Tetrabutylammonium Iodide–Promoted Thiolation of Oxindoles Using Sulfonyl Chlorides as Sulfenylation Reagents

Abstract
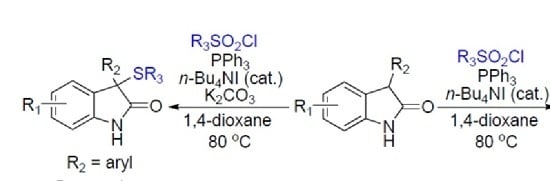
:
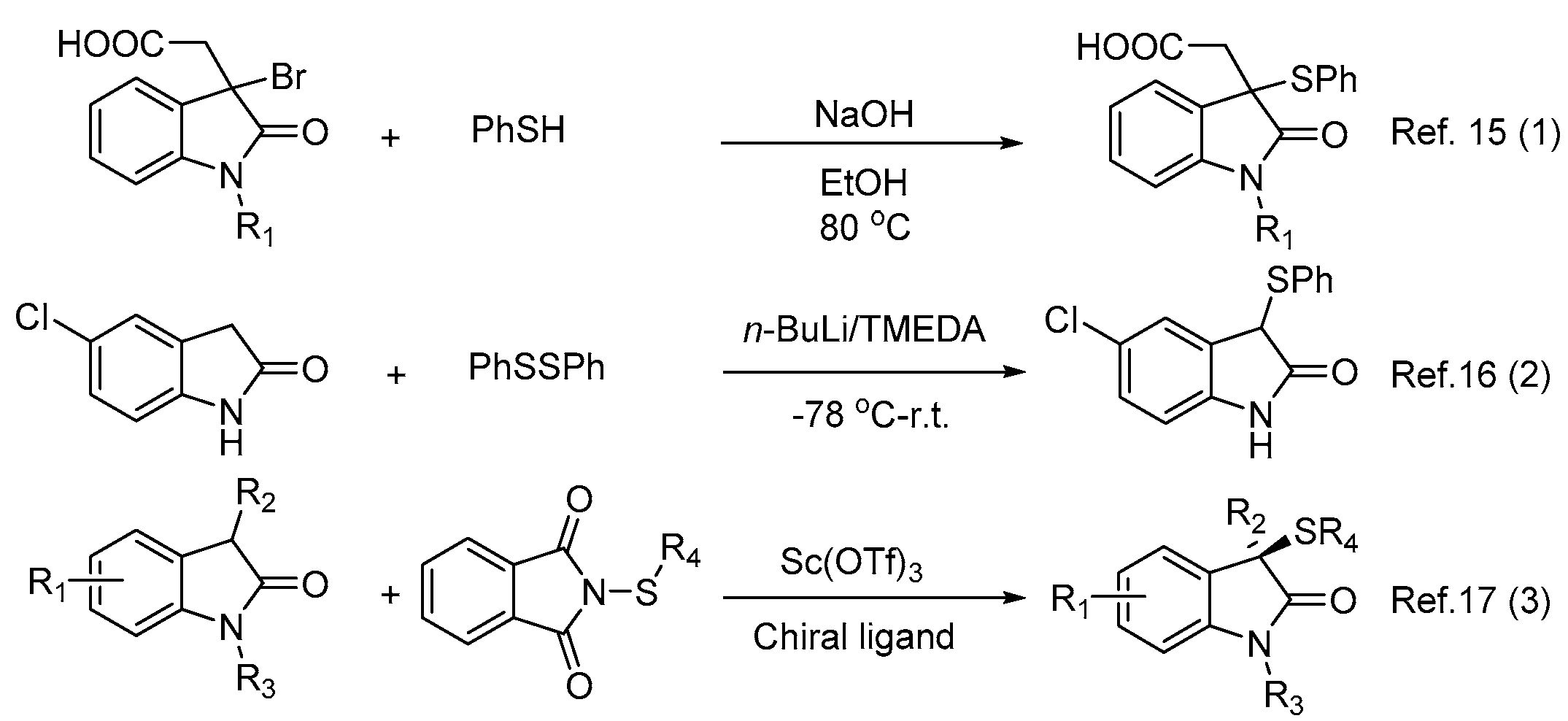
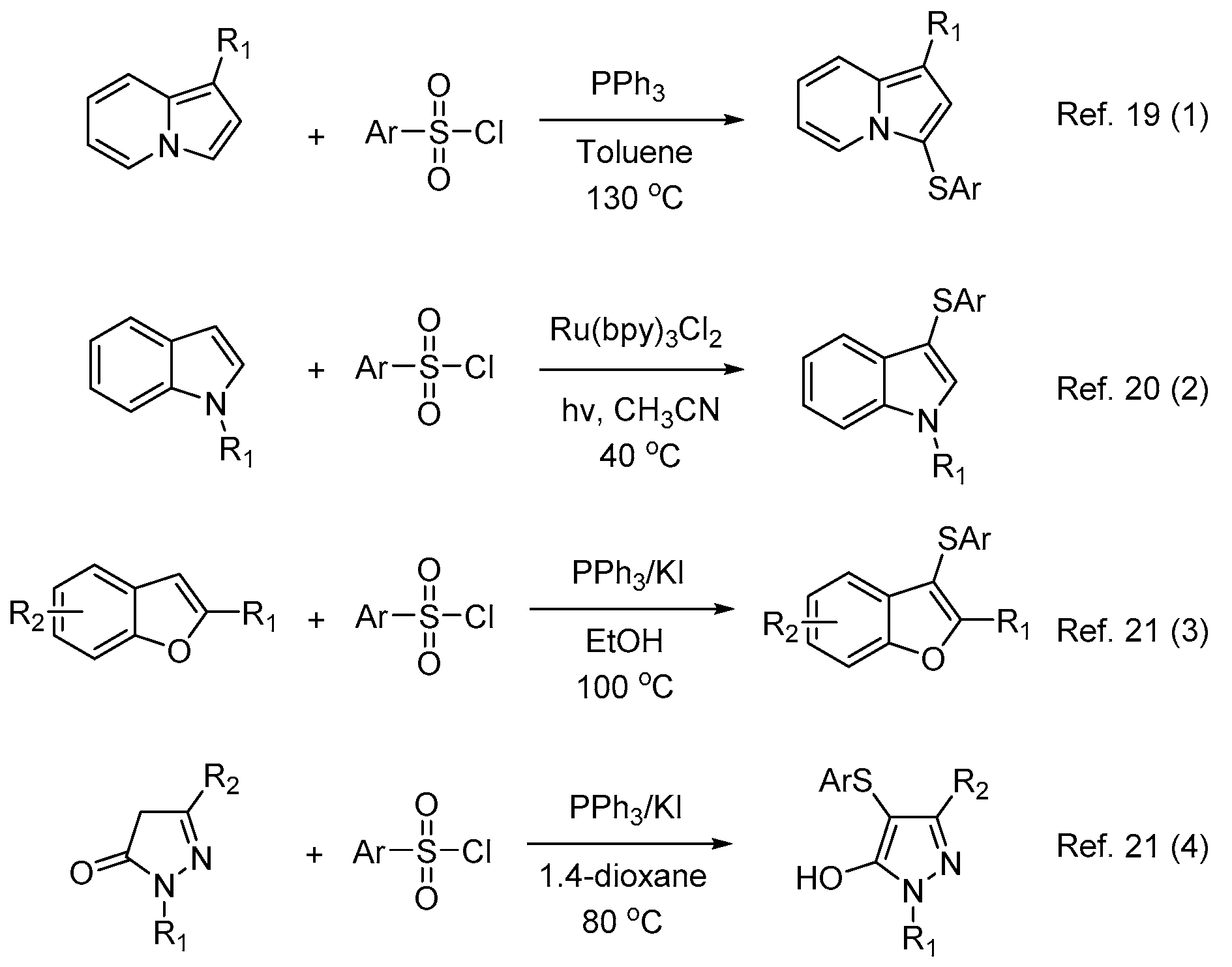
1. Introduction
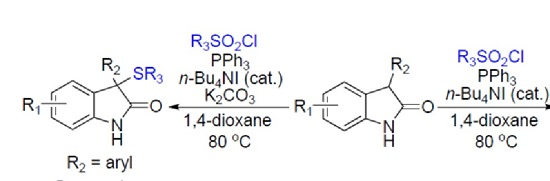
2. Results
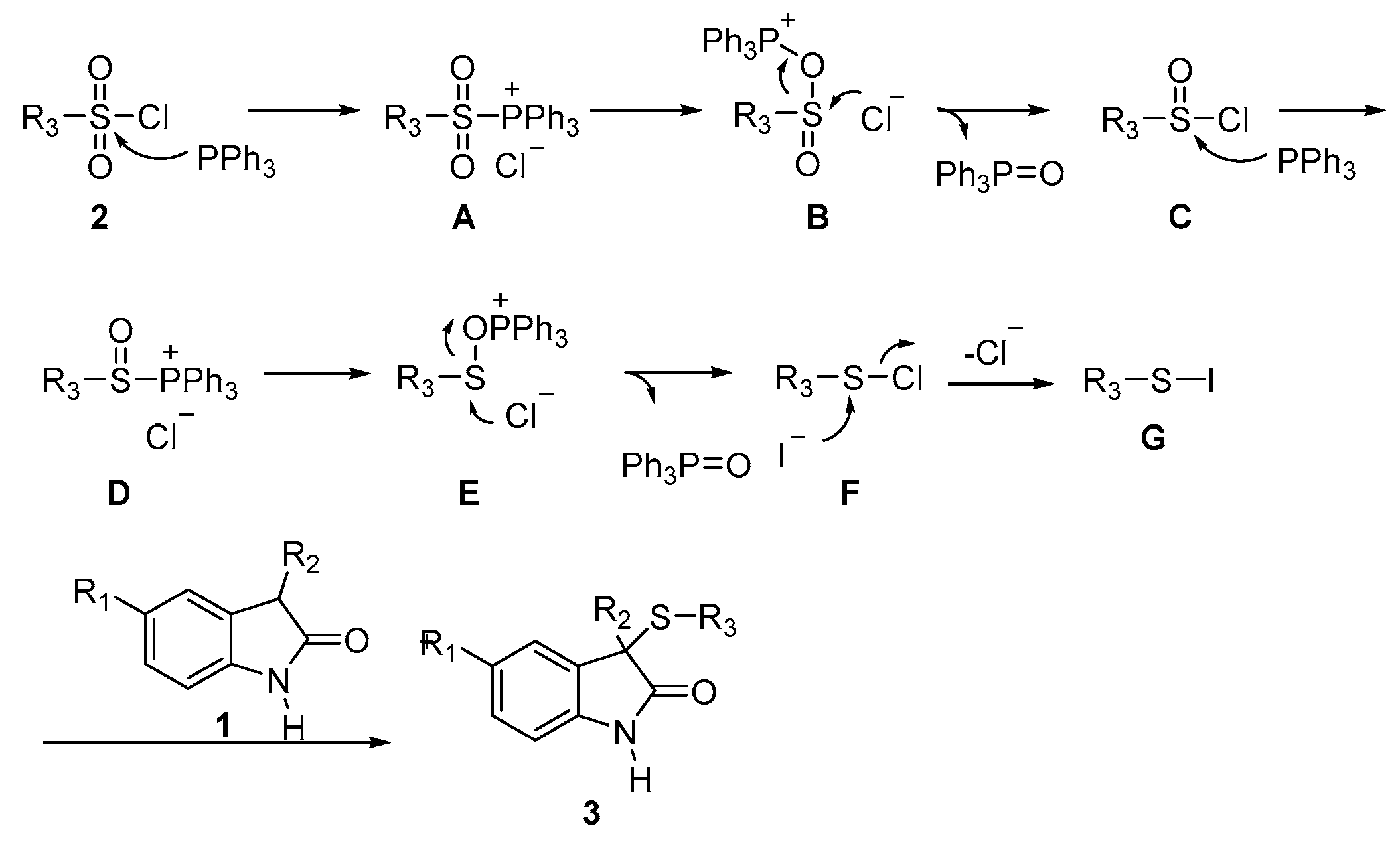
3. Discussion
4. Materials and Methods
4.1. General Methods and Material
4.2. Synthesis of Oxindoles
4.2.1. 5-Bromo-3-methylindolin-2-one (1b)
4.2.2. 3-(p-Tolyl)indolin-2-one (4a)
4.3. General Procedure for the Synthesis of 3aa, 3ab, 3ac, 3ad, 3ae, 3af, 3ag, 3ba, 3ca and 3da
4.4. General Procedure for the Synthesis of 3ea, 3fa, 3ga, 3gb, 3ha and 3ia
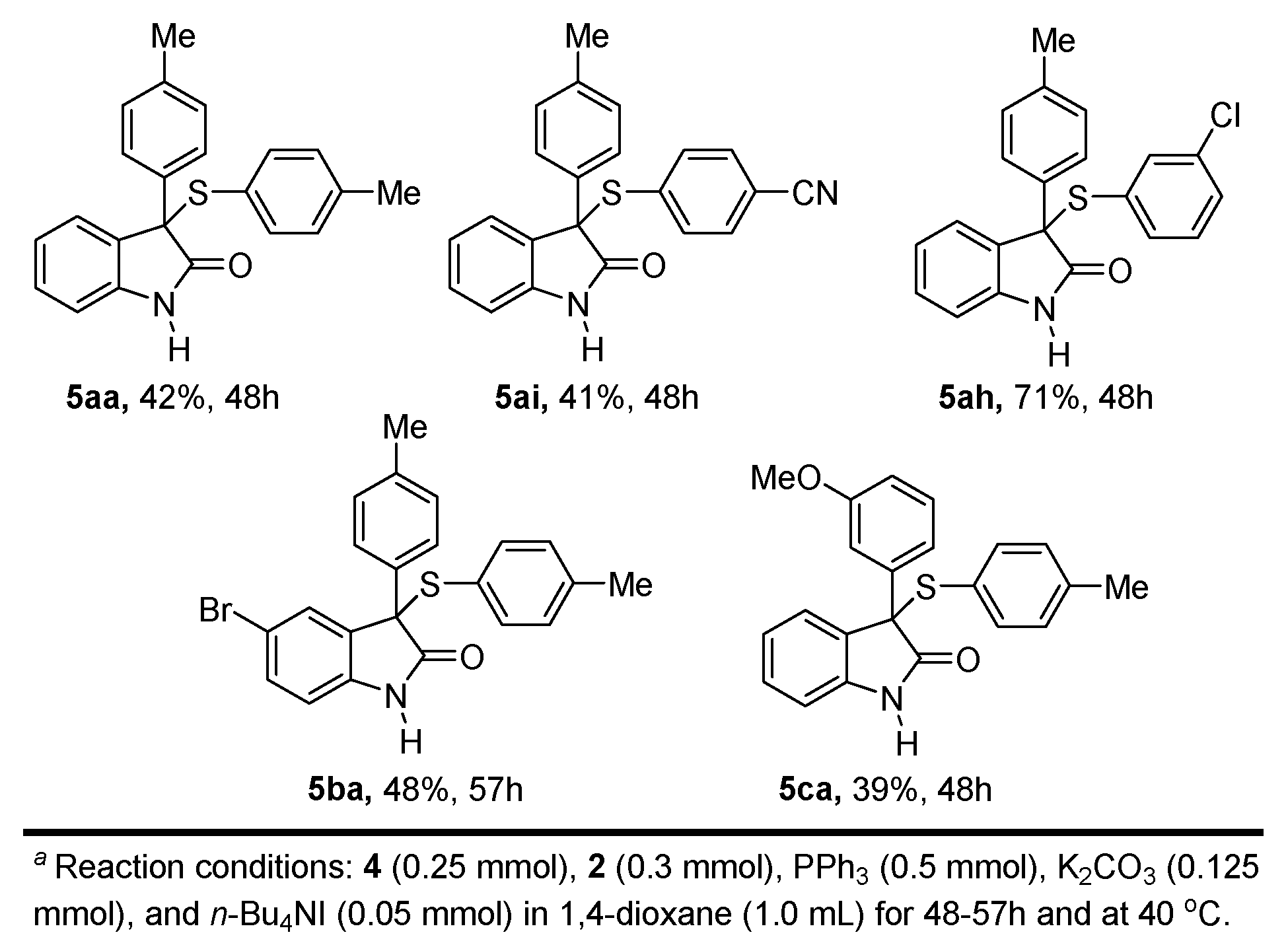
4.5. General Procedure for the Synthesis of 5aa, 5ai, 5ah, 5ba and 5ca
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pedras, M.S.C.; Yaya, E.E.; Glawischnig, E. The phytoalexins from cultivated and wild crucifers: Chemistry and biology. Nat. Prod. Rep. 2011, 28, 1381–1405. [Google Scholar] [CrossRef] [PubMed]
- Millemaggi, A.; Taylor, R.J.K. 3-Alkenyl-oxindoles: Natural products, pharmaceuticals, and recent synthetic advances in tandem/telescoped approaches. Eur. J. Org. Chem. 2010, 4527–4547. [Google Scholar] [CrossRef]
- Trost, B.M.; Brennan, M.K. Asymmetric syntheses of oxindole and indole spirocyclic alkaloid natural products. Synthesis 2009, 3003–3025. [Google Scholar] [CrossRef]
