Tubulin Inhibitor-Based Antibody-Drug Conjugates for Cancer Therapy



Abstract
:1. Introduction
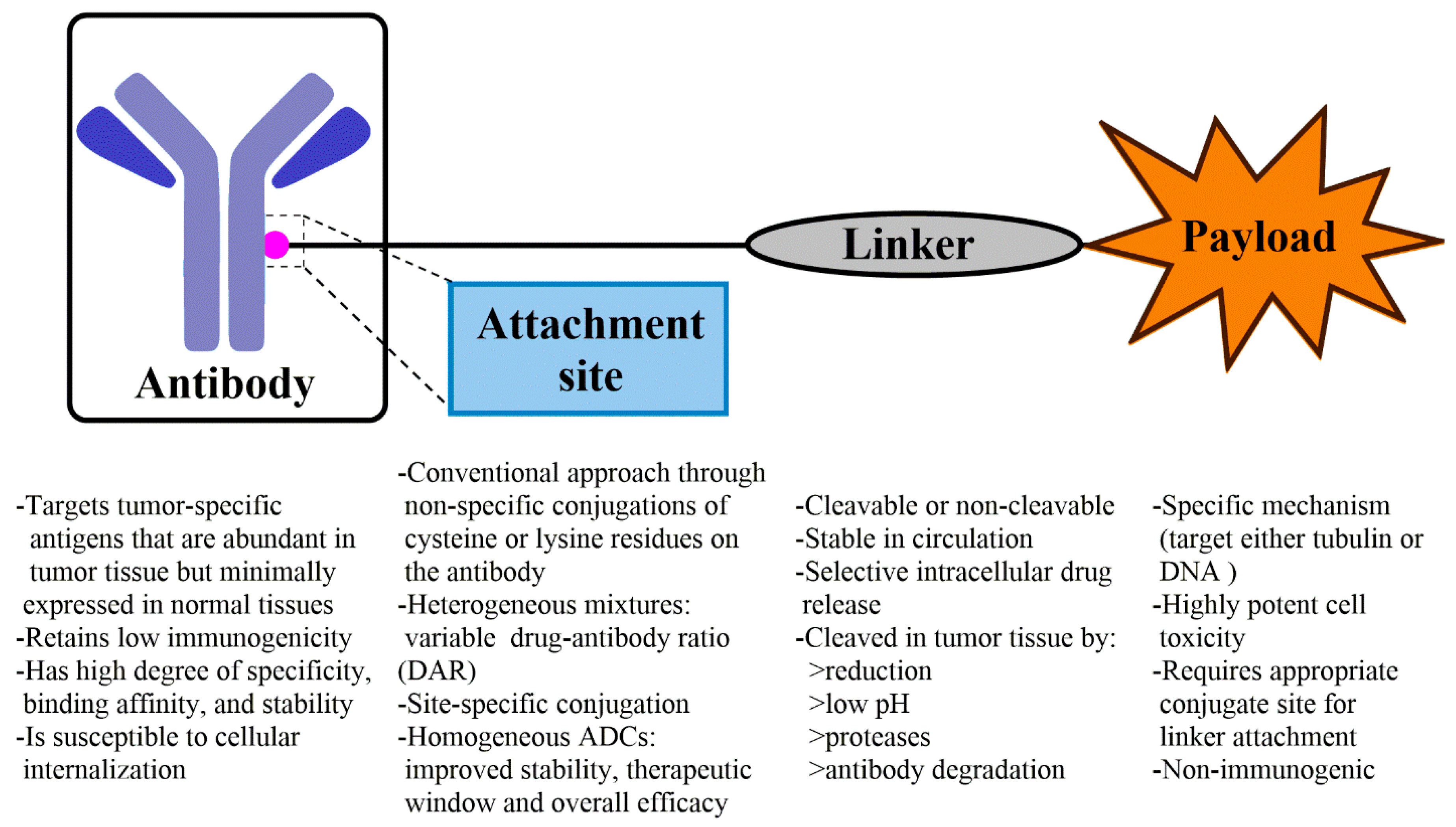
2. General Considerations for ADC
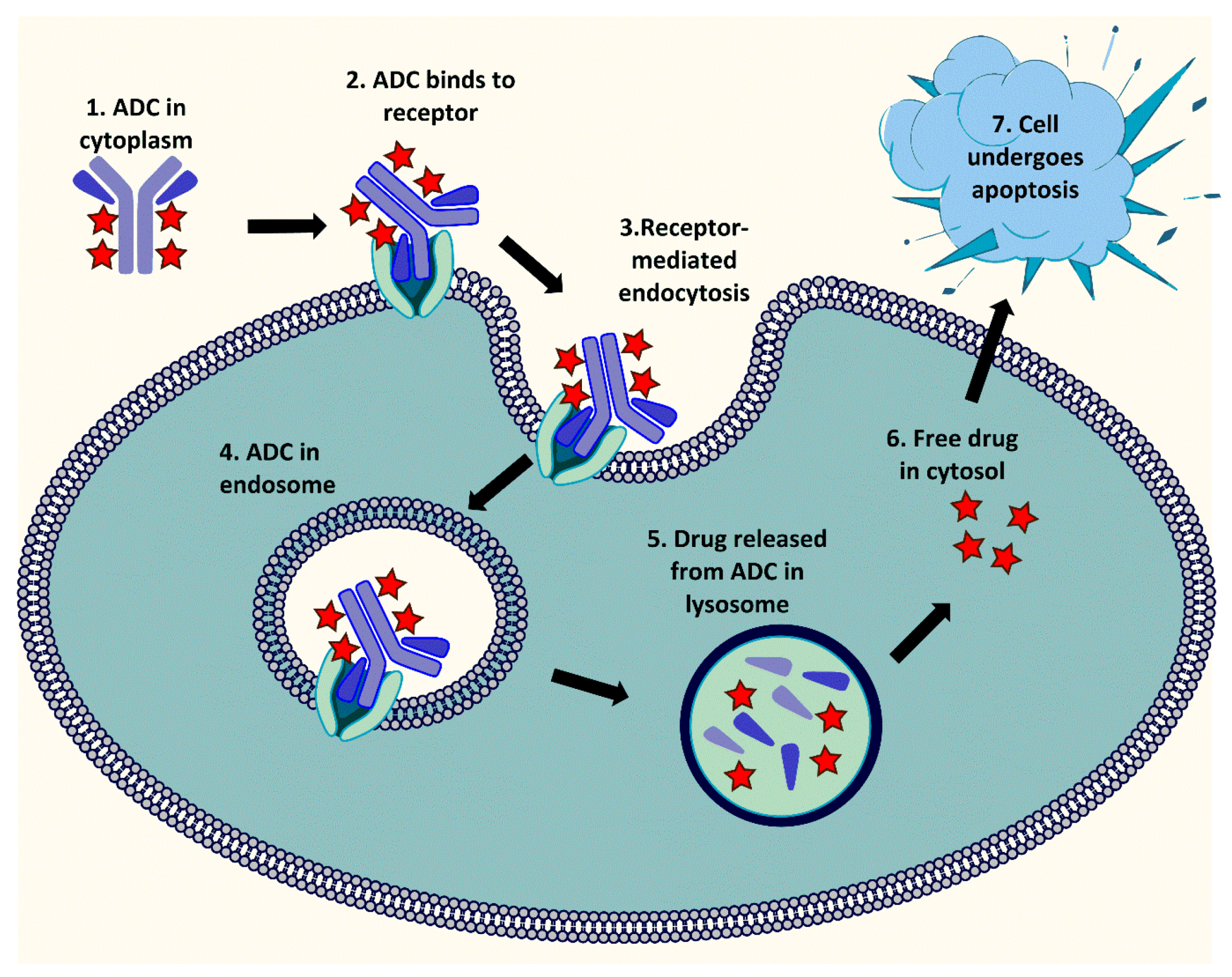
2.1. Drug Release Mechanism of ADC
2.2. Target and Antibody Selection for ADC
2.3. Conjugation Strategy of Payload
2.4. Linker Selection for ADC
2.5. Payload Selection for ADC
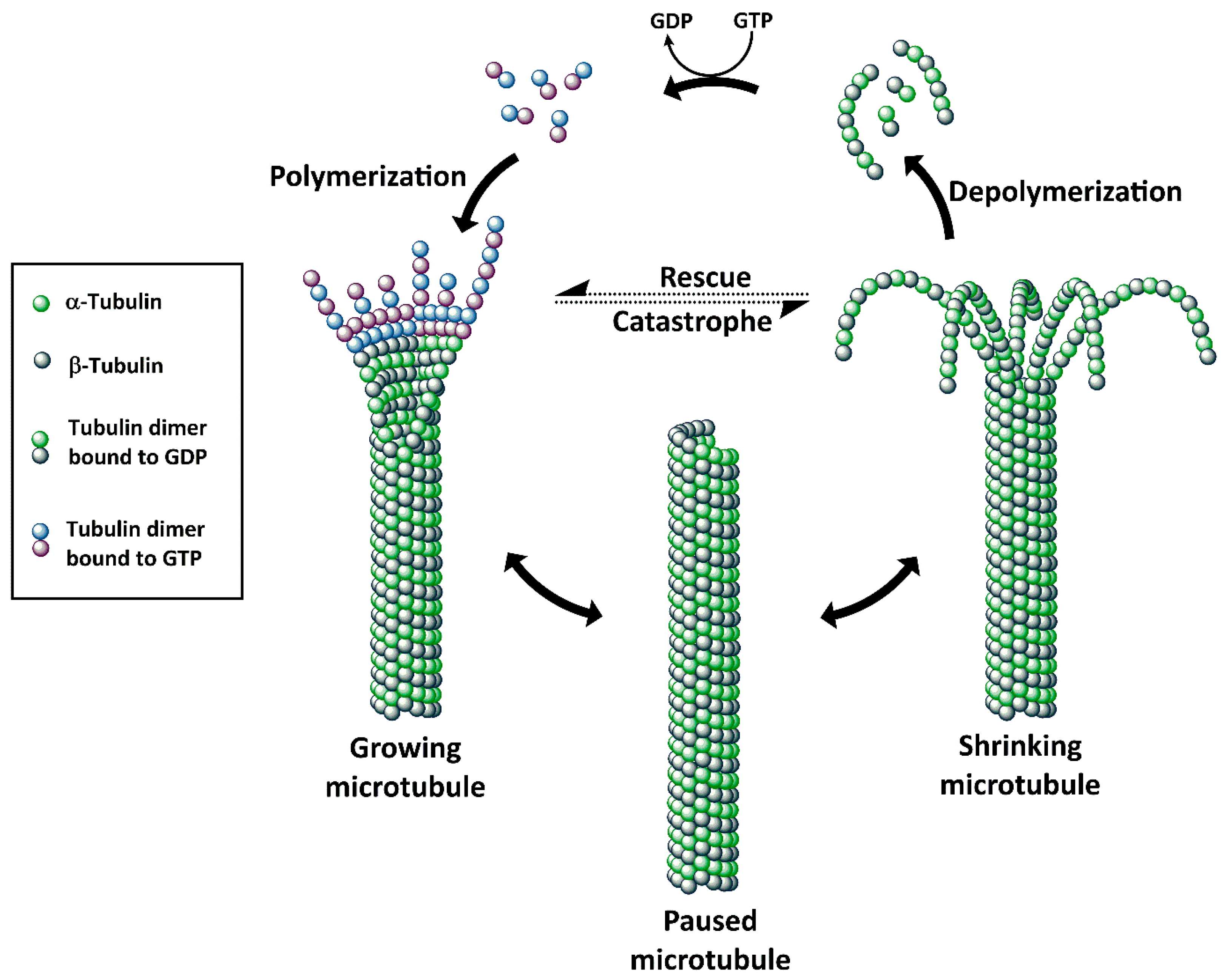
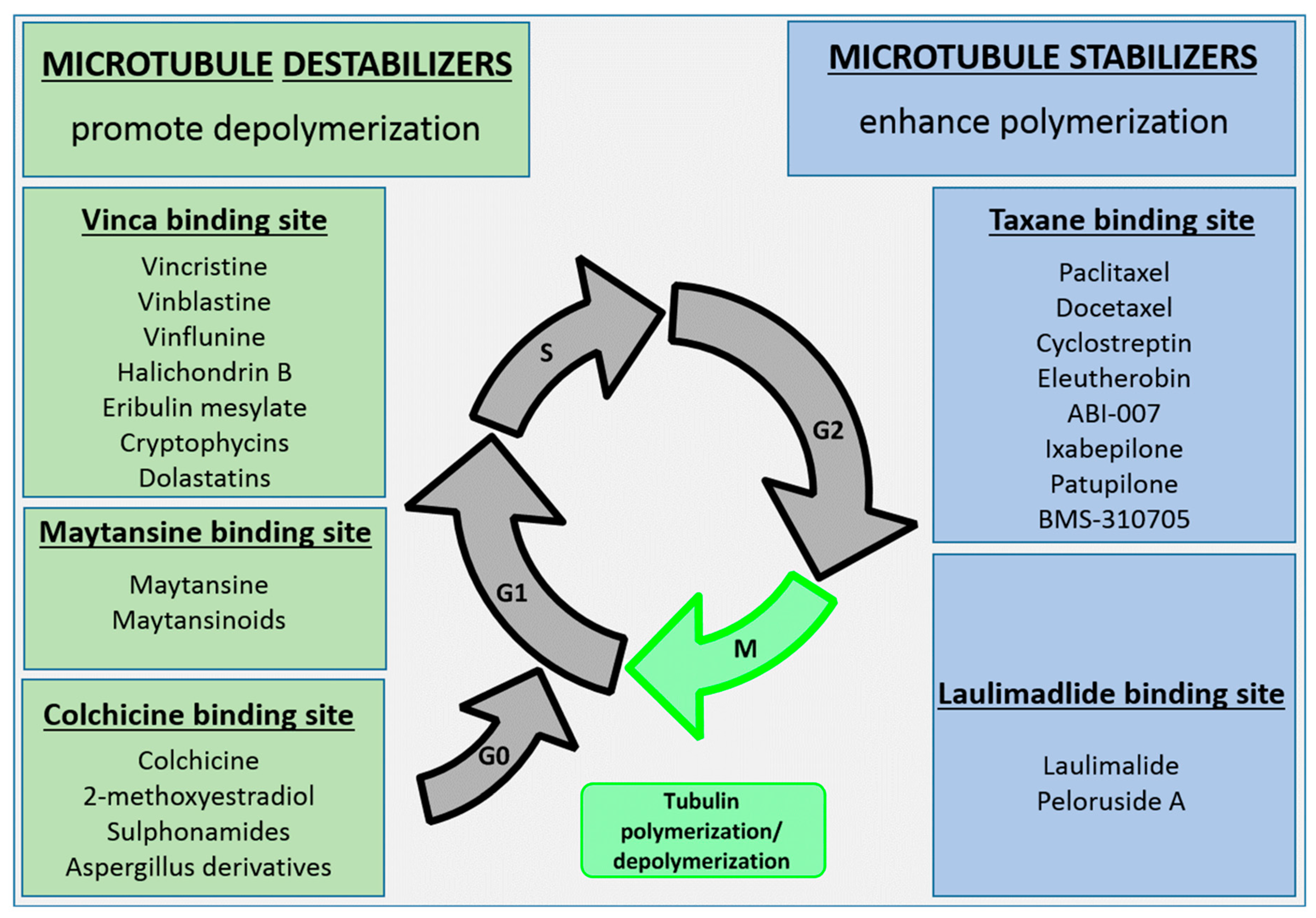
3. Tubulin Inhibitors as Payloads of ADCs
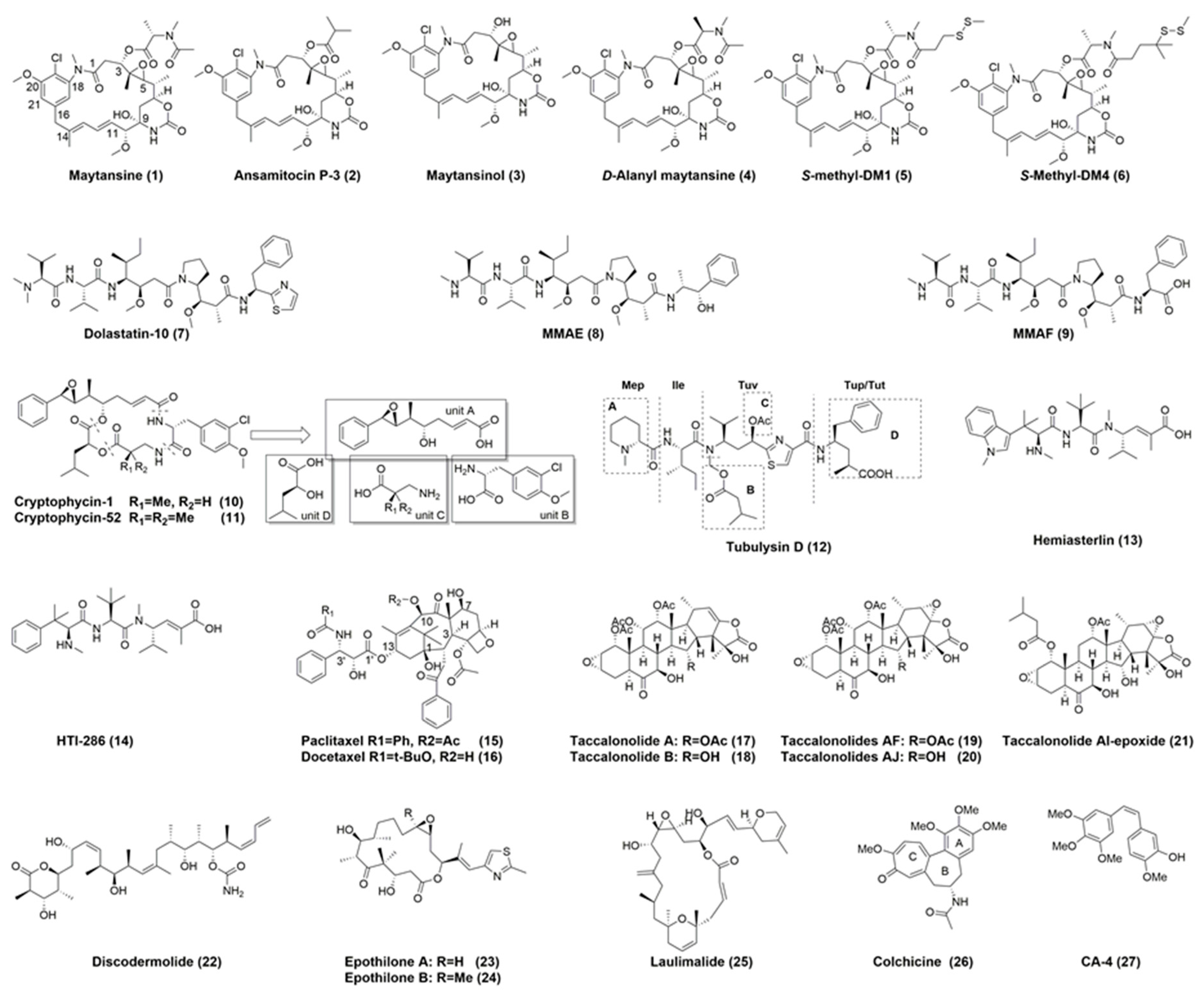
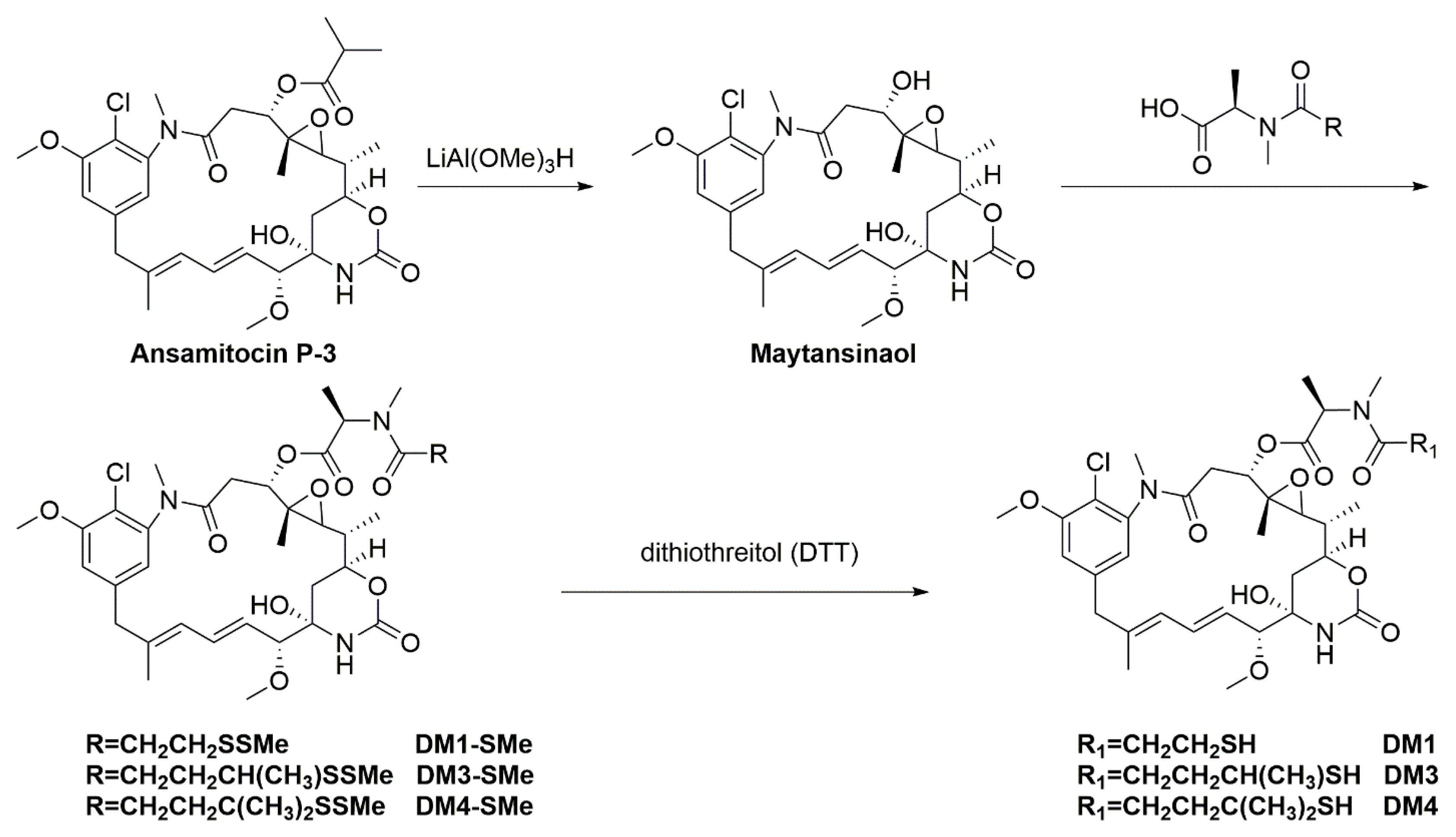
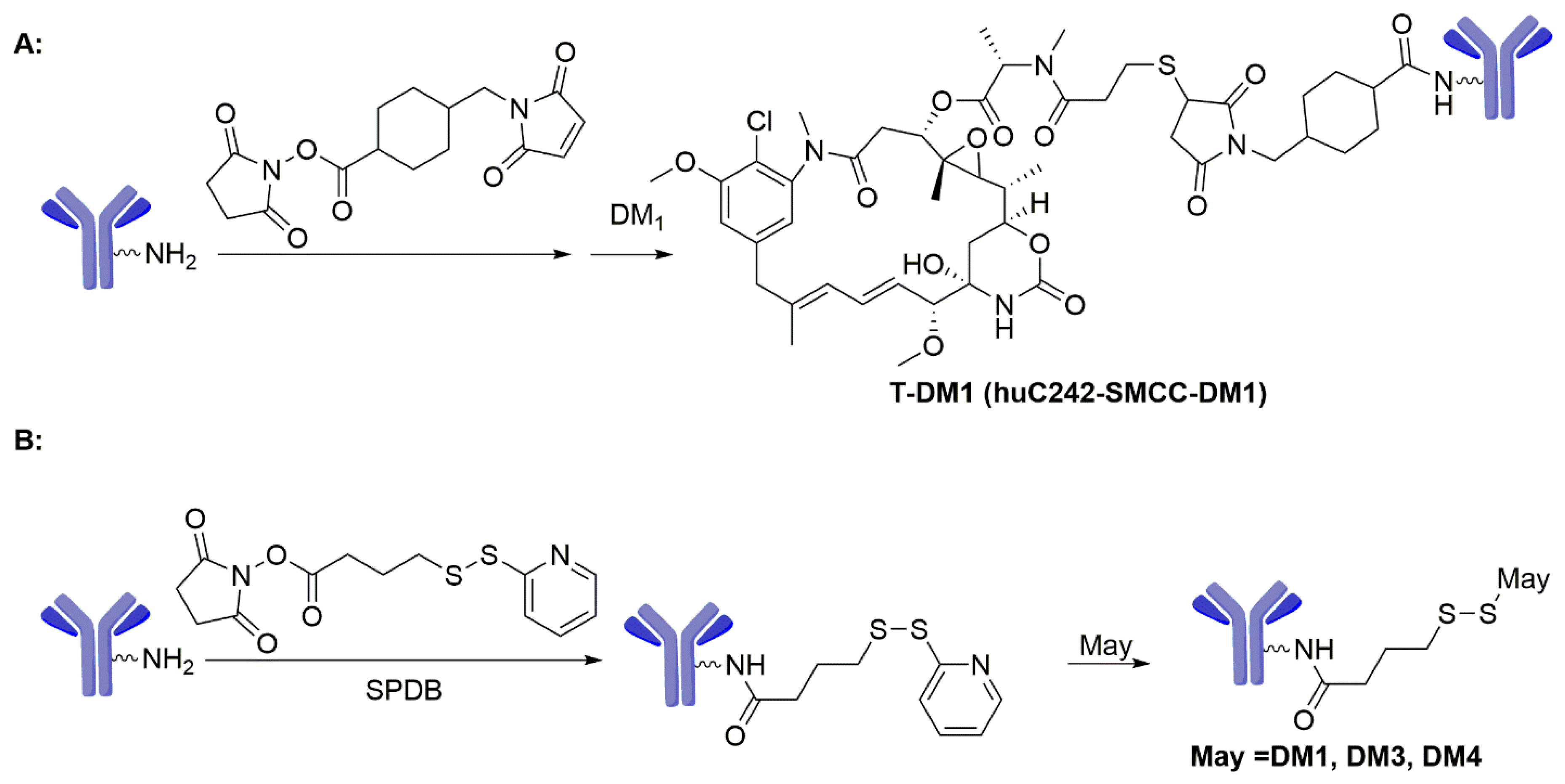
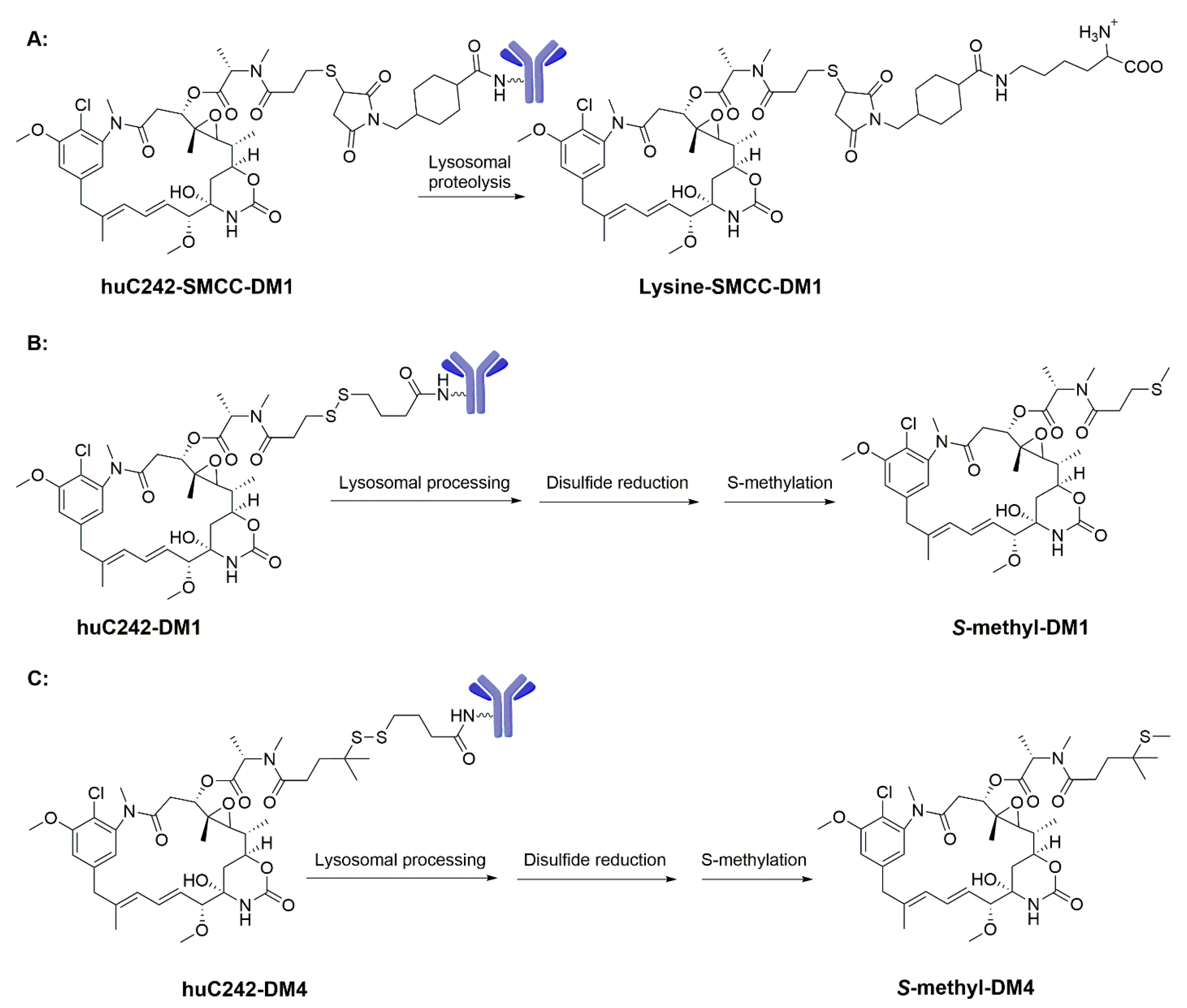
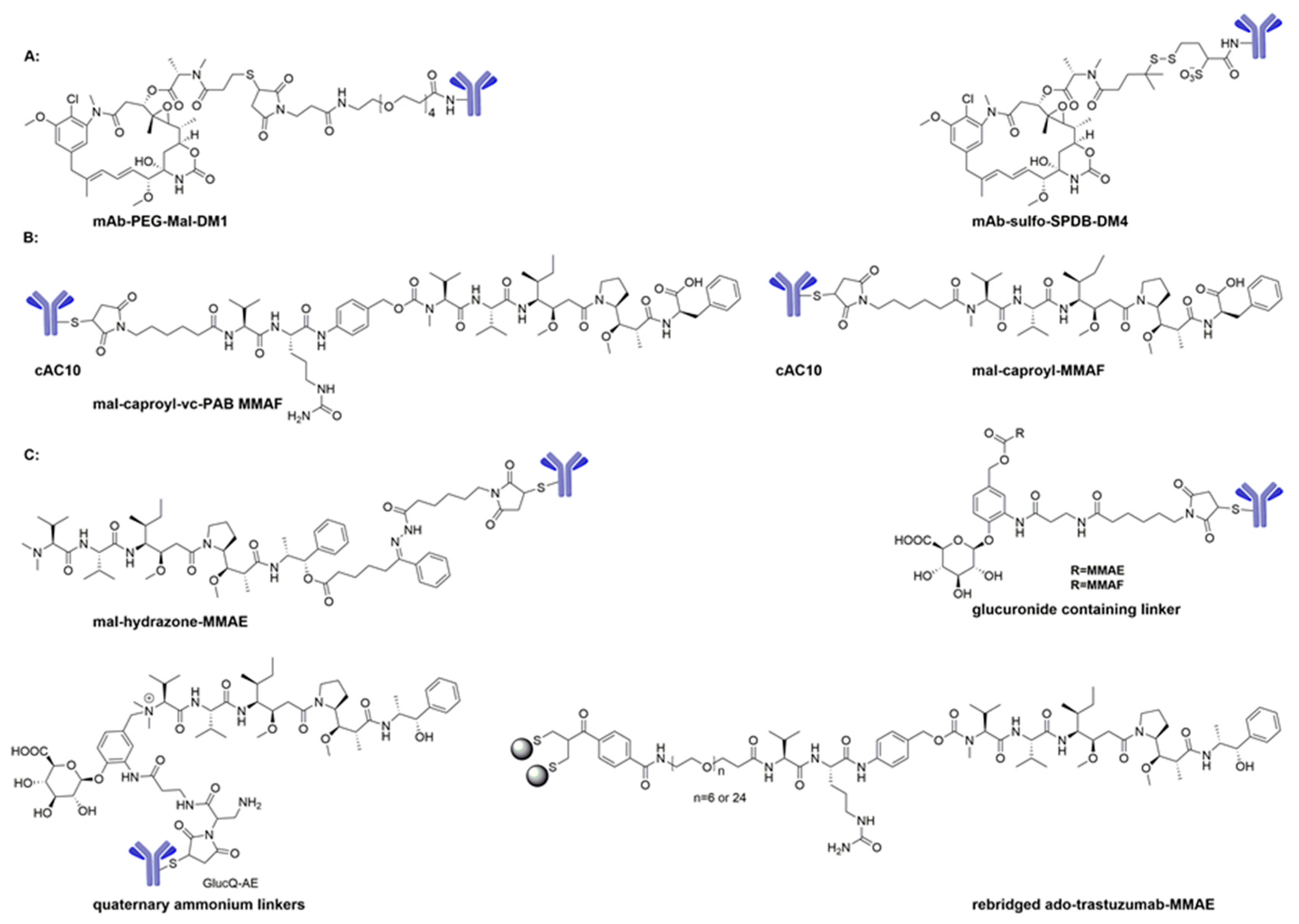
3.1. Maytansinoids as ADC Payloads
3.2. Auristatins as ADC Payloads
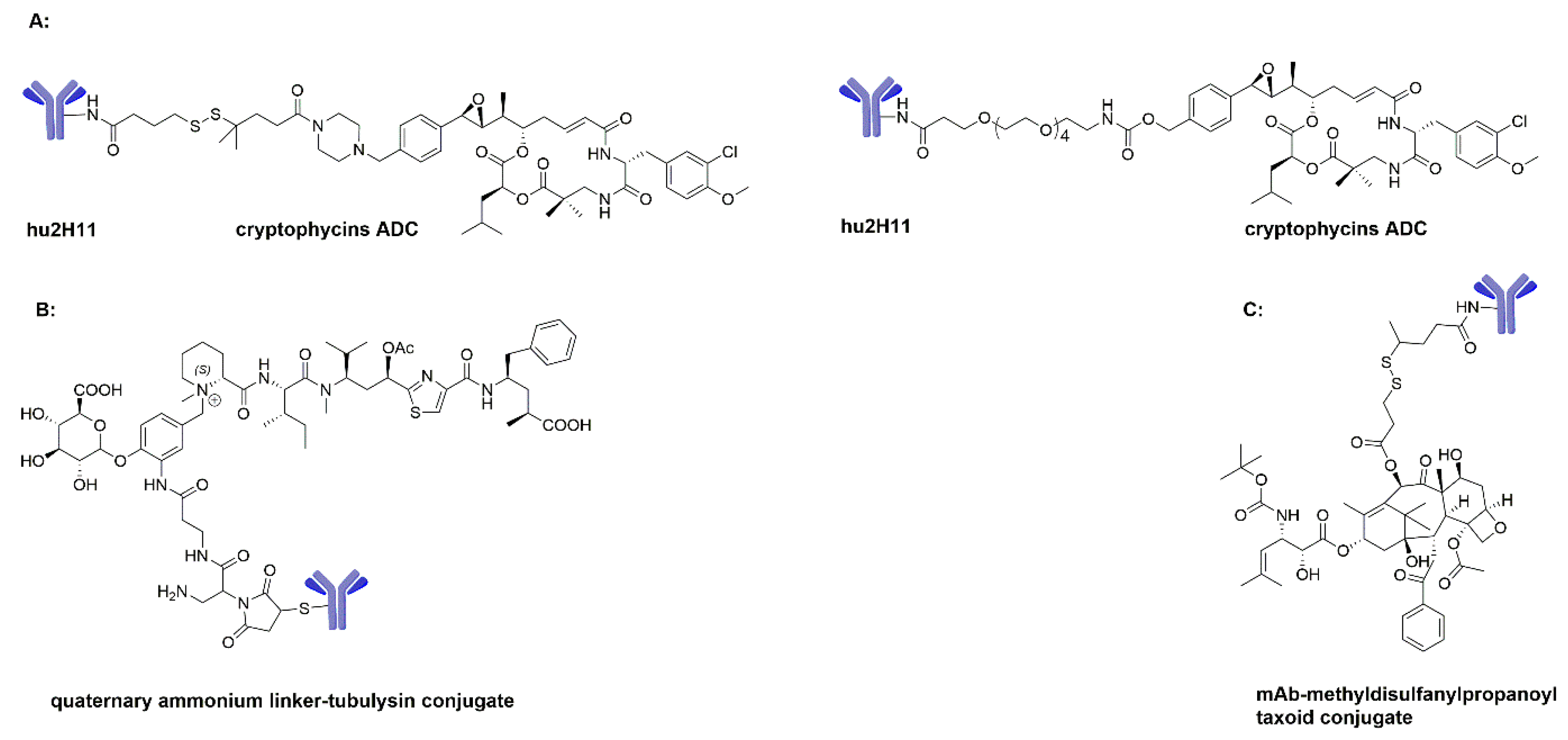
3.3. Cryptophycins as ADC Payloads
3.4. Tubulysins as ADC Payloads
3.5. Hemiasterlins as ADC Payloads
3.6. Paclitaxel and Docetaxel as ADC Payloads
3.7. Other Tubulin Inhibitors as Potential ADC Payloads
4. Conclusions and Final Remarks
Acknowledgments
Conflicts of Interest
Abbreviations
| ADCs | Antibody-drug conjugates |
| ALCL | Anaplastic large-cell lymphoma |
| AML | Acute myelogenous leukemia |
| BCMA | B-cell maturation antigen |
| CA9 | Carbonic anhydrase IX |
| CEA | Carcinoembryonic antigen |
| CLL | Chronic lymphocytic leukemia |
| DAR | Drug-to-antibody ratio |
| DLBCL | Diffuse large B cell lymphoma |
| DTT | Dithiothreitol |
| EDNRB | Endothelin receptor type B |
| EGFR | Epidermal growth factor receptor |
| ENPP3 | Ectonucleotide pyrophosphatase/phosphodiesterase family member 3 |
| EphA2 | Ephrin type-A receptor 2 |
| EphB2 | Ephrin type-B receptor 2 |
| FDA | Food and Drug Administration |
| GCC | Guanyl cyclase C |
| GPNMB | Transmembrane glycoprotein NMB |
| GTP | Guanosine triphosphate |
| HER2 | Human epidermal growth factor receptor 2 |
| HL | Hodgkin’s lymphoma |
| IgG | Immunoglobulin G |
| LBCL | Large B-cell lymphoma |
| LY6E | Lymphocyte antigen 6 complex |
| mAb | Monoclonal antibody |
| MBC | Metastatic breast cancer |
| MC | Maleimidocaproyl |
| MCC | 4-(N-Maleimidomethyl)cyclohexane-1-carboxylate |
| MDR | Multiple-drug resistant |
| MDR1 | Multidrug resistance protein 1 |
| MM | Multiple myeloma |
| MMAE | Monomethyl auristatin E |
| MMAF | Monomethyl auristatin F |
| MTD | Maximum tolerated dose |
| MUC16 | Mucin 16 |
| NHL | Non-Hodgkin’s lymphoma |
| NSCLC | Non-small cell lung cancer |
| PEG | Polyethylene glycol |
| PK/PD | Pharmacokinetic/pharmacodynamics |
