Application of Ammonium Persulfate for Selective Oxidation of Guanines for Nucleic Acid Sequencing

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Single-Strand Oxidation by AP
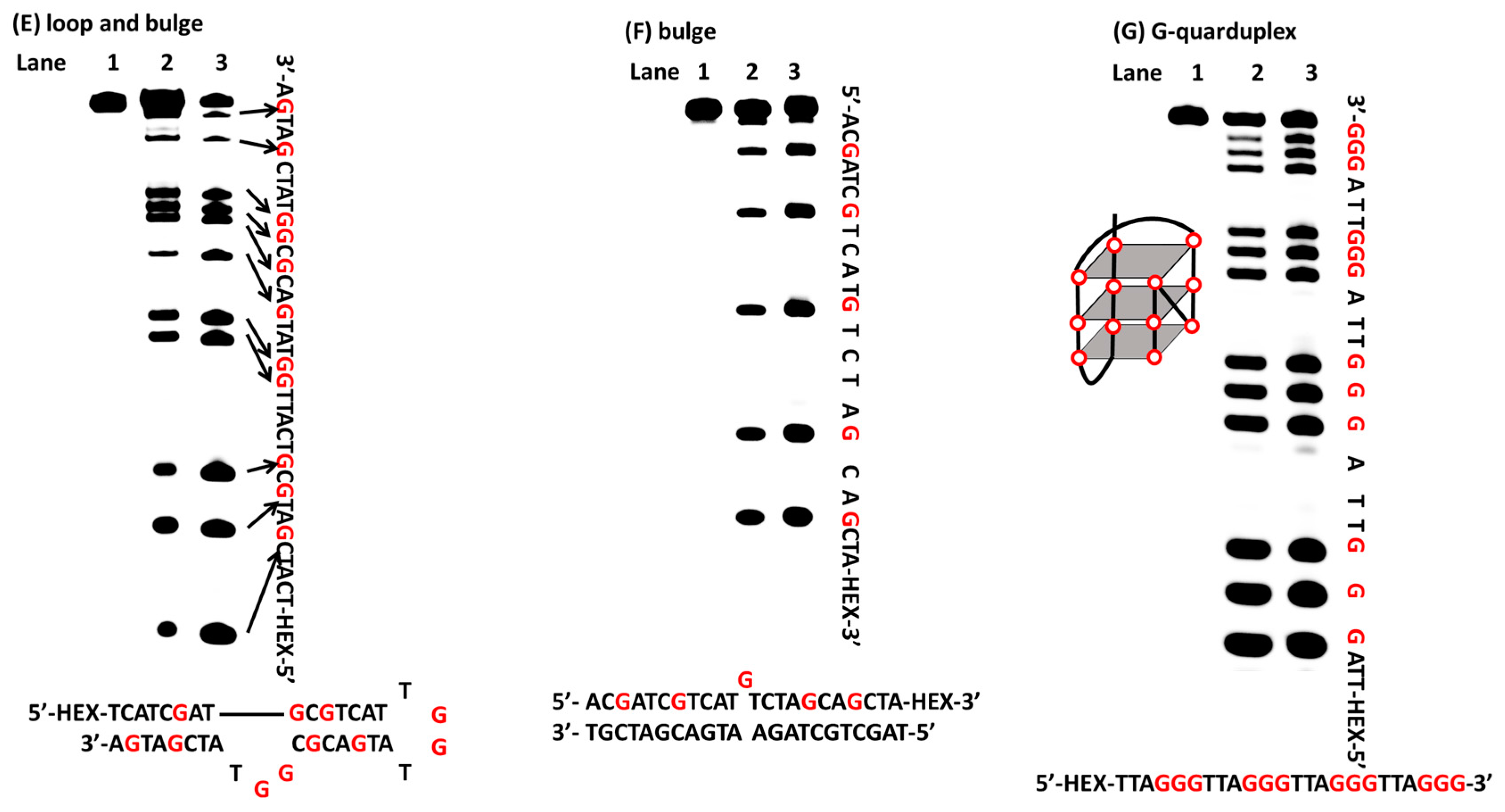
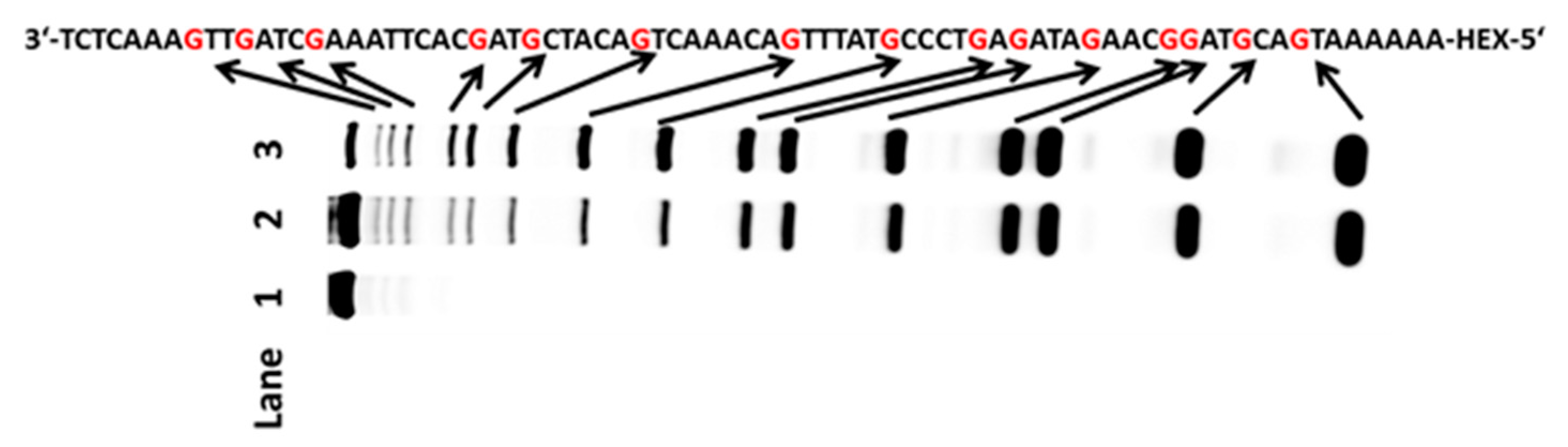
2.2. Oxidation of Guanines with Diverse DNA Structures by AP
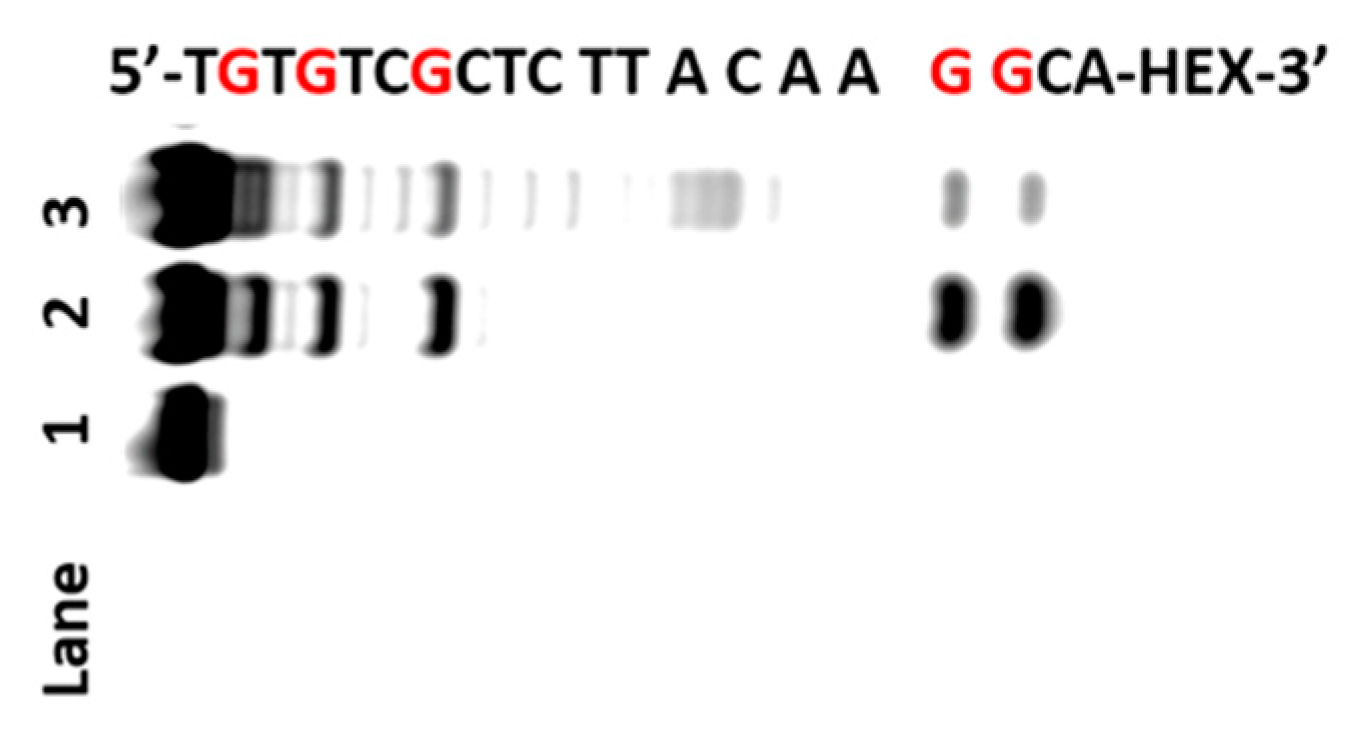
2.3. Oxidation of RNA Sequences by AP
2.4. AP for G-Sequencing Compared with Methylene Blue for G-Sequencing
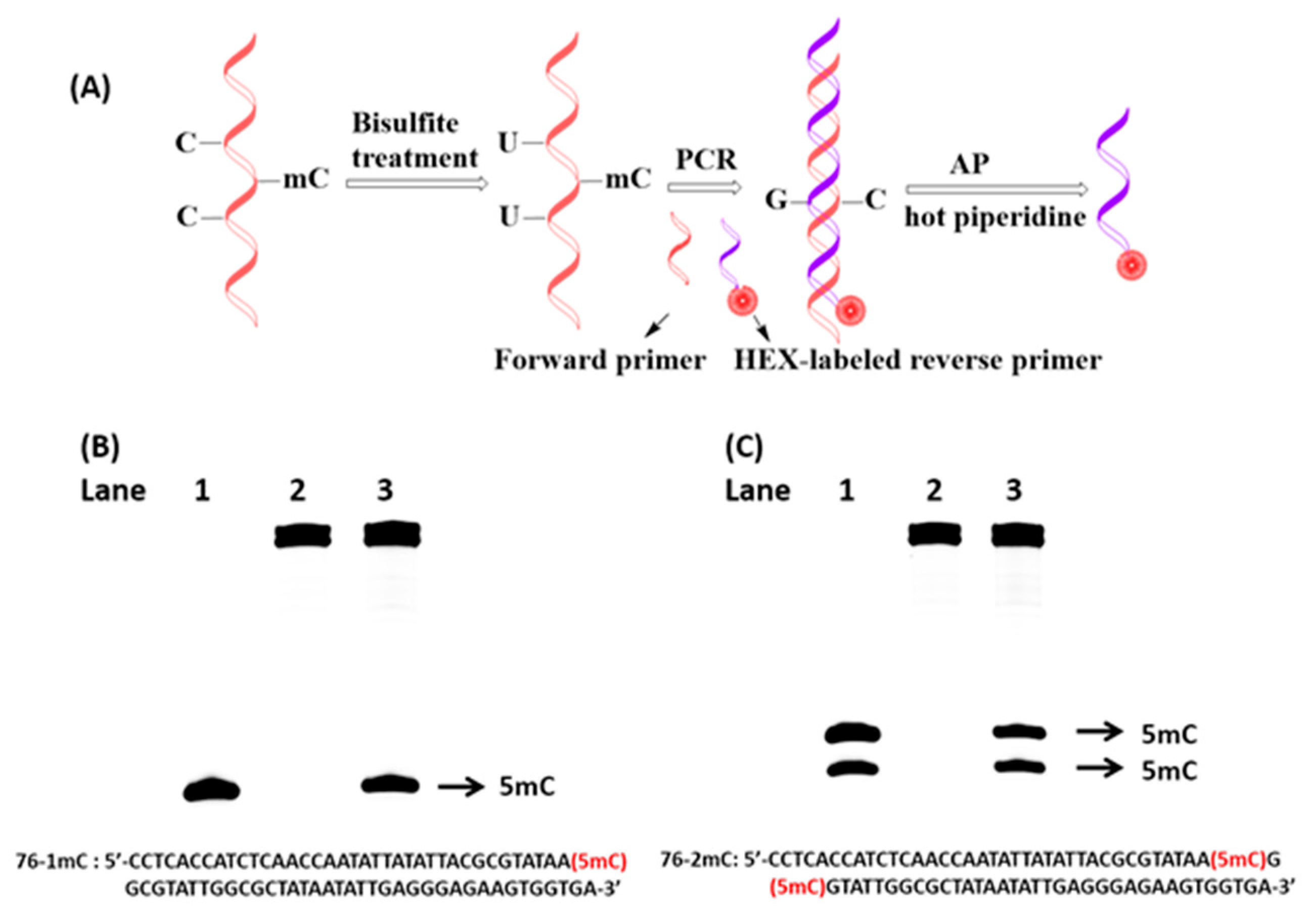
2.5. Detection of 5-Methylcytosine Based on PCR and AP
2.6. LC-MS Analysis of Guanine Oxidized by AP
3. Materials and Methods
3.1. Chemicals and Materials
3.2. Preparation of DNA Containing Different Structures
3.3. Circular Dichroism Studies
3.4. Cleavage of DNA/RNA
3.5. Primer Design and PCR Amplification
3.6. Preparation of G-Ladder
3.7. Photooxidation of DNA by Methylene Blue
3.8. RNA Digestion by RNase T1
3.9. Enzymatic Digestion of AP-Oxidized DNA
3.10. LC-MS Analysis of the AP-Oxidized DNA
3.11. Preparation of Denaturing Polyacrylamide Gel Electrophoresis (PAGE)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rahman, K. Studies on free radicals, antioxidants, and co-factors. Clin. Interv. Aging 2007, 2, 219–236. [Google Scholar] [PubMed]
- Jakhar, R.; Paul, S.; Park, Y.R.; Han, J.; Kang, S.C. 3,5,7,3′,4′-pentamethoxyflavone, a quercetin derivative protects DNA from oxidative challenges: potential mechanism of action. J. Photochem. Photobiol. B. 2014, 131, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Loft, S.; Poulsen, H.E. Cancer