- Galliford, C.V.; Scheidt, K.A. Pyrrolidinyl-spirooxindole natural products as inspirations for the development of potential therapeutic agents. Angew. Chem. Int. Ed. 2007, 46, 8748–8758. [Google Scholar] [CrossRef] [PubMed]
- Marti, C.; Carreira, E.M. Construction of spiro[pyrrolidine-3,3′-oxindoles]-recent applications to the synthesis of oxindole alkaloids. Eur. J. Org. Chem 2003, 2209–2219. [Google Scholar] [CrossRef]
- Mehta, R.G.; Liu, J.; Constantinou, A.; Hawthorne, M.; Pezzuto, J.M.; Moon, R.C.; Moriarty, R.M. Structure-activity relationships of brassinin in preventing the development of carcinogen-induced mammary lesions in organ culture. Anticancer Res. 1994, 14, 1209–1213. [Google Scholar] [PubMed]
- Pedras, M.S.C.; Hossain, M. Metabolism of crucifer phytoalexins in Sclerotinia sclerotiorum: Detoxification of strongly antifungal compounds involves glucosylation. Org. Biomol. Chem. 2006, 4, 2581–2590. [Google Scholar] [CrossRef] [PubMed]
- Dandia, A.; Sati, M.; Arya, K.; Sharma, R.; Loupy, A. Facile one pot microwave induced solvent-free synthesis and antifungal, antitubercular screening of spiro [1,5]-benzothiazepin-2,3′[3′H]indol-2[1′H]-ones. Chem. Pharm. Bull. 2003, 51, 1137–1141. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shi, Y.; Huang, Z.; Wu, X.; Xu, P.; Wang, J.; Zhang, Y. Catalytic thia-sommelet-hauser rearrangement: Application to the synthesis of oxindoles. Org. Lett. 2011, 13, 1210–1213. [Google Scholar] [CrossRef] [PubMed]