| PSMA | Prostate specific membrane antigen |
| RCC | Renal cell carcinoma |
| SAR | Structure-activity relationship |
| SCLC | Small-cell lung cancer |
| SLITRK6 | SLIT and NTRK-like protein 6 |
| SMCC | Succinimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate |
| SPDB | N-Succinimidyl-4-(2-pyridyldithio)butanoate |
| SPP | N-Succinimidyl 4-(2-pyridyldithio) pentanoate |
| sSPDB | Sulfo-SPDB |
| STEAP1 | Six transmembrane epithelial Antigen of prostate 1 |
| TAG-72 | Tumor-associated glycoprotein-72 |
| vc | Valine-citrulline |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- DeVita, V.T., Jr.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [PubMed]
- Chari, R.V. Targeted cancer therapy: Conferring specificity to cytotoxic drugs. Acc. Chem. Res. 2008, 41, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Nishida, T.; Shirao, K.; Sawaki, A.; Koseki, M.; Okamura, T.; Ohtsu, A.; Sugiyama, T.; Miyakawa, K.; Hirota, S. Efficacy and safety profile of imatinib mesylate (ST1571) in Japanese patients with advanced gastrointestinal stromal tumors: A phase II study (STI571B1202). Int. J. Clin. Oncol. 2008, 13, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Katzel, J.A.; Fanucchi, M.P.; Li, Z. Recent advances of novel targeted therapy in non-small cell lung cancer. J. Hematol. Oncol. 2009, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.C. Tamoxifen: Catalyst for the change to targeted therapy. Eur. J. Cancer 2008, 44, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Drebin, J.A.; Link, V.C.; Weinberg, R.A.; Greene, M.I. Inhibition of tumor growth by a monoclonal antibody reactive with an oncogene-encoded tumor antigen. Proc. Natl. Acad. Sci. USA 1986, 83, 9129–9133. [Google Scholar] [CrossRef] [PubMed]
- Oldham, R.K.; Dillman, R.O. Monoclonal antibodies in cancer therapy: 25 years of progress. J. Clin. Oncol. 2008, 26, 1774–1777. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Perez, H.L.; Cardarelli, P.M.; Deshpande, S.; Gangwar, S.; Schroeder, G.M.; Vite, G.D.; Borzilleri, R.M. Antibody-drug conjugates: Current status and future directions. Drug Discov. Today 2014, 19, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Luisi, D.L.; Pak, R.H. Antibody-drug conjugates: Design, formulation and physicochemical stability. Pharm. Res. 2015, 32, 3541–3571. [Google Scholar] [CrossRef] [PubMed]
