3.1. Electronic Structure
We first analyze the geometrical differences between NAPA, NAPMA and NAPAH conformers B. If we consider the NH2–C(=O)– group of NAPA conformer B as the unperturbed system, the perturbation is caused by the substitution of the distal hydrogen atom (Hdist) by the methyl group in NAPMA, or in the case of NAPAH by binding of a water molecule. The perturbation is then transferred to the rest of the molecules mostly by intramolecular dispersive interactions.
The water molecule perturbs the second amide group more than the covalently-attached methyl group in NAPMA. The two hydrogen bonds,
–H
O
(II) and
H
dist–N
(II), cause a 0.014 Å extension of the C=O bond and a 0.013 Å compression of the C–N with respect to the NAPA values. In NAPMA B, these bond distances change by only 0.005 and −0.001 Å respectively. Furthermore, in NAPA B, the second amide NH
2 group is pyramidalized by an angle of
of −159.4 degrees (see
Figure 3). In both NAPMA and NAPAH, the NH
2 group is planar.
Although the differences in the geometry of the second amide group upon methylation or hydration are significant, the effect on the Ac–Phe frame is contained. Root mean square deviations (RMSD) between NAPMA/NAPA and NAPAH/NAPA Ac–Phe frames are only ∼0.05 Å.
The excitation energies are consistent with the geometrical differences between NAPA, NAPMA and NAPAH.
Table 2 compares vertical absorption energies, as well as adiabatic and zero point energy (ZPE)-corrected absorption energies of the four lowest singlet states of the conformer B of NAPA, NAPMA and NAPAH. Results were obtained at the CC2 and multi-state complete-active-space second-order perturbation theory (MS-CASPT2) levels of theory. See also reference [
27] for TDDFT and MS-CASPT2 absorption energies and oscillator strengths in the four most stable NAPA conformers.
In all species, the four lowest states include two
states and two
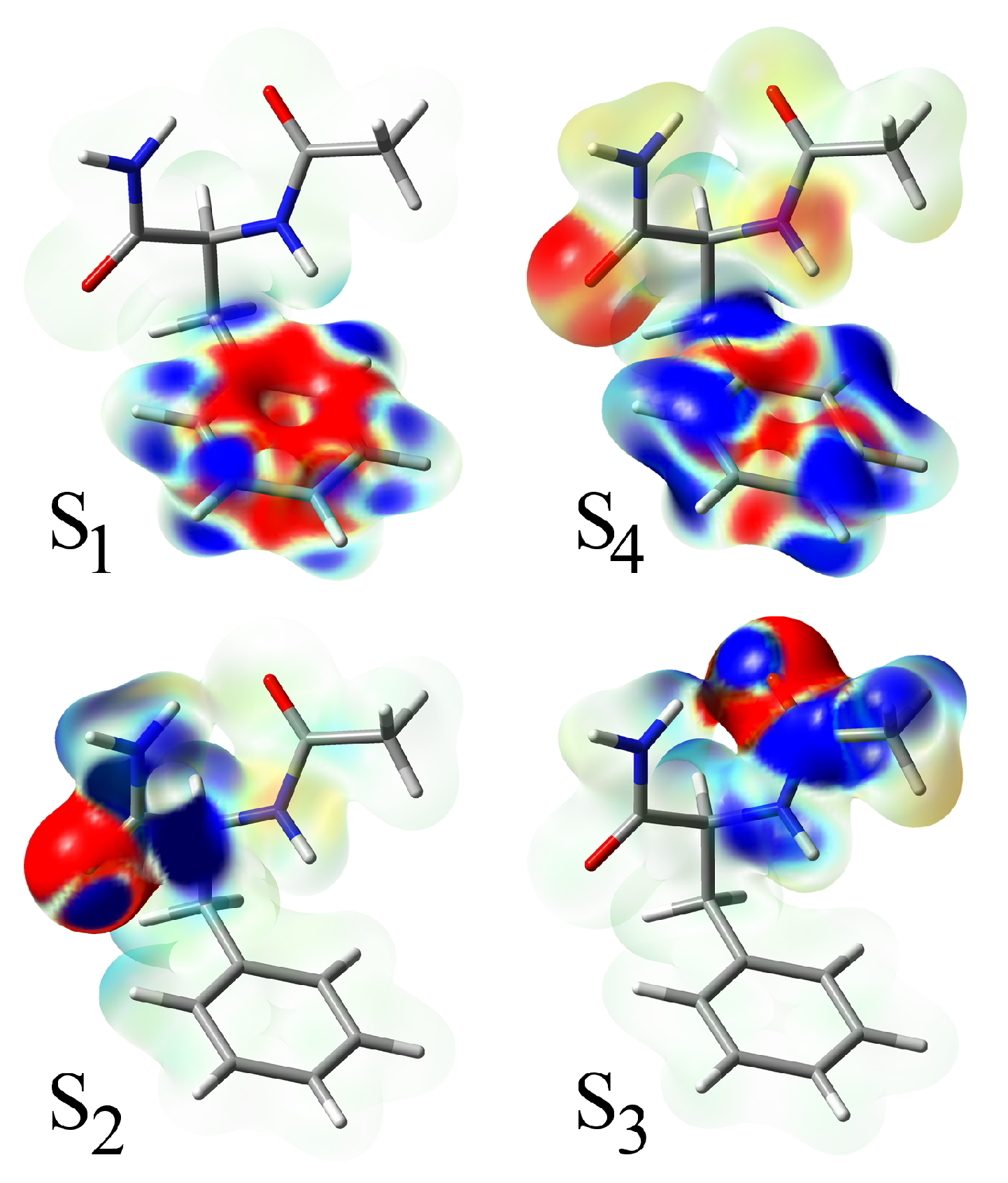
states. The electron density differences between the excited and the ground state are shown in
Figure 4. The lowest excitation of the
type is localized on the phenyl ring and is analogous to the
state of benzene. Comparing the excitation in NAPA, NAPMA and NAPAH, it is evident that the state is virtually unaffected by the chemical motif of the second amide group. The same is true for the second
transition (S
4), the one analogous to the
state of benzene. Density differences reveal that at the CC2 level, this states contains about a 14% contribution of the next S
5 state, which is a charge transfer state located just 0.2 eV above S
4.
