High-Resolution Inhibition Profiling Combined with HPLC-HRMS-SPE-NMR for Identification of PTP1B Inhibitors from Vietnamese Plants



Abstract
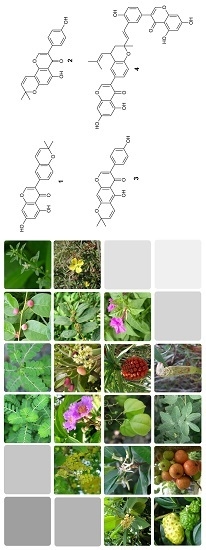
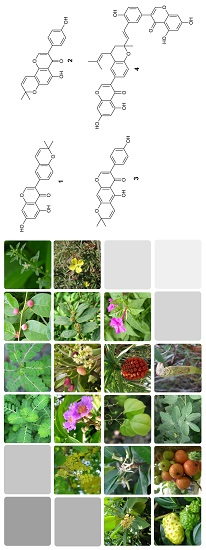
:
1. Introduction
2. Results and Discussion
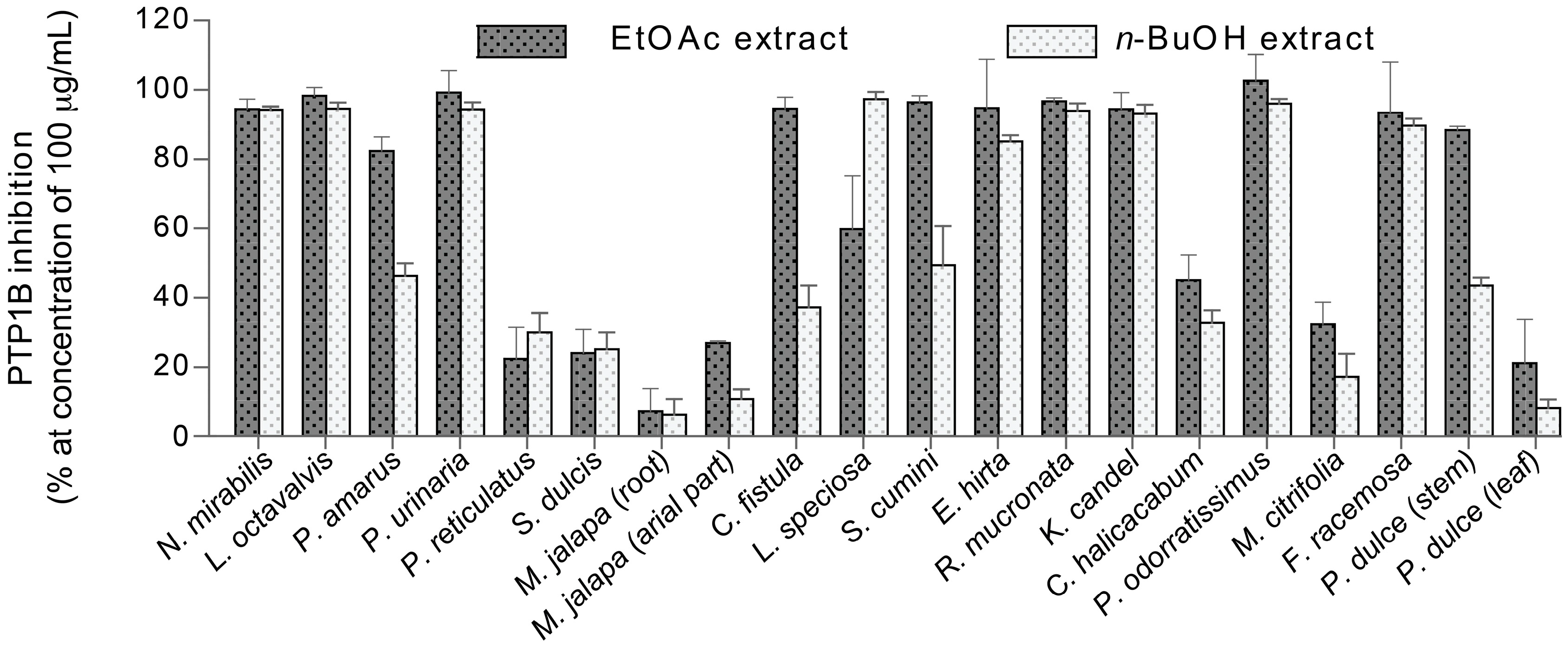
2.1. PTP1B Inhibitory Activity of Crude Extracts
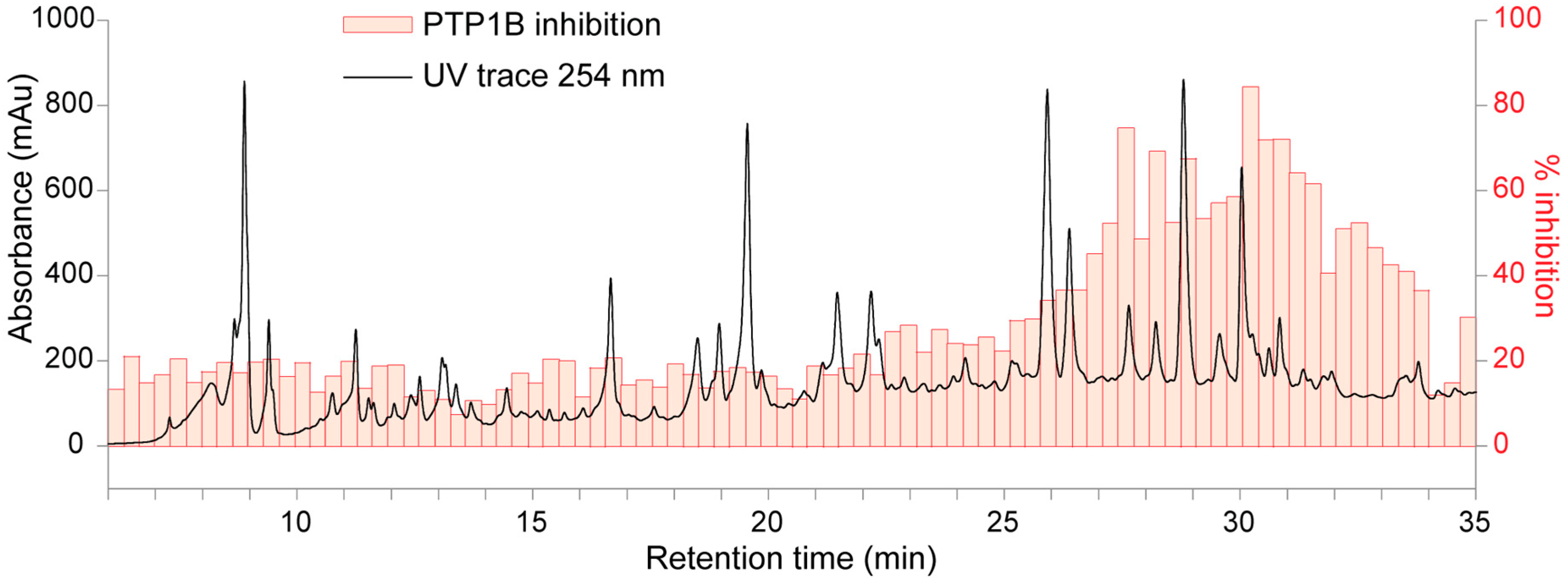
2.2. High-Resolution PTP1B Inhibition Profiles
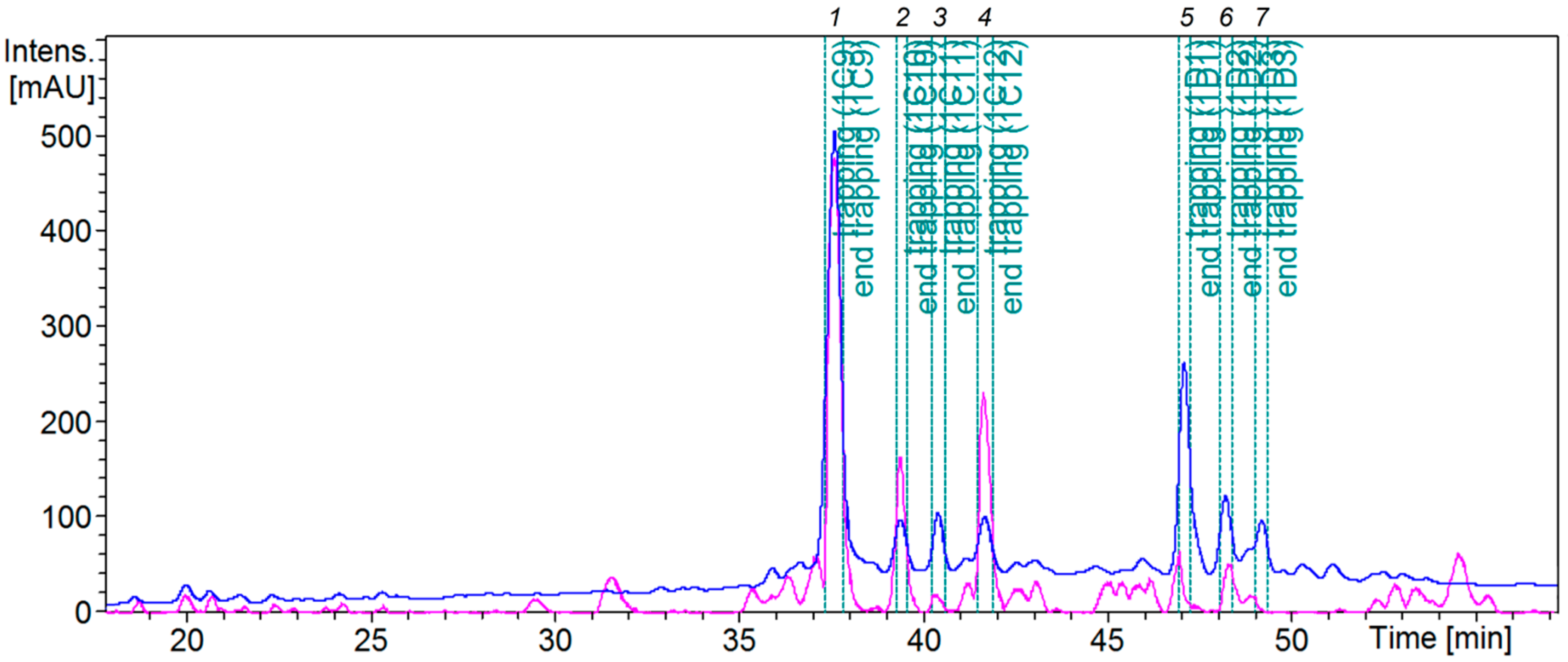
2.3. HPLC-HRMS-SPE-NMR Analysis