- Trail, P.A.; Willner, D.; Lasch, S.J.; Henderson, A.J.; Hofstead, S.; Casazza, A.M.; Firestone, R.A.; Hellstrom, I.; Hellstrom, K.E. Cure of xenografted human carcinomas by BR96-doxorubicin immunoconjugates. Science 1993, 261, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Elias, D.J.; Hirschowitz, L.; Kline, L.E.; Kroener, J.F.; Dillman, R.O.; Walker, L.E.; Robb, J.A.; Timms, R.M. Phase I clinical comparative study of monoclonal antibody KS1/4 and KS1/4-methotrexate immunconjugate in patients with non-small cell lung carcinoma. Cancer Res. 1990, 50, 4154–4159. [Google Scholar] [PubMed]
- Saleh, M.N.; Sugarman, S.; Murray, J.; Ostroff, J.B.; Healey, D.; Jones, D.; Daniel, C.R.; LeBherz, D.; Brewer, H.; Onetto, N.; et al. Phase I trial of the anti-Lewis Y drug immunoconjugate BR96-doxorubicin in patients with lewis Y-expressing epithelial tumors. J. Clin. Oncol. 2000, 18, 2282–2292. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shen, W.-C.; Zaro, J.L. Antibody-Drug Conjugates: The 21st Century Magic Bullets for Cancer; Springer: New York, NY, USA, 2015; Volume 17. [Google Scholar]
- Chari, R.V. Expanding the reach of antibody-drug conjugates. ACS Med. Chem. Lett. 2016, 7, 974–976. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; Brown, S. Antibody-drug conjugates as novel anti-cancer chemotherapeutics. Biosci. Rep. 2015, 35, e00225. [Google Scholar] [CrossRef] [PubMed]
- Panowski, S.; Bhakta, S.; Raab, H.; Polakis, P.; Junutula, J.R. Site-specific antibody drug conjugates for cancer therapy. mAbs 2014, 6, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125, S3–S23. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.M. DNA microarray and cancer. Curr. Opin. Oncol. 2003, 15, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.A.; Sievers, E.L.; Stadtmauer, E.A.; Lowenberg, B.; Estey, E.H.; Dombret, H.; Theobald, M.; Voliotis, D.; Bennett, J.M.; Richie, M.; et al. Final report of the efficacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33-positive acute myeloid leukemia in first recurrence. Cancer 2005, 104, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Thurber, G.M.; Schmidt, M.M.; Wittrup, K.D. Antibody tumor penetration: transport opposed by systemic and antigen-mediated clearance. Adv. Drug Deliv. Rev. 2008, 60, 1421–1434. [Google Scholar] [CrossRef] [PubMed]
- Woof, J.M.; Burton, D.R. Human antibody-Fc receptor interactions illuminated by crystal structures. Nat. Rev. Immunol. 2004, 4, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A. Antibody-drug conjugate targets. Curr. Cancer Drug Targets 2009, 9, 982–1004. [Google Scholar] [CrossRef] [PubMed]
- Mathur, R.; Weiner, G.J. Picking the optimal target for antibody-drug conjugates. Am. Soc. Clin. Oncol. Educ. Book 2013. [Google Scholar] [CrossRef] [PubMed]