Intercalated between the two states are two states, and with excitations localized on the first and second amide group, respectively. In NAPA and NAPMA, the second excited state is . In NAPAH, however, binding of water significantly affects the second amide group and destabilizes the excited state, which exchanges order with the state, both at the CASPT2 and RI-CC2 levels. The weaker perturbation in NAPMA B is not sufficient to cause the same effect.
Adiabatic excitations are more suitable for comparison with experiments. Geometry optimization of the S
1 state at the CC2 level leads to a minimum (further designated as
) that slightly differs from the ground state one. In all three
minima, the C–C bonds of the phenyl ring are longer on average by 0.035 Å with respect to their ground state values. The elongation of the phenyl C–C bonds is slightly smaller than 0.040 Å as obtained in benzene by reference CASPT2 calculations, even though the absolute values of phenyl C–C and C–H bond lengths of 0.011 and 0.014 Å, respectively, are longer than the corresponding benzene values [
30]. The expansion of the phenyl ring accounts for the 0.2 eV energy difference between the vertical and adiabatic excitation energies. The ordering of the other excited states remained unaltered by the geometry optimization of the S
1 state.
The electronic properties of the first
state are completely analogous to the toluene B
2 state, whose experimental 4.65 eV
1A
1 →
1B
2u vibrationless (
) transition is almost isoenergetic to the measured transitions of the NAPA systems [
31]. To reproduce the experimental values, nuclear ZPE contributions were included to simulate the vibronic states. Because of the expansion of the phenyl ring in the
minimum, most harmonic modes are red-shifted with respect to their ground state values. This stabilizes the
vibronic state by ∼0.2 eV and brings the calculated values at the RI-CC2/cc-pVDZ level just 0.1 eV above the measured ones. Furthermore,
vibronic excitations were measured 530 cm
−1 above the UV origin in both NAPA and NAPMA conformers B [
17,
19]. As the corresponding vibronic transition in toluene is found at 532 cm
−1 [
31], we assigned the transitions to the 528 and 521 cm
−1 in-plane distortion normal modes, of NAPA and NAPMA, respectively.
The CASPT2 method yields exactly the same states in all ground and S1 minima. The inversion of the and states in NAPAH B is also reproduced. Compared to corresponding RI-CC2 value, the first excitation is significantly red-shifted, while the fourth excited state is strongly blue-shifted. No state of the CT character was observed among the higher roots at the multireference level, but CC2 calculations suggest that it would be necessary to include the free electron pairs of the Phe nitrogen atom in the active space to adequately describe the CT character.
Focusing on the two
states, population analyses and electron density differences confirm that electron redistribution takes place from the oxygen to the carbon atom of the C=O group. A small increase of the electronic density on the neighboring nitrogen atom is also observed, in contrast to the reported depletion of the electronic charge on the N atom in simple amides by Serrano-Andrés et al. [
2]. Mulliken population analyses at the CASPT2 level confirm the CC2 results and indicate breaking of conjugation between N and C amide atoms upon
excitation. Both
and
states posses a number of local minima in which the corresponding C=O bonds are significantly elongated and the whole amide group deplanarized due to the absence of conjugation between the carbon and the nitrogen atom. These minima, found lower in energy than
, play a central role in the excited state deactivation of the three peptides and will be addressed in the following sections.
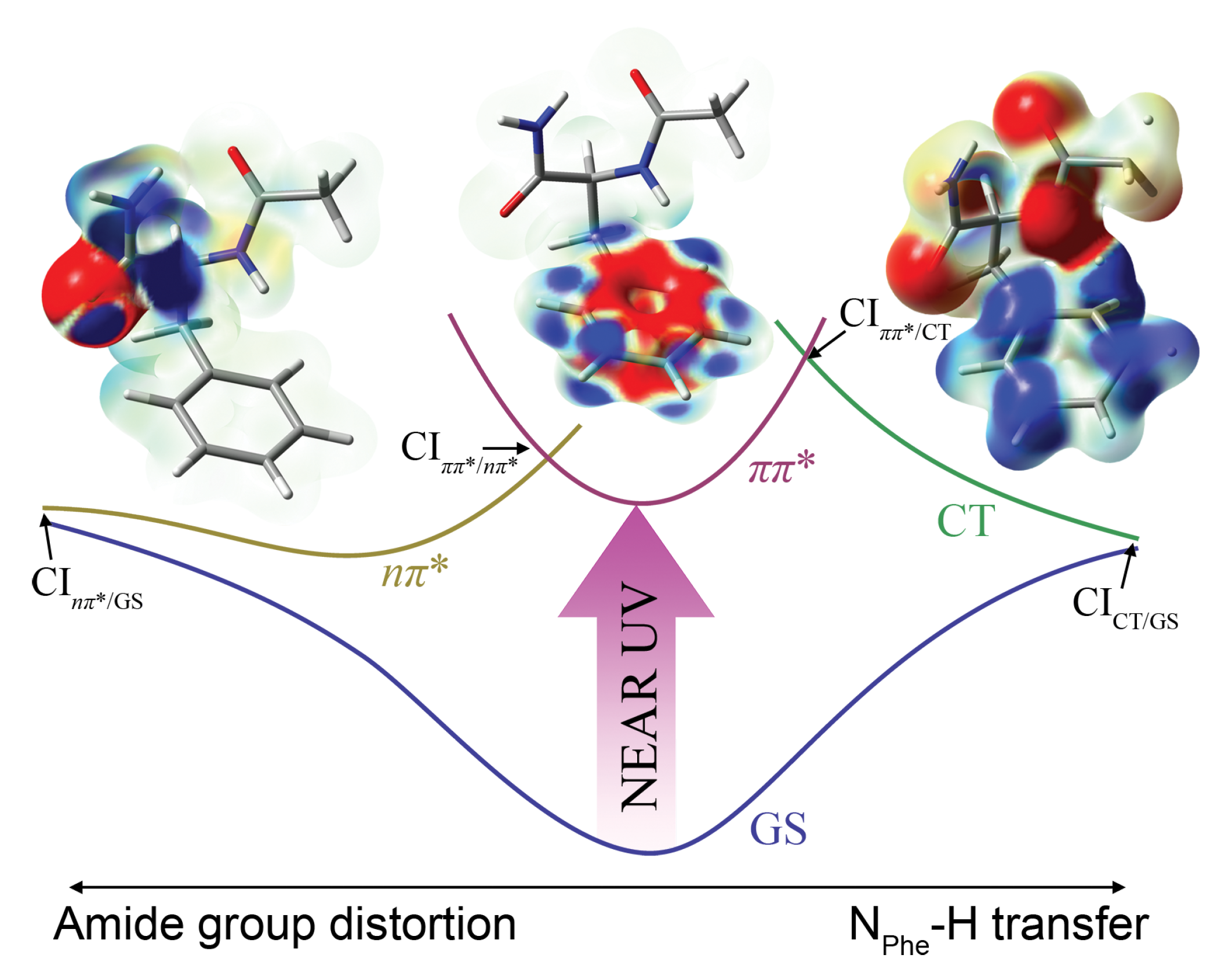
3.2. Investigation of Nonradiative Deactivation Pathways of NAPA B
Before analyzing some of the relaxation pathways that emerged from nonadiabatic dynamics simulations, it is useful to comment on the performance of the TD-PBE0/cc-pVDZ method. The TD-PBE0/cc-pVDZ vertical excitation energy to the two lowest excited states of 5.393 eV and 5.487 eV (ground state optimized at MP2/cc-pVDZ) compares relatively well with the RI-CC2 values of 5.176 and 5.606 eV in NAPA conformer B. However, TD-PBE0 groups the higher excited states more densely than RI-CC2. In particular, the energy of the second state, which has a significant charge transfer contribution arising from the mixing with the S5 state, is significantly underestimated and falls below the state. The problem becomes severe upon optimization of S1 states. The PES is dissociative along the NPhe–H bond stretching coordinate, and the hydrogen atom from the NPhe–H group is transferred to the ortho C atom of the phenyl ring. This causes a rise in the ground state energy, and a S1/S0 CI is reached.
In the dynamics simulations, we propagated a set of 44 NA-TDDFT trajectories starting from the first excited state assuming a vertical excitation of ground state geometries. Depending on the initial geometry, the S
1 state corresponds either to the
or the