risk and oxidative DNA damage in man. J. Mol. Med. 1996, 74, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Nwankwo, D.C.; Abalaka, M.E. Restriction enzymes and their uses in specific sequencing to produce predictable fragment of DNA making genetic engineering simply. J. Pharm. Res. 2011, 1, 148–152. [Google Scholar]
- Zhai, Q.Q.; Xu, L.; Ge, Y.S.; Tian, T.; Wu, W.D.; Yan, S.Y.; Zhou, Y.Y.; Deng, M.G.; Liu, Y.; Zhou, X. Site-specific recognition of guanosine by manganese(III) corroles in DNA non-duplex regions through active oxygen transfer. Chem. Eur. J. 2011, 17, 8890–8895. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.R.; Shilpa, A. Interaction of DNA with small molecules: Role of copper histidyl peptide complexes in DNA binding and hydrolytic cleavage. Ind. J. Chem. 2010, 49, 1003–1015. [Google Scholar]
- Chitranshi, P.; Chen, C.N.; Jones, P.R.; Faridi, J.S.; Xue, L. Investigation on the interactions of NiCR and NiCR-2H with DNA. Bioinorg. Chem. Appl. 2010, 2010, 619436. [Google Scholar] [CrossRef] [PubMed]
- Barve, A.; Kumbhar, A.; Bhat, M.; Joshi, B.; Butcher, R.; Sonawane, U.; Joshi, R. Mixed-ligand copper(II) maltolate complexes: synthesis, characterization, DNA binding and cleavage, and cytotoxicity. Inorg. Chem. 2009, 48, 9120–9132. [Google Scholar] [CrossRef] [PubMed]
- Tto, T.; Thyagarajan, S.; Karlin, K.D.; Rokita, S.E. Recognition of guanines at a double helix-coil junction in DNA by a trinuclear copper complex. Chem. Commun. 2005, 38, 4812–4814. [Google Scholar]