- Mack, F.; Ritchie, M.; Sapra, P. The next generation of antibody drug conjugates. Semin. Oncol. 2014, 41, 637–652. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.; Tchistiakova, L.; Scott, N. Implications of receptor-mediated endocytosis and intracellular trafficking dynamics in the development of antibody drug conjugates. mAbs 2013, 5, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.M.; Wittrup, K.D. A modeling analysis of the effects of molecular size and binding affinity on tumor targeting. Mol. Cancer Ther. 2009, 8, 2861–2871. [Google Scholar] [CrossRef] [PubMed]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, S.I.; Lou, J.; Shaller, C.C.; Tang, Y.; Klein-Szanto, A.J.; Weiner, L.M.; Marks, J.D.; Adams, G.P. Influence of affinity and antigen internalization on the uptake and penetration of Anti-HER2 antibodies in solid tumors. Cancer Res. 2011, 71, 2250–2259. [Google Scholar] [CrossRef] [PubMed]
- Chudasama, V.; Maruani, A.; Caddick, S. Recent advances in the construction of antibody-drug conjugates. Nat. Chem. 2016, 8, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, D.; Hackenberger, C.P.; Leonhardt, H.; Helma, J. Current Status: Site-Specific Antibody Drug Conjugates. J. Clin. Immunol. 2016, 36, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Badescu, G.; Bryant, P.; Bird, M.; Henseleit, K.; Swierkosz, J.; Parekh, V.; Tommasi, R.; Pawlisz, E.; Jurlewicz, K.; Farys, M.; et al. Bridging disulfides for stable and defined antibody drug conjugates. Bioconjug. Chem. 2014, 25, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.W.; Strickland, R.A.; Schumacher, F.F.; Caddick, S.; Baker, J.R.; Gibson, M.I.; Haddleton, D.M. Polymeric dibromomaleimides as extremely efficient disulfide bridging bioconjugation and pegylation agents. J. Am. Chem. Soc. 2012, 134, 1847–1852. [Google Scholar] [CrossRef] [PubMed]
- Maruani, A.; Smith, M.E.; Miranda, E.; Chester, K.A.; Chudasama, V.; Caddick, S. A plug-and-play approach to antibody-based therapeutics via a chemoselective dual click strategy. Nat. Commun. 2015, 6, 6645. [Google Scholar] [CrossRef] [PubMed]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Axup, J.Y.; Bajjuri, K.M.; Ritland, M.; Hutchins, B.M.; Kim, C.H.; Kazane, S.A.; Halder, R.; Forsyth, J.S.; Santidrian, A.F.; Stafin, K.; et al. Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc. Natl. Acad. Sci. USA 2012, 109, 16101–16106. [Google Scholar] [CrossRef] [PubMed]