- Coote, S.C.; Quenum, S.; Procter, D.J. Exploiting Sm(II) and Sm(III) in SmI2-initiated reaction cascades: Application in a tag removal-cyclization approach to spirooxindole scaffolds. Org. Biomol. Chem. 2011, 9, 5104–5108. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.; Vogel, J.C.; Tsang, W.; Merrit, A.; Procter, D.J. Formation of N-heterocycles by the reaction of thiols with glyoxamides: Exploring a connective Pummerer-type cyclization. Org. Biomol. Chem. 2009, 7, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Atobe, M.; Fuchigami, T. Electroorganic synthesis using a fluoride ion mediator under ultrasonic irradiation: Synthesis of oxindole and 3-oxotetrahydroisoquinoline derivatives. Org. Lett. 2004, 6, 2441–2444. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.J.; Wu, Y.L.; Chuang, C.P. Cyclization reactions of methylthioacetanilides. Tetrahedron 2003, 59, 3511–3520. [Google Scholar] [CrossRef]
- Greaney, M.F.; Motherwell, W.B. Studies on the oxidation and fluorination of α-(phenylsulfanyl) acetamides using (difluoroiodo) toluene. Tetrahedron. Lett. 2000, 41, 4467–4470. [Google Scholar] [CrossRef]
- Al-thebeiti, M.S. Synthesis of some new spiro[indoline-3,2′-(thiochroman)]-2,4′-dione derivatives. Phosphoru, Sulfur Silicon Relat. Elem. 1998, 141, 89–95. [Google Scholar] [CrossRef]
- Young, S.D.; Amblard, M.C.; Britcher, S.F.; Grey, V.E.; Tran, L.O.; Lumma, W.C.; Huff, J.R.; Schleif, W.A.; Emini, E.E.; O’Brien, J.A. 2-Heterocyclic indole-3-sulfones as inhibitors of HIV-1 reverse transcriptase. Bioorg. Med. Chem. Lett. 1995, 5, 491–496. [Google Scholar] [CrossRef]
- Cai, Y.; Li, J.; Chen, W.; Xie, M.; Liu, X.; Lin, L.; Feng, X. Catalytic asymmetric sulfenylation of unprotected 3-substituted oxindoles. Org. Lett. 2012, 14, 2726–2729. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, C.; Xue, X.S.; Cheng, J.P. Enantioselective organocatalyzed sulfenylation of 3-substituted oxindoles. Org. Lett. 2012, 14, 4374–4377. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Zhao, D.; Qin, X.; Lan, J.; You, J. Synthesis of di(hetero) aryl sulfides by directly using arylsulfonyl chlorides as a sulfur source. Chem. Commun. 2011, 47, 9188–9190. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Huang, Z.T.; Zheng, Q.Y. Visible light-induced 3-sulfenylation of N-methylindoles with arylsulfonyl chlorides. Chem. Commun. 2012, 48, 11686–11688. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Lu, X.; Wei, A.; Jia, X.; Chen, J.; Lu, K. Potassium iodide promoted thiolation of pyrazolones and benzofurans using aryl sulfonyl chlorides as sulfenylation reagents. Tetrahedron Lett. 2016, 57, 5330–5333. [Google Scholar] [CrossRef]