state. Taking into account the limitations of TDDFT, nonadiabatic dynamics simulations have an exploratory character, and the deactivation mechanisms indicated by the simulations need to be refined at the more adequate CC2 level. Mechanism I involves H-transfer to the closest C atom of the phenyl ring. While there is a growing body of evidence indicating that H-transfer to carbon atoms of aromatic rings may be a very efficient photochemical reaction [
32,
33], in NAPA
, this pathway is likely to be overrepresented in the simulation (11 out of 44 trajectories) owing to the shortcomings of TD-PBE0. Mechanism II involves as a crucial step a CI between the initially excited
state and the
with excitation localized on the second amide group. This is the dominant deactivation mechanism according to nonadiabatic dynamics simulations encountered in 25 out of 44 trajectories. Mechanism III, encountered in four trajectories, operates similarly to Mechanism II, but involves internal conversion from the
state to the
with excitation localized on the first amide group (N-terminus). Once in the
state, deactivation to the ground state proceeds either by H-transfer along the N
Phe–H⋯O=C
Ac bond or by elongation of the C
Ac=O bond. The former is actually the realization of the electron driven H-transfer mechanism of Domcke and Sobolewski in NAPA [
14]. In both Mechanisms II and III, after leaving the initially excited
state, the system requires no additional energy to return to the ground state. The three deactivation mechanisms are illustrated in
Scheme 2. The electron density differences characteristic for Mechanism I and Mechanism II are shown in the insets.
Let us discuss Mechanism II in more detail. A characteristic NA trajectory deactivating by Mechanism II is shown in
Figure 5. The trajectory starts from a well-defined
state. The population transfer to the
state occurs after 150 fs.
Figure 6 shows the details of the first 200 fs of the dynamics. In this time span, surface hopping between the neighboring S
1 and S
2 states had occurred six times paired in three separate events of the S
1 → S
2 → S
1 type. In the first two, the change of states is so rapid that the electronic populations just swap between the two adiabatic states. This can be seen from the instantaneous increase of dynamical coupling between the states. In these two regions, the electronic population and the characters of the states behave diabatically, and after the second interaction region (
85 fs), the system still remains in the initial
state. However, the third encounter exhibits a steadily monotonic increase of the dynamical coupling as a consequence of a smooth change of the character of the electronic states. As the electronic density difference shows, the initial
state acquires a
type excitation due to mixing between the two states. This gradual mixing arises from the trajectory passing through the S
1/S
2 CI, but in a region of the configuration space where the nonadiabatic coupling is smaller than in the previous four cases. Due to the gradual change of dynamical couplings in this region, the S
1 electronic population displays a monotonic decrease, and the whole transition between states is adiabatic. At 145 fs, the characters of the S
1 and S
2 states are almost indistinguishable due to equal mixing of adiabatic electronic states. This equal contribution changes in the following few steps, and the last S
2 → S
1 surface hop is more diabatic, as the narrower dynamical coupling shows. This diabaticity of surface hop is necessary to preserve the character of the new state. After 150 fs, the S
1 state has completely evolved into
. The energy of the other excited states rises abruptly after 150 fs (see
Figure 5a) and soon exceeds the total energy of the system. Thus, they do not influence the dynamics in any way. Once in the
state, the benzene ring C–C bonds contract by 0.02 Å, while the C
Phe=O and C
Phe–N bonds expand by 0.18 and 0.12 Å, respectively (
Figure 5b). At this point, the H-bond is still preserved. The modest gain in kinetic energy of 0.25 eV is initially channeled into the C
Phe=O and C
Phe–N stretching modes and then redistributed into distortion modes. The initial planarity of the second amide group is lost due to breaking of the conjugation between the C
Phe and N atoms. Local functional groups involving the two atoms deplanarize, and the O atom moves away from the phenyl ring. Although these geometrical changes only weakly affect the energy of the S
1 state, the energy of the ground state exhibits a sudden rise of almost 2 eV. At around 300 fs, the NH
2 twisting and waging motions become so excited that they eventually break the
-ring H-bond. The NH
2 group can now almost freely rotate around the C
Phe–N bond. After the next 100 fs, part of the C
Phe=O vibrational energy is transferred to the C
Phe–N stretch vibration. The combination of backbone strain and heavy distortion of the second amide group contributes to the ground state instability, due to which the S
0 state shows large oscillations, and on a few occasions, the S
1-S
0 energy gap decreases to less than 0.5 eV. This is also the situation in the last point of the dynamics in which the excited state calculation is aborted due to numerical instabilities in the solution of Cassida’s equation near a true intersection point [
34].