- Zheng, K.W.; Zhang, D.; Zhang, L.X.; Hao, Y.H.; Zhou, X.; Tan, Z. Dissecting the strand folding orientation and formation of G-quadruplexes in single- and double-stranded nucleic acids by ligand-induced photocleavage footprinting. J. Am. Chem. Soc. 2011, 133, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Starrs, S.M.; Davies, R.J.H. Sequence Specificity of Alkali-labile DNA Damage Photosensitized by Suprofen. Photochem. Photobiol. 2000, 72, 291–297. [Google Scholar] [CrossRef]
- Simon, M.T.; Vunakis, H.V. The photodynamic reaction of methylene blue with deoxyribonucleic acid. J. Mol. Biol. 1962, 4, 488–499. [Google Scholar] [CrossRef]
- Friedmann, T.; Brown, D.M. Base-specific reactions useful for DNA sequencing: Methylene blue–sensitized photooxidation of guanine and osmium tetraoxide modification of thymine. Nucleic Acids Res. 1978, 5, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, T.W. RNA structure determination using nuclease digestion. Cold Spring Harb. Protoc. 2013, 4, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Heinemann, U.; Hahn, U.; Saenger, W. Ribonuclease T1: structure, function, and stability. Angew. Chem. Int. Ed. 1991, 30, 343–360. [Google Scholar] [CrossRef]
- Maxam, A.M.; Gilbert, W. A new method for sequencing DNA. Proc. Natl. Acad. Sci. USA 1977, 74, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Mao, W.X.; Xu, X.W.; He, H.; Huang, R.; Chen, X.; Xiao, H.; Yu, Z.D.; Liu, Y.; Zhou, X. Specific recognition of guanines in non-duplex regions of nucleic acids with potassium tungstate and hydrogen peroxide. Nucleic Acids Res. 2015, 43, e3. [Google Scholar] [CrossRef] [PubMed]