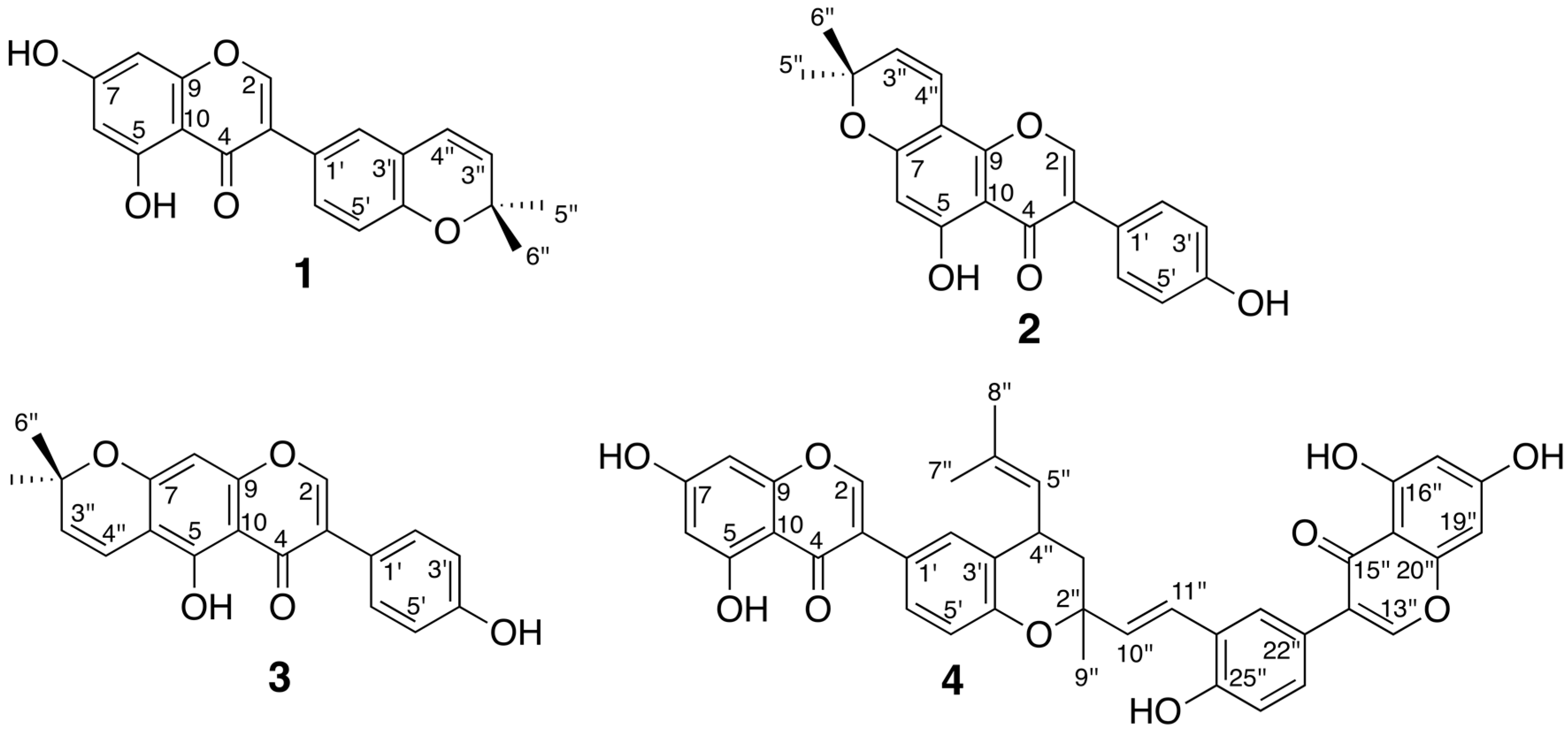
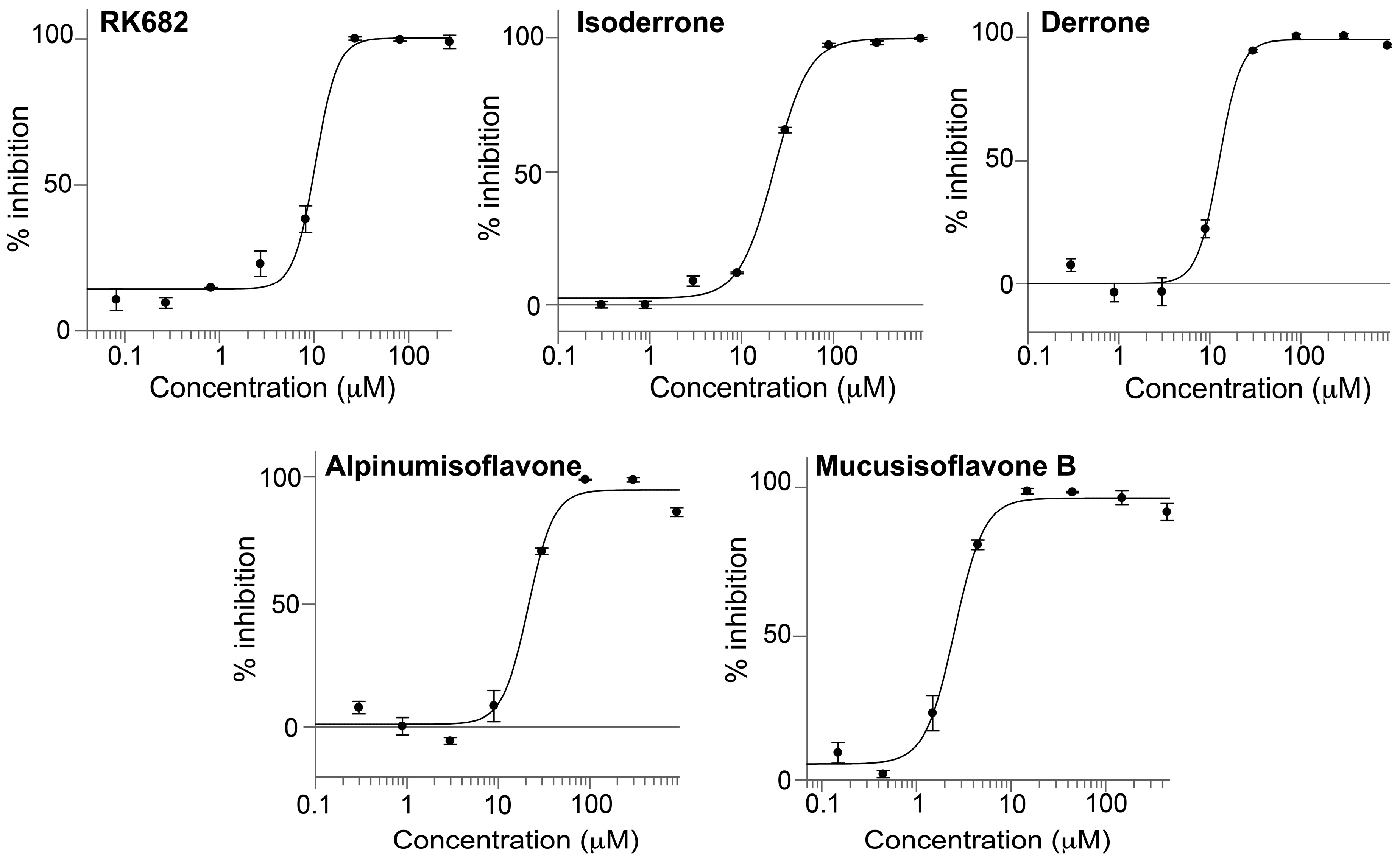
2.4. Isolation and Evaluation of Bioactive Compounds
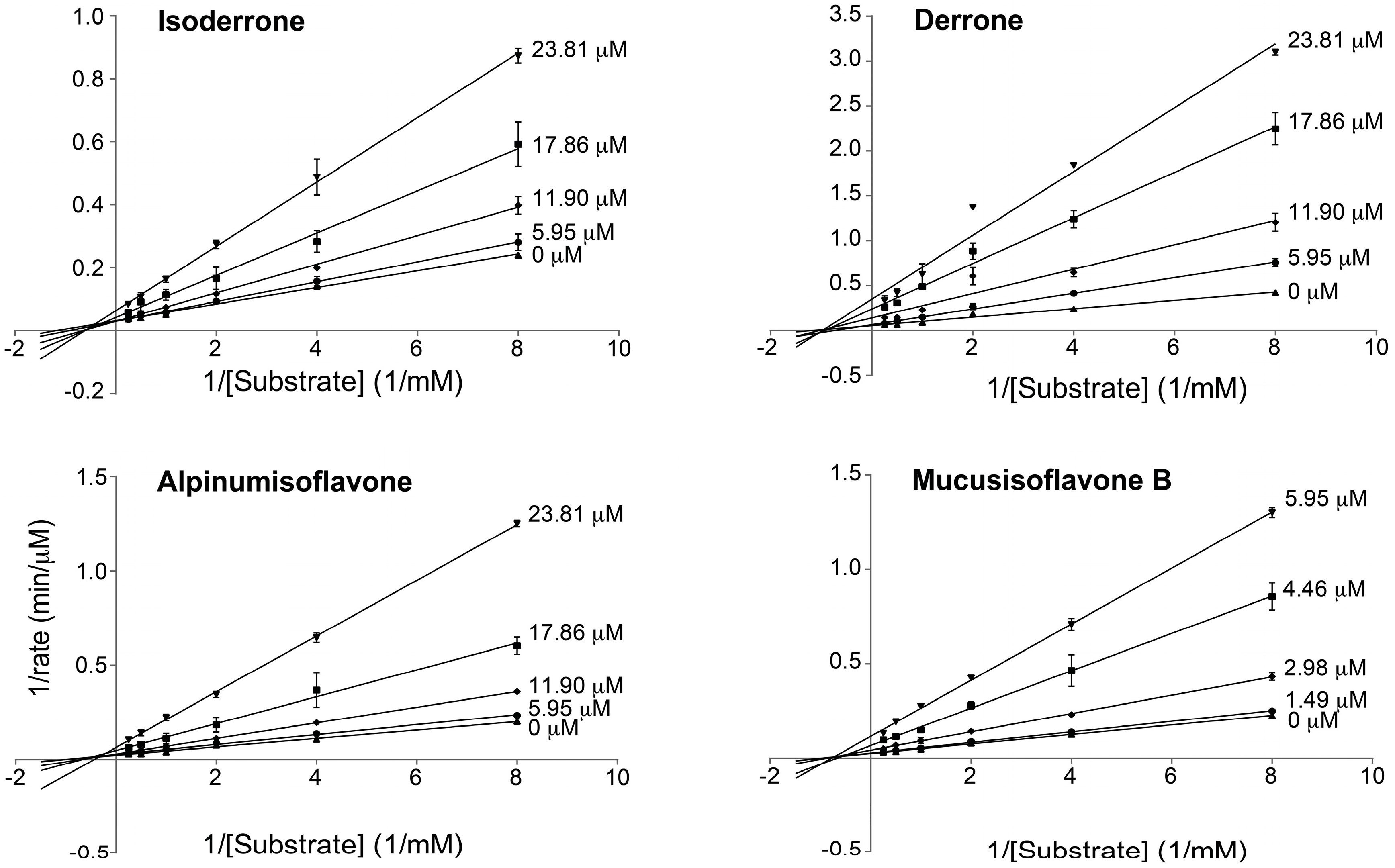
2.5. Kinetics of PTP1B Inhibition
3. Materials and Methods
3.1. Reagents
3.2. Plant Material and Sample Preparation
3.3. In Vitro PTP1B Inhibition Assay
3.4. Analytical-Scale HPLC Separation
3.5. High-Resolution PTP1B Inhibition Profiling
3.6. Preparative-Scale HPLC Separation
3.7. HPLC-HRMS-SPE-NMR Analysis
3.8. NMR Experiments
3.9. Kinetics of PTP1B Inhibition
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- International Diabetes Federation (IDF). IDF Diabetes Atlas, 7th ed.; IDF: Brussels, Belgium, 2015; pp. 1–144. ISBN 978-2-930229-81-2. Available online: http://www.diabetesatlas.org/ (accessed on 9 June 2017).