3.3. Comparative Analysis of the States of NAPA B and NAPMA B
Nonadiabatic molecular dynamics simulations of NAPA B have revealed three deactivation mechanisms starting from the state. We made no attempts to compute the excited state lifetimes associated with the three mechanisms. The experimental excitation at the origin of the transition results in an excited state lifetime of 1.5 ns in the case of NAPA B and thus makes such computation unfeasible, even at the TDDFT level. Thus, simulations could not indicate the mechanism responsible for the remarkably shorter lifetime of NAPA B. The absence of any deuteration effect on the experimental lifetimes and the known underestimation of the energy of charge transfer states in TD-PBE0 led us to exclude Mechanism I. To discriminate between Mechanisms II and III, we adopted a different strategy. We supposed that if Mechanism II was the one responsible for the short lifetime of NAPA B, then a chemical modification of the second amide group would lead to changes in the excited state lifetime. Accordingly, we focused on NAPMA B.
In
Table 1, we reported the lifetimes of the three NAPMA conformers measured at the origin of the S
1 state. For NAPA conformers A, B and C, pump-probe experiments with nanosecond (picosecond) resolution provided lifetimes of 70 ns, <7 ns (1.5 ns) and 35 ns, respectively. Comparing these values to NAPMA A, B and C lifetimes of 67, 48 and 62 ns, it becomes clear that the mechanism responsible for the very short lifetime of NAPA B is likely to be Mechanism II. As the difference in the lifetimes of NAPA B and NAPMA B is striking, it is worth investigating how methylation of the second amide group actually hampers the deactivation of NAPMA via Mechanism II.
Mechanism II involves a transfer of excitation from the
state to the
state followed by a set of geometry deformations of the second amide group, which lead to the CI with the ground state. The second part is comparable to acetamide deactivation from the
state for which a variety of deactivation mechanism have been found [
35]. In NAPA and NAPMA, however, the gain in kinetic energy for reaching the
state is approximatively half the one in acetamide, implying that only deactivation channels with a low activation barrier contribute to the deactivation of the
state. RI-CC2 reaction path calculations indicate that in both NAPA B and NAPMA B, the CI with the ground state is most easily reached by elongation of the C
Phe=O bond. In both systems, this path is barrierless.
Thus, the difference between NAPA B and NAPMA B originates from the first part of Mechanism II governing the population transfer from the
to the
state via the
/
CI seam. To map the seam, we start by searching for local minima on the
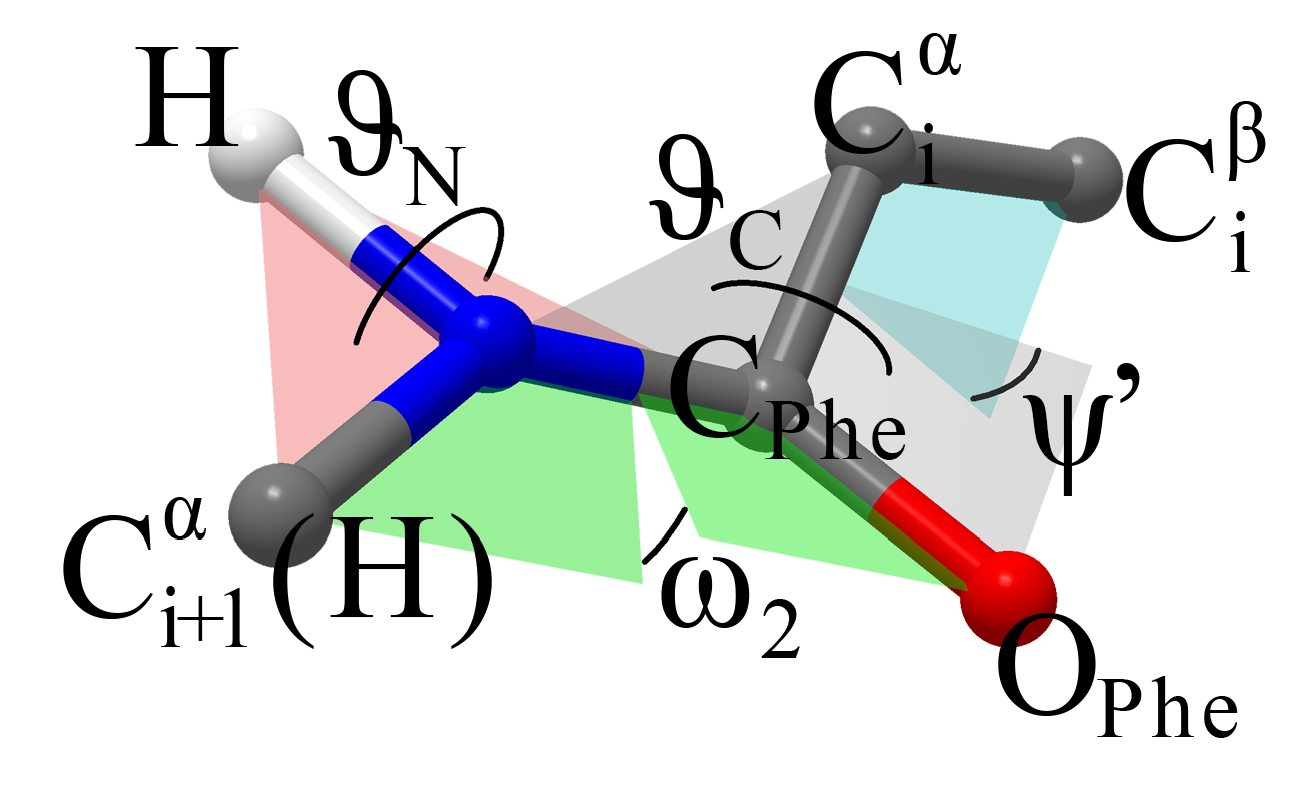
PESs of NAPA B and NAPMA B. By scanning the three torsional, angles
,
and
, defining the second amide group (see
Figure 3), we obtained six minima with preserved
secondary structure. These were grouped into two categories based on the pyramidalization of the C
Phe=O group defined by the
angle. The
angle basically determines the direction in which the C
Phe=O bond moves from the plane defined by the initial
structure. Positive (negative) values indicate that the O
Phe atom moves toward (away from) the phenyl ring. Structures with negative and positive
angle values are designated as M
a and M
b, respectively, while numerical notations 1, 2 and 3 are used for structures within the set.