- Lu, K.; Deng, Z.; Li, M.; Li, T.; Zhao, X. Transition metal-free direct trifluoromethylthiolation of indoles using trifluoromethanesulfonyl chloride in the presence of triphenylphosphine. Org. Biomol. Chem. 2017, 15, 1254–1260. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wei, A.; Li, T.; Su, Z.; Chen, J.; Lu, K. Transition-metal free direct difluoromethylthiolation of electron-rich aromatics with difluoromethanesulfonyl chloride. Org. Chem. Font. 2017, 4, 232–235. [Google Scholar] [CrossRef]
- Zhao, X.; Li, T.; Yang, B.; Qiu, D.; Lu, K. Transition-metal-free trifluoromethylthiolation and difluoromethylthiolation of thiols with trifluoromethanesulfonyl chloride and difluoromethanesulfonyl chloride. Tetrahedron 2017, 73, 3112–3117. [Google Scholar] [CrossRef]
- Zhao, X.; Deng, Z.; Wei, A.; Li, B.; Lu, K. Iodine-catalysed regioselective thiolation of flavonoids using sulfonyl hydrazides as sulfenylation reagents. Org. Biomol. Chem. 2016, 14, 7304–7312. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Li, T.; Zhang, L.; Lu, K. Iodine-catalyzed thiolation of electron-rich aromatics using sulfonyl hydrazides as sulfenylation reagents. Org. Biomol. Chem. 2016, 14, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhang, L.; Lu, X.; Li, T.; Lu, K. Synthesis of 2-aryl and 3-aryl benzo[b]furan thioethers using aryl sulfonyl hydrazides as sulfenylation reagents. J. Org. Chem. 2015, 80, 2918–2924. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhang, L.; Li, T.; Liu, G.; Wang, H.; Lu, K. p-Toluenesulphonic acid-promoted, I2-catalysed sulphenylation of pyrazolones with aryl sulphonyl hydrazides. Chem. Commun. 2014, 50, 13121–13123. [Google Scholar] [CrossRef] [PubMed]
- Dua, T.P.; Zhu, G.G.; Zhou, J. A facile method for the synthesis of 3-alkyloxindole. Lett. Org. Chem. 2012, 9, 225–232. [Google Scholar] [CrossRef]
Sample Availabilty: Samples of the compounds 3aa–3gb and 5aa–5ca are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
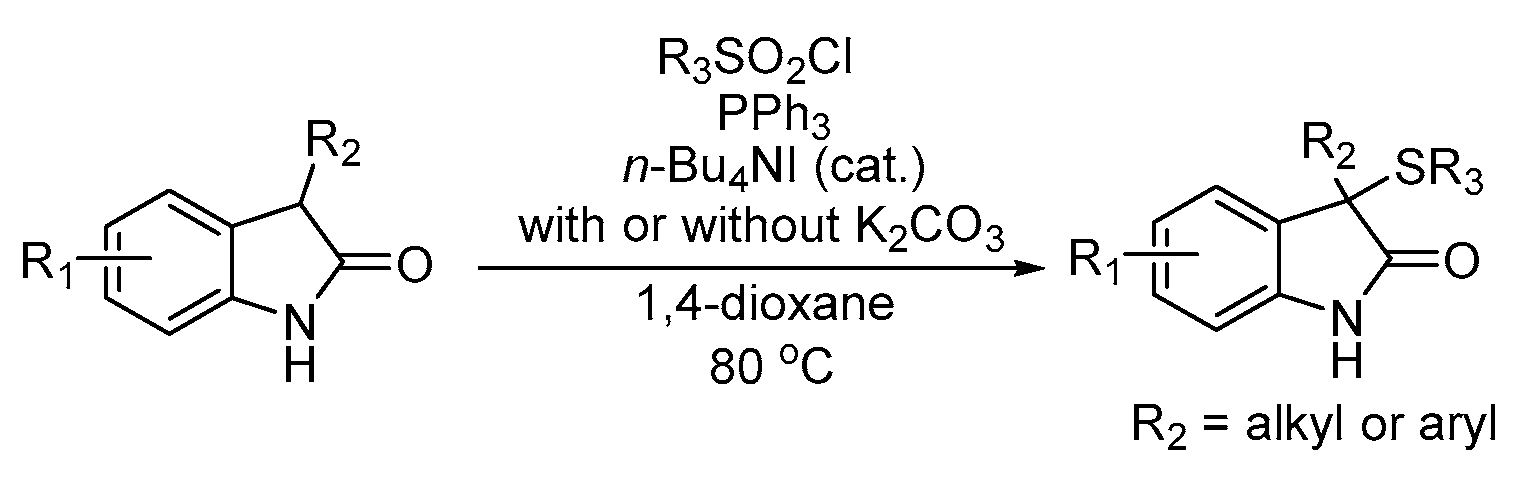{kind=link}
{kind=link}

| Entry | Additive/eq. | Temperature (°C) | Solvent/Volume (mL) | Yield (%) b |
|---|---|---|---|---|
| 1 | - | 80 | 1,4-dioxane/1.0 | 56 |
| 2 | KI/0.2 | 80 | 1,4-dioxane/1.0 | 56 |
| 3 | NH4I/0.2 | 80 | 1,4-dioxane/1.0 | 73 |
| 4 | n-Bu4NI/0.2 | 80 | 1,4-dioxane/1.0 | 82 |
| 5 | n-Bu4NI/0.2 | 80 | DCE/1.0 | 75 |
| 6 | n-Bu4NI/0.2 | 80 | toluene/1.0 | 66 |
| 7 | n-Bu4NI/0.2 | 80 | CH3CN/1.0 | 46 |
| 8 | n-Bu4NI/0.2 | 80 | DMF/1.0 | 45 |
| 9 | n-Bu4NI/0.2 | 70 | 1,4-dioxane/1.0 | 79 |
| 10 | n-Bu4NI/0.2 | 90 | 1,4-dioxane/1.0 | 28 |
| 11 | n-Bu4NI/0.2 | 80 | 1,4-dioxane/1.5 | 68 |
| 12 | n-Bu4NI/0.2 | 80 | 1,4-dioxane/0.5 | 86 |