- Alberti, G.; Zimmet, P.; Shaw, J.; Bloomgarden, Z.; Kaufman, F.; Silink, M. Type 2 diabetes in the young: The evolving epidemic. Diabetes Care 2004, 27, 1798–1811. [Google Scholar] [CrossRef] [PubMed]
- Montalibet, J.; Kennedy, B.P. Therapeutic strategies for targeting PTP1B in diabetes. Drug Discov. Today Ther. Strateg. 2005, 2, 129–135. [Google Scholar] [CrossRef]
- Fowler, M.J. Microvascular and macrovascular complications of diabetes. Clin. Diabetes 2008, 26, 77–82. [Google Scholar] [CrossRef]
- American Diabetes Association. Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 2014, 37, S81–S90. [Google Scholar]
- Marcovecchio, M.L.; Lucantoni, M.; Chiarelli, F. Role of chronic and acute hyperglycemia in the development of diabetes complications. Diabetes Technol. Ther. 2011, 13, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.M.; Anand, I.S.; Suva, M.A. Role of protein tyrosine phosphatase-1B inhibitors in type 2 diabetes mellitus. J. Pharm. Sci. Technol. 2014, 4, 2–6. [Google Scholar]
- Zhang, Z.-Y.; Lee, S.-Y. PTP1B inhibitors as potential therapeutics in the treatment of type 2 diabetes and obesity. Expert Opin. Investig. Drugs 2003, 12, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, Z.-Y. PTP1B as a drug target: Recent developments in PTP1B inhibitor discovery. Drug Discov. Today 2007, 12, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Tamrakar, A.K.; Maurya, C.K.; Rai, A.K. PTP1B inhibitors for type 2 diabetes treatment: A patent review (2011–2014). Expert Opin. Ther. Pat. 2014, 24, 1101–1115. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.-S.; Liang, L.-F.; Guo, Y.-W. Natural products possessing protein tyrosine phosphatase 1B (PTP1B) inhibitory activity found in the last decades. Acta Pharmacol. Sin. 2012, 33, 1217–1245. [Google Scholar] [CrossRef] [PubMed]
- Kongstad, K.T.; Wubshet, S.G.; Johannesen, A.; Kjellerup, L.; Winther, A.-M.L.; Jäger, A.K.; Staerk, D. High-resolution screening combined with HPLC-HRMS-SPE-NMR for identification of fungal plasma membrane H+-ATPase inhibitors from plants. J. Agric. Food Chem. 2014, 62, 5595–5602. [Google Scholar] [CrossRef] [PubMed]
- Kongstad, K.T.; Özdemir, C.; Barzak, A.; Wubshet, S.G.; Staerk, D. Combined use of high-resolution α-glucosidase inhibition profiling and HPLC-HRMS-SPE-NMR for investigation of antidiabetic principles in crude plant extracts. J. Agric. Food Chem. 2015, 63, 2257–2263. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Kongstad, K.T.; Qinglei, S.; Nyberg, N.T.; Jäger, A.K.; Staerk, D. Dual high-resolution α-glucosidase and radical scavenging profiling combined with HPLC-HRMS-SPE-NMR for identification of minor and major constituents directly from the crude extract of Pueraria lobata. J. Nat. Prod. 2015, 78, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Kongstad, K.T.; Wiese, S.; Jäger, A.K.; Staerk, D. Edible seaweed as future functional food: Identification of α-glucosidase inhibitors by combined use of high-resolution α-glucosidase inhibition profiling and HPLC-HRMS-SPE-NMR. Food Chem. 2016, 203, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Wubshet, S.G.; Moresco, H.H.; Tahtah, Y.; Brighente, I.M.C.; Staerk, D. High-resolution bioactivity profiling combined with HPLC-HRMS-SPE-NMR: α-Glucosidase inhibitors and acetylated ellagic acid rhamnosides from Myrcia palustris DC. (Myrtaceae). Phytochemistry 