Table 3 compiles the relative energies and most important geometrical parameters of the minima.
Six almost isoenergetic M(II) minima were found 0.62–0.76 eV lower in energy than the reference minimum. This large stabilization stems from a major distortion of the second amide group, which encompasses torsion () and pyramidalization of the amide N and carbonyl C atoms, as well as by a 0.14–0.23 Å elongation of the C=O bond. Apart from the distortion of the second amide group, the rest of the molecular framework deviates only slightly from , implying that the seam of / CIs is geometrically close to the structure.
Table 4 compiles the energy of six CIs geometries (
a1–
a3 and
b1–
b3) obtained by linear interpolation (LIP) between the
and the
minima. In addition critical CI points, such as minimum energy CI (MECI) and minimum distance CI (MDCI) from
, are given [
36]. All geometries belong to the same
/
CI seam. In NAPA B, two MECI structures differing in the direction of the N atom pyramidalization (
) are located 0.13 and 0.16 eV above
. In NAPMA B, two analogous, but isoenergetic structures are found 0.17 eV above
. The two MDCI structures, which are expected to represent a dynamically important portion of the CI seam, are located 0.22 eV and 0.28 eV above
in NAPA B and NAPMA B, respectively. Comparing the MDCI and
structures, a major difference is found only in the C=O bond elongation. Altogether, the structural analysis reveals that NAPA B and NAPMA B share a quite similar topography of the
/
CI seam.
In terms of energy, one sees that as a consequence of smaller RMSD from , the barriers in the basin a are lower than the ones in b. Furthermore, the barriers to equivalent CI geometries are systematically lower in NAPA B than in NAPMA B. In particular, the lowest barriers corresponding to MECIa, CIa1 and CIa2 increase from 0.13, 0.22 and 0.19 eV in NAPA B to 0.17, 0.32 and 0.25 eV in NAPMA B. However, these differences are not large enough to exclude the possibility of NAPMA B deactivation by Mechanism II.
Before addressing the problem of the accessibility of the
/
CI seam, it is important to verify that the CC2 method describes adequately this region of the configuration space.
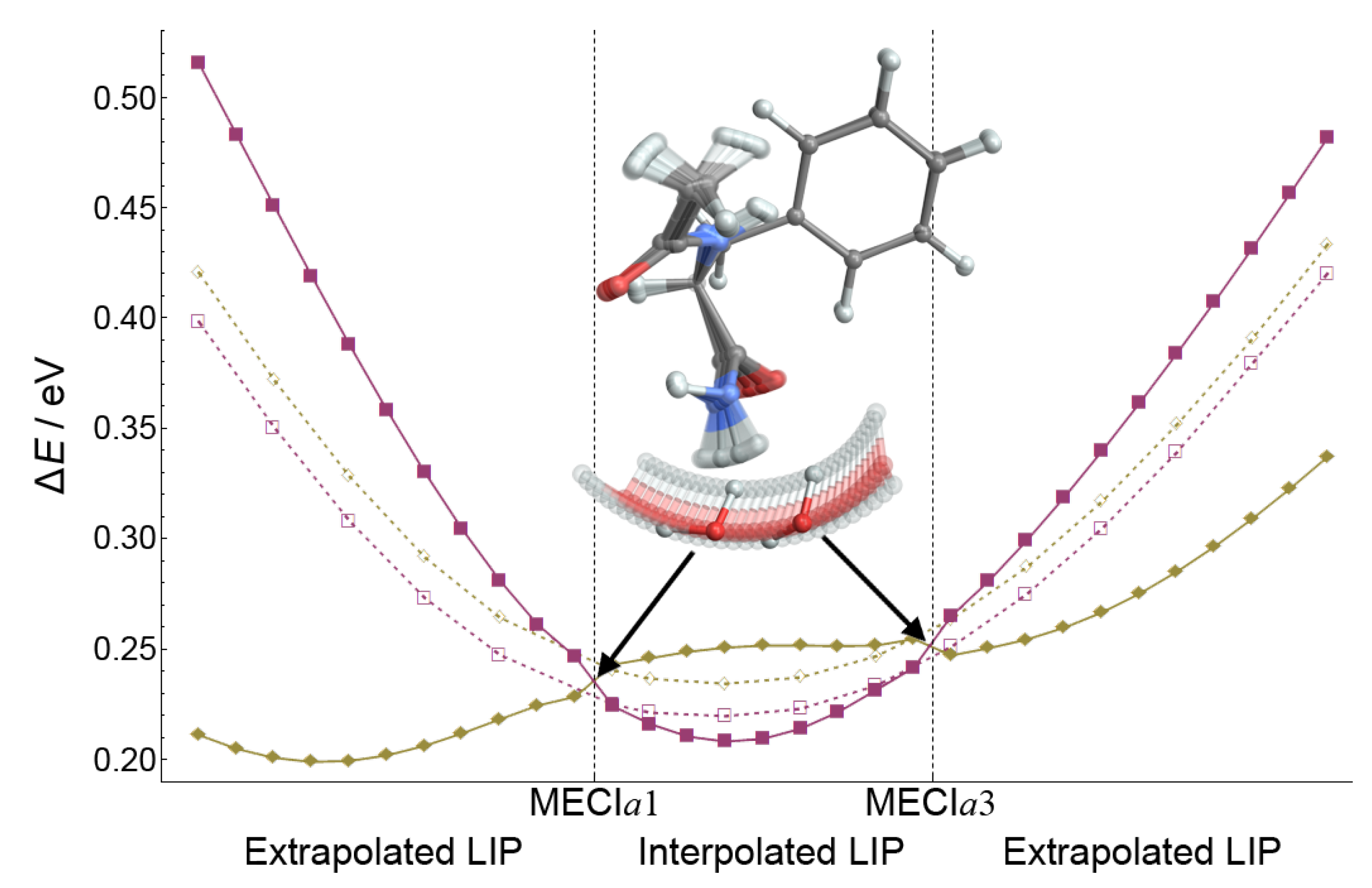
Figure 7 compares CASPT2 (solid lines) and CC2 (dashed lines) results for an LIP constructed between the
and MECI
a geometries. Both
and MECI
a geometries have been optimized at the CC2/cc-pVDZ level. The energy profiles of the
,
and
states are shown. It is evident that CC2 and CASPT2 LIPs follow the same trend. However, the CC2 method overestimates the energy of the
state by 0.4 eV (see also
Table 2) and consequently underestimates the barrier to the
/
CI. This means that the problem of the accessibility of the
/
seam is actually more acute at the CASPT2 level and needs to be carefully investigated. We will do this in the next section.