- Zeglis, B.M.; Davis, C.B.; Aggeler, R.; Kang, H.C.; Chen, A.; Agnew, B.J.; Lewis, J.S. Enzyme-mediated methodology for the site-specific radiolabeling of antibodies based on catalyst-free click chemistry. Bioconjug. Chem. 2013, 24, 1057–1067. [Google Scholar] [CrossRef] [PubMed]
- Jeger, S.; Zimmermann, K.; Blanc, A.; Grunberg, J.; Honer, M.; Hunziker, P.; Struthers, H.; Schibli, R. Site-specific and stoichiometric modification of antibodies by bacterial transglutaminase. Angew. Chem. Int. Ed. Engl. 2010, 49, 9995–9997. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Hart, S.A.; Schink, A.; Pollok, B.A. Sortase-mediated protein ligation: A new method for protein engineering. J. Am. Chem. Soc. 2004, 126, 2670–2671. [Google Scholar] [CrossRef] [PubMed]
- Qasba, P.K. Glycans of antibodies as a specific site for drug conjugation using glycosyltransferases. Bioconjug. Chem. 2015, 26, 2170–2175. [Google Scholar] [CrossRef] [PubMed]
- Drake, P.M.; Albers, A.E.; Baker, J.; Banas, S.; Barfield, R.M.; Bhat, A.S.; de Hart, G.W.; Garofalo, A.W.; Holder, P.; Jones, L.C.; et al. Aldehyde tag coupled with HIPS chemistry enables the production of ADCs conjugated site-specifically to different antibody regions with distinct in vivo efficacy and PK outcomes. Bioconjug. Chem. 2014, 25, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Albers, A.E.; Garofalo, A.W.; Drake, P.M.; Kudirka, R.; de Hart, G.W.; Barfield, R.M.; Baker, J.; Banas, S.; Rabuka, D. Exploring the effects of linker composition on site-specifically modified antibody-drug conjugates. Eur. J. Med. Chem. 2014, 88, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, D.; Helma, J.; Mann, F.A.; Pichler, G.; Natale, F.; Krause, E.; Cardoso, M.C.; Hackenberger, C.P.; Leonhardt, H. Versatile and efficient site-specific protein functionalization by tubulin tyrosine ligase. Angew. Chem. Int. Ed. Engl. 2015, 54, 13787–13791. [Google Scholar] [CrossRef] [PubMed]
- Janke, C. The tubulin code: Molecular components, readout mechanisms, and functions. J. Cell Biol. 2014, 206, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Prota, A.E.; Magiera, M.M.; Kuijpers, M.; Bargsten, K.; Frey, D.; Wieser, M.; Jaussi, R.; Hoogenraad, C.C.; Kammerer, R.A.; Janke, C.; et al. Structural basis of tubulin tyrosination by tubulin tyrosine ligase. J. Cell Biol. 2013, 200, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Bertozzi, C.R. Site-specific antibody-drug conjugates: The nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconjug. Chem. 2015, 26, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Smith, S.W.; Ghone, S.; Tomczuk, B. Current ADC linker chemistry. Pharm. Res. 2015, 32, 3526–3540. [Google Scholar] [CrossRef] [PubMed]