2015, 116, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Wubshet, S.G.; Tahtah, Y.; Heskes, A.M.; Kongstad, K.T.; Pateraki, I.; Hamberger, B.; Møller, B.M.; Staerk, D. Identification of PTP1B and α-glucosidase inhibitory serrulatanes from Eremophila spp. by combined use of dual high-resolution PTP1B and α-glucosidase inhibition profiling and HPLC-HRMS-SPE-NMR. J. Nat. Prod. 2016, 79, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Hoang, M.D.; Trinh, N.T.; Phan, T.T.H.; Do, T.H.; Hoang, V.L.; Lee, M.; Bae, K. Screening of protein tyrosine phosphatase 1B inhibitory activity from some Vietnamese medicinal plants. Nat. Prod. Sci. 2010, 16, 239–244. [Google Scholar]
- Veerapur, V.P.; Prabhakar, K.R.; Thippeswamy, B.S.; Bansal, P.; Srinivasan, K.K.; Unnikrishnan, M.K. Antidiabetic effect of Ficus racemosa Linn. stem bark in high-fat diet and low-dose streptozotocin-induced type 2 diabetic rats: A mechanistic study. Food Chem. 2012, 132, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Yuan, T.; Liu, W.; Ma, H.; Seeram, N.P.; Li, Y.; Xu, L.; Mu, Y.; Huang, X.; Li, L. Phloroglucinol derivatives with protein tyrosine phosphatase 1B inhibitory activities from Eugenia jambolana seeds. J. Nat. Prod. 2017, 80, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Sawant, L.; Singh, V.K.; Dethe, S.; Bhaskar, A.; Balachandran, J.; Mundkinajeddu, D.; Agarwal, A. Aldose reductase and protein tyrosine phosphatase 1B inhibitory active compounds from Syzygium cumini seeds. Pharm. Biol. 2015, 53, 1176–1182. [Google Scholar] [CrossRef] [PubMed]
- Maximo, P.; Lourenco, A. A pterocarpan from Ulex parviflorus. Phytochemistry 1998, 48, 359–362. [Google Scholar] [CrossRef]
- Tanaka, T.; Ohyama, M.; Iinuma, M.; Shirataki, Y.; Komatsu, M.; Burandt, C.L. Isoflavonoids from Sophora secundiflora, S. arizonica and S. gypsophila. Phytochemistry 1998, 48, 1187–1193. [Google Scholar] [CrossRef]
- Ndemangou, B.; Sielinou, V.T.; Vardamides, J.C.; Ali, M.S.; Lateef, M.; Iqbal, L.; Afza, N.; Nkengfack, A.E. Urease inhibitory isoflavonoids from different parts of Calopogonium mucunoides (Fabaceae). J. Enzyme Inhib. Med. Chem. 2013, 28, 1156–1161. [Google Scholar] [CrossRef] [PubMed]
- Bankeu, J.J.K.; Khayala, R.; Lenta, B.N.; Noungoué, D.T.; Ngouela, S.A.; Mustafa, S.A.; Asaad, K.; Choudhary, M.I.; Prigge, S.T.; Hasanov, R.; et al. Isoflavone dimers and other bioactive constituents from the figs of Ficus mucuso. J. Nat. Prod. 2011, 74, 1370–1378. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Li, Z.; Song, W.; Wang, Y.; Liang, W.; Li, K.; Tang, S.; Wang, Q.; Qiao, X.; Zhou, D.; Yu, S.; Ze, M. Bioactive constituents of Glycyrrhiza uralensis (Licorice): Discovery of the effective components of a traditional herbal medicine. J. Nat. Prod. 2016, 79, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Kim, C.H.; Hoang, M.D.; Kim, B.Y.; Sohn, C.B.; Kim, M.R.; Ahn, J.S. Genistein-derivatives from Tetracera scandens stimulate gucose-uptake in L6 Myotubes. Biol. Pharm. Bull. 2009, 32, 504–508. [Google Scholar] [CrossRef] [PubMed]
- Tahtah, Y.; Wubshet, S.G.; Kongstad, K.T.; Heskes, A.M.; Pateraki, I.; Møller, B.L.; Jäger, A.K.; Staerk, D. High-resolution PTP1B inhibition profiling combined with high-performance liquid chromatography-high-resolution mass spectrometry-solid-phase extraction-nuclear magnetic resonance spectroscopy: Proof-of-concept and antidiabetic constituents in crude extract of Eremophila lucida. Fitoterapia 2016, 110, 52–58. [Google Scholar] [PubMed]