3.4. Accessibility of the / Seam of CI in NAPA B and NAPMA B
To investigate the depopulation of the vibrationless phenyl
state, it is better to switch to the diabatic representation in which a
state nuclear wave packet can be examined as a standing wave function in the minimum well of the
PES. By neglecting the nuclear kinetic energy term, the time-dependent Schrödinger equation for an arbitrary number of states in diabatic representation can be easily transformed to the following form:
which describes the probability density
of the initial
state in terms of
and
diabatic nuclear functions of
and remaining
j states, respectively. The bar designates complex conjugation, and
denotes the Cartesian coordinate vector with components
.
are the diabatic coupling terms describing the interaction strength between
and
j electronic states and, as the adiabatic terms, are most relevant around CI points. However, contrary to adiabatic, the diabatic couplings vanish at CI points. By integrating the above expression over the entire nuclear configuration space, the time-dependent population of the
state is obtained:
On the right-hand side, the integration is restricted only to the portion of the configuration space around the CI seams where the couplings do not vanish. Because the
values are proportional to the
wave function values in the diabatic Schrödinger equation, the expression reduces to:
By approximating the wave functions as the product of their space and time independent parts,
, the previous expression further simplifies to:
with only the spatial part of the
wave function left under the integral. This shows that the time-dependent population of the
state satisfies the first order rate law with the above integrals as the rate constants
The obtained first order rate is consistent with the monoexponential decay of the phenyl
first excited state in NAPA and NAPMA conformers as observed in the experiments [
17,
18]. As stated, the decay rate depends on the two terms, the coupling factor between the electronic states and the probability
of the starting
state to access the interaction region. Unfortunately, for the systems under investigation, the coupling terms are not available. However, owing to the similarity of the electronic states, large differences in the couplings cannot be assumed. Furthermore, the distinctive sharp crossings obtained between
and
PES indicates that they are small and localized around CI points, which further emphasize the importance of the accessibility of the nuclear wave function to the corresponding CI seam.
Let us remind that lifetimes of the three NAPA/NAPMA conformers were measured at the origin of the S
1 state (see also
Scheme 2). Because of vibrationless excitation, the ZPE is the only energy available to the system. Therefore, we need to estimate whether a given CI geometry is classically-accessible or not to the vibrationless nuclear wave function. We retain only the harmonic terms in the normal mode expansion of the
PES and compute the extension of the normal mode coordinates at the CI geometries. The Cartesian coordinate vector of the CI geometry is denoted as
and the mass-weighted Cartesian coordinate as
with
,
. The normal mode vector
is given in terms of mass-weighted Cartesian displacements from a reference geometry
as:
where
is the unitary transformation matrix containing the eigenvectors of the Hessian matrix evaluated at the reference geometry
. In our case, the reference geometry is
, i.e., the initially excited geometry. The full dimensional harmonic
PES is then given as:
where
is the frequency of the
i-th normal mode. In principle, the quality of the harmonic approximation is questionable, but in both NAPA and NAPMA, it was found adequate, at least for the purpose of our investigation. This can be seen from the relatively small differences between the exact (LIP) and approximate (HAR) energies of the CI geometries given in
Table 4.
The potential energy along each normal mode coordinate evaluated at the CI geometry (
) can now be compared to the ZPE and the ratio (atomic units):
This determines whether the normal mode coordinate
lies in a classically-accessible or inaccessible part of the configuration space. It is important to stress that in order to be classically-accessible, a CI geometry needs to be accessible along all normal modes. A representative example is given in
Figure 8 where the MECI
a geometry is analyzed in terms of the
ratio. It is apparent that in NAPA B (red), the MECI
a geometry is classically-accessible along all of the modes as the ration
. In contrast, four low frequency normal modes of NAPMA (blue) lie in the classically inaccessible region. In the same fashion, we assess the accessibility of the key CI geometries compiled in
Table 4. Those that are classically-accessible are given in bold.
The question that naturally arises is what are the consequences of the reduced accessibility of some CI in NAPMA. The probability to reach the CI seam and the rate of decay of the
state are clearly connected. Semiclassically, when
for one of the normal modes, the probability,
, to reach the CI geometry every time the particle “hits” the classical turning point,
, is [
37]:
where
S is the action integral through the barrier:
Expressing the integral in terms of
, one easily obtains the probability that the system reaches the CI along the
i-th normal mode as:
For n modes in the tunneling regime, the total probability to reach the CI is the product of probabilities along individual modes, .
Let us return to the example of
Figure 8. If we assume that the probability to reach MECI
a in NAPA B is
, then the probability to reach MECI
a in NAPMA is only
. In the case of the
a1 CI geometry, it is
. Of course, these are very rough approximations for the 32-times increase of the lifetime of NAPMA with respect to NAPA, but they illustrate how an energy difference of only 0.04 eV (MECI
a) can lead to a 10-fold reduction of the probability of internal conversion.
