSSYEAAMELFSPDQDMREAGAQLKKLVDTLPQKPRESIIKLMEKIAQSSLCN | FPR1 FPR2 | [85] [86] |
| Cyclosporin H (CsH) |  | FPR1 | [87] |
| Cyclized uPAR88-92 | [88Ser-Arg-Ser-Arg-Tyr92]( cyclized SRSRY) | FPR1, N.D for FPR2 | [88] |
| |||
| WRW4 | WRWWWW (Trp-Arg-Trp-Trp-Trp-Trp) | FPR2, FPR3 | [89] |
| PBP10 | RhoB-QRLFQVKGRR | FPR2, others | [90] |
| Lipopeptides | |||
| F1Pal16 | Pam-KIHKQGMIKSSRPLRV | FPR2 | [91] |
| Pam-(Lys-βNSpe)6-NH2 |  | FPR2, mFpr2 | [92,93] |
| Non-peptide molecules | |||
| Isoflavone analog |  | FPR1 | [94] |
| 1754-31 |  | FPR2 | [84] |
| Quin-C7 |  | FPR2 | [95] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, H.-Q.; Ye, R.D. The Formyl Peptide Receptors: Diversity of Ligands and Mechanism for Recognition. Molecules 2017, 22, 455. https://doi.org/10.3390/molecules22030455
He H-Q, Ye RD. The Formyl Peptide Receptors: Diversity of Ligands and Mechanism for Recognition. Molecules. 2017; 22(3):455. https://doi.org/10.3390/molecules22030455
Chicago/Turabian StyleHe, Hui-Qiong, and Richard D. Ye. 2017. "The Formyl Peptide Receptors: Diversity of Ligands and Mechanism for Recognition" Molecules 22, no. 3: 455. https://doi.org/10.3390/molecules22030455
APA StyleHe, H. -Q., & Ye, R. D. (2017). The Formyl Peptide Receptors: Diversity of Ligands and Mechanism for Recognition. Molecules, 22(3), 455. https://doi.org/10.3390/molecules22030455