Sample Availability: Samples of the compounds 1–4 are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (µg/mL) | |||||
|---|---|---|---|---|---|
| Scientific Name | Family | Part Used | Voucher No. | EtOAc Extract | n-BuOH Extract |
| Nepenthes mirabilis (Lour.) Druce | Nepenthaceae | Whole plant | VN-01 | 1.4 ± 0.05 | 0.4 ± 0.1 |
| Ludwigia octovalvis (Jacq.) P.H. Raven | Onagraceae | Aerial part | VN-02 | 16.9 ± 3.2 | 3.3 ± 0.3 |
| Phyllanthus amarus Schumach. & Thonn. | Phyllanthaceae | Whole plant | VN-03 | 74.4 ± 3.9 | n.t. |
| Phyllanthus urinaria L. | Phyllanthaceae | Whole plant | VN-04 | 14.0 ± 0.8 | 10.8 ± 0.9 |
| Phyllanthus reticulatus Poir. | Phyllanthaceae | Stem, leaf | VN-05 | n.t. | n.t. |
| Scoparia dulcis L. | Scrophulariaceae | Whole plant | VN-06 | n.t. | n.t. |
| Mirabilis jalapa L. | Nyctaginaceae | Root | VN-07 | n.t. | n.t. |
| Arial part | n.t. | n.t. | |||
| Cassia fistula L. | Leguminosae | Leaf | VN-08 | 24.1 ± 8.4 | n.t. |
| Lagerstroemia speciosa (L.) Pers. | Lythraceae | Leaf | VN-09 | n.t. | 19.6 ± 5.1 |
| Syzygium cumini (L.) Skeels | Myrtaceae | Fruit | VN-10 | 27.5 ± 7.8 | n.t. |
| Euphorbia hirta L. | Euphorbiaceae | Whole plant | VN-11 | 29.2 ± 6.2 | 38.3 ± 1.9 |
| Rhizophora mucronata Lam. | Rhizophoraceae | Bark | VN-12 | 17.2 ± 1.2 | 1.8 ± 0.4 |
| Kandelia candel (L.) Druce | Rhizophoraceae | Bark | VN-14 | 12.9 ± 2.2 | 0.02 ± 0.01 |
| Cardiospermum halicacabum L. | Sapindaceae | Whole plant | VN-14 | n.t. | n.t. |
| Pandanus odoratissimus L.f. | Pandanaceae | Fruit | VN-15 | 20.8 ± 5.6 | 40.4 ± 7.9 |
| Morinda citrifolia L. | Rubiaceae | Fruit | VN-16 | n.t. | n.t. |
| Ficus racemosa L. | Moraceae | Fruit | VN-17 | 38.3 ± 10.6 | 3.6 ± 1.4 |
| Pithecellobium dulce (Roxb.) Benth. | Leguminosae | Stem | VN-18 | 26.1 ± 2.5 | n.t. |
| Leaf | n.t. | n.t. | |||
| Compound | IC50 (µM) | Ki (µM) | Mode of Inhibition |
|---|---|---|---|
| Isoderrone (1) | 22.7 ± 1.7 | 21.3 ± 2.8 | Non-competitive |
| Derrone (2) | 12.6 ± 1.6 | 7.9 ± 1.9 | Non-competitive |
| Alpinumisoflavone (3) | 21.2 ± 3.8 | 14.3 ± 2.0 | Non-competitive |
| Mucusisoflavone B (4) | 2.5 ± 0.2 | 3.0 ± 0.5 | Non-competitive |
| RK682 a | 10.4 ± 1.6 | n.t. | n.t. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trinh, B.T.D.; Jäger, A.K.; Staerk, D. High-Resolution Inhibition Profiling Combined with HPLC-HRMS-SPE-NMR for Identification of PTP1B Inhibitors from Vietnamese Plants. Molecules 2017, 22, 1228. https://doi.org/10.3390/molecules22071228
Trinh BTD, Jäger AK, Staerk D. High-Resolution Inhibition Profiling Combined with HPLC-HRMS-SPE-NMR for Identification of PTP1B Inhibitors from Vietnamese Plants. Molecules. 2017; 22(7):1228. https://doi.org/10.3390/molecules22071228
Chicago/Turabian StyleTrinh, Binh Thi Dieu, Anna K. Jäger, and Dan Staerk. 2017. "High-Resolution Inhibition Profiling Combined with HPLC-HRMS-SPE-NMR for Identification of PTP1B Inhibitors from Vietnamese Plants" Molecules 22, no. 7: 1228. https://doi.org/10.3390/molecules22071228
APA StyleTrinh, B. T. D., Jäger, A. K., & Staerk, D. (2017). High-Resolution Inhibition Profiling Combined with HPLC-HRMS-SPE-NMR for Identification of PTP1B Inhibitors from Vietnamese Plants. Molecules, 22(7), 1228. https://doi.org/10.3390/molecules22071228