| 13 | n-Bu4NI/0.2 | 80 | 1,4-dioxane/0.3 | 74 |
| 14 | n-Bu4NI/0.2 | 80 | 1,4-dioxane/0.5 | 86 c |

| Entry | Oxindole | R1 | R2 | Sulfonyl Chloride | R3 | Product | Yield (%) |
|---|---|---|---|---|---|---|---|
| 1 | 1a | H | Me | 2b | p-MeOC6H4 | 3ab | 90 |
| 2 | 1a | H | Me | 2c | m-MeC6H4 | 3ac | 62 |
| 3 | 1a | H | Me | 2d | 3,5-Cl2C6H3 | 3ad | 68 |
| 4 | 1a | H | Me | 2e | p-BrC6H4 | 3ae | 82 |
| 5 | 1a | H | Me | 2f | cyclopropyl | 3af | 44 |
| 6 | 1a | H | Me | 2g | n-Butyl | 3ag | 56 |
| 7 | 1b | Br | Me | 2a | p-MeC6H4 | 3ba | 90 |
| 8 | 1c | H | Et | 2a | p-MeC6H4 | 3ca | 79 |
| 9 | 1d | H | Pr | 2a | p-MeC6H4 | 3da | 81 |
| 10 | 1e | H | i-Pr | 2a | p-MeC6H4 | 3ea | 63 b |
| 11 | 1f | H | i-Bu | 2a | p-MeC6H4 | 3fa | 78 b |
| 12 | 1g | H | cyclohexyl | 2a | p-MeC6H4 | 3ga | 67 b |
| 13 | 1g | H | cyclohexyl | 2b | p-MeOC6H4 | 3gb | 65 b |
| 14 | 1h | H | p-NCC6H4CH2 | 2a | p-MeC6H4 | 3ha | 60 b |
| 15 | 1i | H | p-ClC6H4CH2 | 2a | p-MeC6H4 | 3ia | 79 b |

| Entry | Base/eq. | Temperature (°C) | Reaction Time (h) | Solvent/Volume (mL) | Yield (%) b |
|---|---|---|---|---|---|
| 1 | - | 80 | 15 | 1,4-dioxane/0.5 | 0 |
| 2 | - | 60 | 15 | 1,4-dioxane/0.5 | 44 |
| 3 | - | 60 | 14 | 1,4-dioxane/1.0 | 48 |
| 4 | - | 60 | 20 | 1,4-dioxane/1.5 | 39 |
| 5 | K2CO3/0.5 | 60 | 15 | 1,4-dioxane/1.0 | 54 |
| 6 | K2CO3/0.5 | 50 | 34 | 1,4-dioxane/1.0 | 45 |