- McCombs, J.R.; Owen, S.C. Antibody drug conjugates: Design and selection of linker, payload and conjugation chemistry. AAPS J. 2015, 17, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar] [PubMed]
- Chari, R.V.; Miller, M.L.; Widdison, W.C. Antibody-drug conjugates: An emerging concept in cancer therapy. Angew. Chem. Int. Ed. Engl. 2014, 53, 3796–3827. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Jiang, F.; Lu, A.; Zhang, G. Linkers having a crucial role in antibody-drug conjugates. Int. J. Mol. Sci. 2016, 17, 561. [Google Scholar] [CrossRef] [PubMed]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2016, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nogales, E.; Wang, H.W. Structural mechanisms underlying nucleotide-dependent self-assembly of tubulin and its relatives. Curr. Opin. Struct. Biol. 2006, 16, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Akhmanova, A.; Steinmetz, M.O. Tracking the ends: A dynamic protein network controls the fate of microtubule tips. Nat. Rev. Mol. Cell Biol. 2008, 9, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Nogales, E.; Wolf, S.G.; Downing, K.H. Structure of the alpha beta tubulin dimer by electron crystallography. Nature 1998, 391, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Sham, H.L.; Rosenberg, S.H. Antimitotic Agents. Annu. Rep. Med. Chem. 1999, 34, 139–148. [Google Scholar]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Kupchan, S.M.; Komoda, Y.; Court, W.A.; Thomas, G.J.; Smith, R.M.; Karim, A.; Gilmore, C.J.; Haltiwanger, R.C.; Bryan, R.F. Maytansine, a novel antileukemic ansa macrolide from Maytenus ovatus. J. Am. Chem. Soc. 1972, 94, 1354–1356. [Google Scholar] [CrossRef] [PubMed]
- Remillard, S.; Rebhun, L.I.; Howie, G.A.; Kupchan, S.M. Antimitotic activity of the potent tumor inhibitor maytansine. Science 1975, 189, 1002–1005. [Google Scholar] [CrossRef] [PubMed]
- Issell, B.F.; Crooke, S.T. Maytansine. Cancer Treat. Rev. 1978, 5, 199–207. [Google Scholar] [CrossRef]
- Benechie, M.; Khuong-Huu, F. Total synthesis of (−)-maytansinol. J. Org. Chem. 1996, 61, 7133–7138. [Google Scholar] [CrossRef] [PubMed]