- Ehresmann, C.; Baudin, F.; Mougel, M.; Romby, P.; Ebel, J.P.; Ehresmann, B. Probing the structure of RNAs in solution. Nucleic Acids Res. 1987, 15, 9109–9127. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Chen, Y.C.; Chong, J.; Fin, A.; McCoy, L.S.; Xu, J.; Zhang, C.; Wang, D. Pyrene-based quantitative detection of the 5-formylcytosine loci symmetry in the CpG duplex content during TET-dependent demethylation. Angew. Chem. Int. Ed. 2014, 126, 11223–11227. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.G.; Graff, J.R.; Myӧhänen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9821–9826. [Google Scholar] [CrossRef] [PubMed]
- Hickerson, R.P.; Ferran, P.; Muller, J.G.; Foore, C.S.; Burrows, C.J. Sequence and stacking dependence of 8-oxoguanine oxidation: Comparison of one-electron vs singlet oxygen mechanisms. J. Am. Chem. Soc. 1999, 121, 9423–9428. [Google Scholar] [CrossRef]
- Li, X.M.; Zheng, K.W.; Hao, Y.H.; Tan, Z. Exceptionally selectiveand tunable sensing of guanine derivatives and analogues by structural complementation in a G-quadruplex. Angew. Chem. Int. Ed. 2016, 128, 13963–13968. [Google Scholar] [CrossRef]
- Bont, R.D.; Larebeke, N. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis 2004, 19, 169–185. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.C.; Muller, J.G.; Rachlin, E.M.; Burrows, C.J. Characterization of spiroiminodihydantoin as a product of one-electron oxidation of 8-Oxo-7,8-dihydroguanosine. Org. Lett. 2000, 2, 613–616. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Xiong, J.; Jiang, H.P.; Zheng, S.J.; Feng, Y.Q.; Yuan, B.F. Determination of oxidation products of 5-Methylcytosine in plants by chemical derivatization coupled with liquid chromatography/tandem mass spectrometry analysis. Anal. Chem. 2014, 86, 7764–7772. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds used in this paper are available from the authors. |







© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Liu, C.; Hong, T.; Wu, F.; Yu, S.; He, Z.; Mao, W.; Zhou, X. Application of Ammonium Persulfate for Selective Oxidation of Guanines for Nucleic Acid Sequencing. Molecules 2017, 22, 1222. https://doi.org/10.3390/molecules22071222
Wang Y, Liu C, Hong T, Wu F, Yu S, He Z, Mao W, Zhou X. Application of Ammonium Persulfate for Selective Oxidation of Guanines for Nucleic Acid Sequencing. Molecules. 2017; 22(7):1222. https://doi.org/10.3390/molecules22071222
Chicago/Turabian StyleWang, Yafen, Chaoxing Liu, Tingting Hong, Fan Wu, Shuyi Yu, Zhiyong He, Wuxiang Mao, and Xiang Zhou. 2017. "Application of Ammonium Persulfate for Selective Oxidation of Guanines for Nucleic Acid Sequencing" Molecules 22, no. 7: 1222. https://doi.org/10.3390/molecules22071222
APA StyleWang, Y., Liu, C., Hong, T., Wu, F., Yu, S., He, Z., Mao, W., & Zhou, X. (2017). Application of Ammonium Persulfate for Selective Oxidation of Guanines for Nucleic Acid Sequencing. Molecules, 22(7), 1222. https://doi.org/10.3390/molecules22071222