| 7 | K2CO3/0.5 | 40 | 38 | 1,4-dioxane/1.0 | 71 |
| 8 | K2CO3/0.5 | 25 | 114 | 1,4-dioxane/1.0 | 68 |
| 9 | Na2CO3/0.5 | 40 | 34 | 1,4-dioxane/1.0 | 70 |
| 10 | Cs2CO3/0.5 | 40 | 39 | 1,4-dioxane/1.0 | 26 |
| 11 | K2CO3/1.0 | 40 | 11 | 1,4-dioxane/1.0 | 70 |
| 12 | K2CO3/0.5 | 40 | 21 | 1,4-dioxane/1.0 | 66 c |
| 13 | K2CO3/0.5 | 40 | 51 | 1,4-dioxane/1.0 | 27 c |
| 14 | K2CO3/0.5 | 40 | 32 | 1,4-dioxane/1.0 | 44 d |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.; Wei, A.; Lu, X.; Lu, K. Tetrabutylammonium Iodide–Promoted Thiolation of Oxindoles Using Sulfonyl Chlorides as Sulfenylation Reagents. Molecules 2017, 22, 1208. https://doi.org/10.3390/molecules22081208
Zhao X, Wei A, Lu X, Lu K. Tetrabutylammonium Iodide–Promoted Thiolation of Oxindoles Using Sulfonyl Chlorides as Sulfenylation Reagents. Molecules. 2017; 22(8):1208. https://doi.org/10.3390/molecules22081208
Chicago/Turabian StyleZhao, Xia, Aoqi Wei, Xiaoyu Lu, and Kui Lu. 2017. "Tetrabutylammonium Iodide–Promoted Thiolation of Oxindoles Using Sulfonyl Chlorides as Sulfenylation Reagents" Molecules 22, no. 8: 1208. https://doi.org/10.3390/molecules22081208
APA StyleZhao, X., Wei, A., Lu, X., & Lu, K. (2017). Tetrabutylammonium Iodide–Promoted Thiolation of Oxindoles Using Sulfonyl Chlorides as Sulfenylation Reagents. Molecules, 22(8), 1208. https://doi.org/10.3390/molecules22081208