- Kupchan, S.M.; Sneden, A.T.; Branfman, A.R.; Howie, G.A.; Rebhun, L.I.; McIvor, W.E.; Wang, R.W.; Schnaitman, T.C. Structural requirements for antileukemic activity among the naturally occurring and semisynthetic maytansinoids. J. Med. Chem. 1978, 21, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Widdison, W.C.; Wilhelm, S.D.; Cavanagh, E.E.; Whiteman, K.R.; Leece, B.A.; Kovtun, Y.; Goldmacher, V.S.; Xie, H.; Steeves, R.M.; Lutz, R.J.; et al. Semisynthetic maytansine analogues for the targeted treatment of cancer. J. Med. Chem. 2006, 49, 4392–4408. [Google Scholar] [CrossRef] [PubMed]
- Cassady, J.M.; Chan, K.K.; Floss, H.G.; Leistner, E. Recent developments in the maytansinoid antitumor agents. Chem. Pharm. Bull. 2004, 52, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Amiri-Kordestani, L.; Blumenthal, G.M.; Xu, Q.C.; Zhang, L.; Tang, S.W.; Ha, L.; Weinberg, W.C.; Chi, B.; Candau-Chacon, R.; Hughes, P.; et al. FDA approval: Ado-trastuzumab emtansine for the treatment of patients with HER2-positive metastatic breast cancer. Clin. Cancer Res. 2014, 20, 4436–4441. [Google Scholar] [CrossRef] [PubMed]
- Erickson, H.K.; Lewis Phillips, G.D.; Leipold, D.D.; Provenzano, C.A.; Mai, E.; Johnson, H.A.; Gunter, B.; Audette, C.A.; Gupta, M.; Pinkas, J.; et al. The effect of different linkers on target cell catabolism and pharmacokinetics/pharmacodynamics of trastuzumab maytansinoid conjugates. Mol. Cancer Ther. 2012, 11, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Erickson, H.K.; Park, P.U.; Widdison, W.C.; Kovtun, Y.V.; Garrett, L.M.; Hoffman, K.; Lutz, R.J.; Goldmacher, V.S.; Blattler, W.A. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res. 2006, 66, 4426–4433. [Google Scholar] [CrossRef] [PubMed]
- Polson, A.G.; Calemine-Fenaux, J.; Chan, P.; Chang, W.; Christensen, E.; Clark, S.; de Sauvage, F.J.; Eaton, D.; Elkins, K.; Elliott, J.M.; et al. Antibody-drug conjugates for the treatment of non-Hodgkin’s lymphoma: target and linker-drug selection. Cancer Res. 2009, 69, 2358–2364. [Google Scholar] [CrossRef] [PubMed]
- Erickson, H.K.; Widdison, W.C.; Mayo, M.F.; Audette, C.; Wilhelm, S.D. Tumor delivery and in vivo processing of disulfide-linked and thioether-linked antibody—Maytansinoid conjugates. Bioconjug. Chem. 2010, 21, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Lewis Phillips, G.D.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blattler, W.A.; Lambert, J.M.; Chari, R.V.; Lutz, R.J.; et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Widdison, W.; Mayo, M.; Wilhelm, S.; Leece, B.; Chari, R.; Singh, R.; Erickson, H. Design of antibody-maytansinoid conjugates allows for efficient detoxification via liver metabolism. Bioconjug. Chem. 2011, 22, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Dosio, F.; Brusa, P.; Cattel, L. Immunotoxins