Owing to the high dimensionality of the seam space, it is desirable to go beyond the analysis of individual CI geometries. A glimpse into the reduction of the classically-accessible volume of the CI seam space in NAPMA can be obtained by considering two-dimensional (2D) projections of the seam space on the torsional angles
and
and the
d(C=O) stretching coordinate. In total, 153 NAPA and 63 NAPMA CI geometries have been generated first by interpolating between pairs and triplets of reference CI geometries (
Table 4) and then by including in the search the newly-obtained CI geometries that satisfied the ZPE accessibility condition.
Figure 9 shows the 2D projections of the classically-accessible portion of
/
CI seams in NAPA B (left) and NAPMA B (right). ZPE-accessible areas are shown in gray with the two shades indicating the size of the energy gap, i.e., the accuracy of the calculation.
The difference between the classically-accessible portion of the seam in NAPA B and NAPMA B is stunning. The characteristics of the CI geometries given in
Table 4 are captured in
Figure 9. In NAPA B, MECI
a and
a1 are on the edge of the classically-accessible area, while
a2 and
a3 CI are deep in the ZPE-accessible region. For NAPMA, MECI
a and
a1 are far from the classically-accessible area.
Altogether, we found that the NH2 group of NAPA B, already distorted in the minimum, requires a small extension of the torsional normal modes to reach the CI seam. In contrast, the second amide group in NAPMA B is planar in , and the extension of several low frequency normal modes beyond the classical turning point is needed to reach the CI seam. The classically-accessible portion of the NAPMA CI seam is reduced, and the decay of the state is slower.
3.5. Alternative Nonradiative Deactivation Pathways: Phenyl Ring Puckering and Intersystem Crossing
The ring-puckering mechanism of benzene [
38] was reconstructed in NAPA B to investigate the possibility of direct deactivation from the
state to the electronic ground state. First, the phenyl moiety was isolated and optimized to the lowest prefulvene symmetrical MECI between the S
1 and S
0 states using the method of Levine et al. [
36] starting from structural parameters from [
38]. Since six distinctive positions and two puckering direction are possible in the bounded phenyl ring of NAPA, only three sites,
ortho,
meta and
para, closest and in the direction of the N
Phe–H bond were investigated. The puckered benzene ring was then attached to the peptide backbone and the system reoptimized to obtained the new S
1/S
0 MECI structure. We found that structurally, the puckered phenyl ring in NAPA B does not deviate much from the starting benzene CI structure regardless of the puckering position. The LIPs constructed between CIs and the
minimum revealed barriers of 0.65, 0.63 and 0.74 eV for puckering at the
ortho,
meta and
para positions, respectively. Since the estimated barriers are larger than the reported values for benzene [
38,
39], we optimized the S
1 transition states (TS).
Figure 10 displays the obtained TS structure together with the MECI geometry along the ring-puckering pathway at the
para C position. The vibrational analysis confirmed one imaginary frequency in the TS structure. In all three ring-puckering mechanisms, TSs were found 0.28 eV above the
minimum, which is ∼0.1–0.2 eV lower than the MC-SCF values for benzene [
38,
39]. Apart from the difference in barrier height, no significant effects of the backbone on the ring-puckering mechanisms were observed. Altogether, we found that all three ring-puckering pathways are classically inaccessible from the vibrationless states of NAPA B and NAPMA B, as was actually expected from previous theoretical studies of benzene [
38,
39,
40,
41,
42,
43].
Next, the contribution of the intersystem crossing (ISC) is considered. ISC is the main deactivation channel of vibrationless
states of benzene and toluene. Specifically, the lifetime of toluene is 86.4 ns [
31,
44], which is very similar to the lifetime of NAPA and NAPMA conformers A, as well as of NAPMA conformer C (see
Table 1). These observations agree with the El-Sayed result that the ISC rate for singlet
to triplet
state transfer between phenyl and N atom containing groups might be on the order of a few tens of ns [
45]. However, the 1.5-ns lifetime of NAPA B is significantly shorter.
To include the triplet states in the description of NRD mechanisms of NAPA and NAPMA, spin coupling terms between states of different multiplicity would be required. Unfortunately, these are unavailable at the CC2 level of theory. Therefore, we compared only the energy profiles of the triplet and singlet states.
Figure 11 shows the LIP between the
and M
a1 minimum of NAPA B. Low lying singlet and triplet states are indicated with filled and unfilled symbols, respectively. Focusing on the triplet
3 state, one sees that it intersects with the phenyl singlet
1 prior to the singlet
1 PES. The
1/
3 crossing is found barely 0.01 eV above the
minimum and is classically-accessible, indicating a potential contribution of ISC to NAPA B deactivation. However, this observation should be taken with caution. Namely, due to the more pronounced stability of triplet states, most of the
1/
3 crossing seam is classically-accessible in all NAPA and NAPMA conformers. As no difference in the spin-orbit couplings between structurally equivalent NAPA and NAPMA conformers should be a priori anticipated, the ISC mechanism actually fails to explain the experimental difference in the lifetimes of NAPA B and NAPMA B. Therefore, Mechanism II remains the more plausible deactivation pathway of NAPA and NAPMA conformer B.
3.6. Investigation of Nonradiative Deactivation Pathways in NAPA Monohydrate
In NAPAH B, the monohydrate of NAPA conformer B, water is H-bonded to the second amide group. On the basis of the results obtained for NAPA and NAPMA, we concentrate on the
state. From the vertical excitation energies reported in
Table 2, it is evident that hydration destabilizes the
state. At the geometry of vertical excitation, the energy gap between the
and the
states increased from 0.43 (0.52) eV in NAPA (NAPMA) to 0.635 in NAPAH. Thus, unless a new deactivation mechanism sets in, NAPAH should relax more slowly than NAPA and NAPMA. Upon geometry optimization of the
state, several minima have been found. A one to one comparison between NAPA and NAPAH minima is, however, not possible. On the one hand, not all NAPA (NAPMA) minima are realizable in NAPAH, as some cannot accommodate a hydrogen-bonded water molecule. On the other hand, NAPAH M
(II) minima are characterized by the high flexibility of the water molecule. Water can occupy a range of positions independently of the distortion of the second amide group, meaning that for a given NAPA minimum, one finds several M
(II) minima in NAPAH.
Figure 12 shows the most stable NAPAH B M
(II) minimum, which is also geometrically the closest to the starting
structure. In this particular M
(II) minimum, the water molecule is attached with two hydrogen bonds to the second amide group, but the co-linearity of the O–H
O
(II) hydrogen bond, and thus, its strength, is reduced due to the decreased electronic density on the O
(II) atom.
To understand how microsolvation affects the deactivation of the state, we investigated two possible relaxation pathways. Both start from the minimum and reach one of the M(II) minima, but differ in the mechanism of deactivation.
In the first, shown in
Figure 12, the reaction coordinate is the elongation of the
–H bond in the
–H
O
(II) moiety. It is evident that
–H bond elongation leads to a continuum increase of the energies of S
0 and S
1 states. After the double hydrogen transfer, a sudden decrease of the energy of the
state resulted in a CI that intersects with the ground state at a point almost isoenergetic to the
minimum. A reaction barrier of 0.7 eV is found on the H-transfer path. At the CI geometry, whose energy slightly exceeds the energy of the
minimum, the hydrogen atom is transferred to the O
(II) atom, and the distal H atom of the amino group is transferred to water to compensate for the missing H atom. The intersection geometry can be viewed as a highly deformed structure of the imidic acid form of the second amide group. The changes in other bonds are very small, below 0.02 Å.
The second mechanism is shown in
Figure 13. Here, in analogy to the deactivation of NAPA and NAPMA, we elongated the C
Phe=O bond. One immediately sees that the extension of the C
Phe=O bond leads only to a marginal increase in the energy of the
state. However, the geometrical changes along the path are significant as the two overlapped geometries clearly show. Already a 0.16 Å extension of the C
Phe=O induces a major change in the structure of the monohydrate as water repositions to preserve the two hydrogen bonds. To reach the CI with the ground state, the C
Phe=O bond needs to be extended by 0.48 Å. The RMSD between the intersection and the M
(II) minimum structures is 2 Å while in the hydrogen transfer mechanism, the RMSD is ten-times smaller. However, the system needs to preserve both hydrogen bonds in order to deactivate via hydrogen transfer, whereas C
Phe=O pre-dissociation can potentially proceed from any
structure.
To explore the initial steps of NAPAH B deactivation on the surface, we have launched 10, 350 fs-long, RI-CC2/cc-pVDZ trajectories from the / MECI. The initial velocities were obtained from the Wigner distribution function for the harmonic ZPE modes of the minimum evaluated at the MECI geometry. Because of the initial C=O bond extension, the potential energy of the ground state rose very sharply, reducing the gap between S1 and S0 to ∼1.0 eV. After a few fs, the energy of S2 increased, and the state remained well separated from S1 for the whole duration of the propagation. The water-amino group hydrogen bond has been preserved in all trajectories. In contrast, the hydrogen bond with the C=O oxygen was easily disrupted. A twisting motion around the H−N(II) hydrogen bond was observed, but we have found no evidence of significant vibrational excitations of the water O−H bonds. Within the short propagation period, only one trajectory reached the S1/S0 CI. Compared to , in the CI geometry, the C=O bond was elongated by more than 0.55 Å. Altogether, combined reaction path calculations and dynamics simulations show that once in the state, NAPAH deactivates by CPhe=O bond extension. We have found no evidence that intermolecular H-transfer may speed up the deactivation of the state.
In analogy to NAPA B and NAPMA B, the key step in the deactivation of NAPA monohydrates is the transfer of excitation from the
to the
state. Following the procedure detailed in previous sections, a set of
/
CI geometries was constructed by linear interpolation between the
and different M
(II) minima. Since CI geometries are linear combinations of two minima, their geometrical changes relative to the starting
minimum are a fraction of the difference between the two minima. However, all CI geometries are structurally closer to
. Among the nine CI geometries, six belonged to the
a class of CI structures and the remaining three to class
b. In both sets, CI geometries are at least 0.27 eV higher than the
structure. Compared to NAPA and NAPMA CI structures, they display a smaller energy variation (0.29 ± 0.02 eV). The constructed CI geometries have been used to obtain MECIs.
Table 5 compiles the relative energies of four MECI geometries, one of the MECI
b type (see
Table 3 and
Table 4) and three of the MECI
a type. All four are found higher in energy than their NAPA and NAPMA counterparts. MECI
a1 and MECI
a3 differ in the position of the water molecule, which changes from one side of the plane defined by the second amide group to the other with a difference of 25° in the torsional angle
(
−N−C−O). This indicates that the CI seam topography may be rather flat with respect to the water motion.
The topography of the CI seam was investigated by interpolating the structures between two MECI
a geometries, as well as extrapolating from them. As shown with full lines in
Figure 14, motion along the LIP coordinate causes a degeneracy breaking between the electronic states, even in the interpolated region between the two MECIs. Apart from the huge change in the position and orientation of water, the second amide group exhibits a change of the
torsion angle by 7° from one MECI to the other. The remaining covalent bonds change by less than 0.2%.
Dashed lines show the energy changes of the true CI seam. The geometries on the CI seam have been obtained as minimum distance CI geometries from the linearly interpolated/extrapolated geometries. From the change in the energy profile of the state (bordeaux) and of the seam (dashed line), it is evident that the state is the one strongly affected by the disruption of the hydrogen bonds. While the state is only marginally affected, the energy of the state changes rapidly, causing the splitting of the two PESs. Thus, coordinates describing the position and orientation of water with respect to the second amide group contribute to the branching coordinates.
The results obtained for the NAPA monohydrate fit well within the broader picture of peptide deactivation in which the flexibility of the second amide group promotes the deactivation. We have shown that the remarkable flexibility of the terminal NH2 group is the cause of the faster deactivation of NAPA with respect to NAPMA. In the monohydrate, the NH2 flexibility is hampered by two hydrogen bonds preventing efficient transfer from the Phe side chain to the peptide backbone.




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}