and anticancer drug conjugate assemblies: The role of the linkage between components. Toxins 2011, 3, 848–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovtun, Y.V.; Audette, C.A.; Mayo, M.F.; Jones, G.E.; Doherty, H.; Maloney, E.K.; Erickson, H.K.; Sun, X.; Wilhelm, S.; Ab, O.; et al. Antibody-maytansinoid conjugates designed to bypass multidrug resistance. Cancer Res. 2010, 70, 2528–2537. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Y.; Wilhelm, S.D.; Audette, C.; Jones, G.; Leece, B.A.; Lazar, A.C.; Goldmacher, V.S.; Singh, R.; Kovtun, Y.; Widdison, W.C.; et al. Synthesis and evaluation of hydrophilic linkers for antibody-maytansinoid conjugates. J. Med. Chem. 2011, 54, 3606–3623. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Pettit, G.R.; Hamel, E. Dolastatin 10, a powerful cytostatic peptide derived from a marine animal. Inhibition of tubulin polymerization mediated through the vinca alkaloid binding domain. Biochem. Pharmacol. 1990, 39, 1941–1949. [Google Scholar] [CrossRef]
- Aherne, G.W.; Hardcastle, A.; Valenti, M.; Bryant, A.; Rogers, P.; Pettit, G.R.; Srirangam, J.K.; Kelland, L.R. Antitumour evaluation of dolastatins 10 and 15 and their measurement in plasma by radioimmunoassay. Cancer Chemother. Pharmacol. 1996, 38, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Garteiz, D.A.; Madden, T.; Beck, D.E.; Huie, W.R.; McManus, K.T.; Abbruzzese, J.L.; Chen, W.; Newman, R.A. Quantitation of dolastatin-10 using HPLC/electrospray ionization mass spectrometry: Application in a phase I clinical trial. Cancer Chemother. Pharmacol. 1998, 41, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Krug, L.M.; Miller, V.A.; Kalemkerian, G.P.; Kraut, M.J.; Ng, K.K.; Heelan, R.T.; Pizzo, B.A.; Perez, W.; McClean, N.; Kris, M.G. Phase II study of dolastatin-10 in patients with advanced non-small-cell lung cancer. Ann. Oncol. 2000, 11, 227–228. [Google Scholar] [CrossRef] [PubMed]
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B.; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003, 21, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Doronina, S.O.; Mendelsohn, B.A.; Bovee, T.D.; Cerveny, C.G.; Alley, S.C.; Meyer, D.L.; Oflazoglu, E.; Toki, B.E.; Sanderson, R.J.; Zabinski, R.F.; et al. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: Effects of linker technology on efficacy and toxicity. Bioconjug. Chem. 2006, 17, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Francisco, J.A.; Cerveny, C.G.; Meyer, D.L.; Mixan, B.J.; Klussman, K.; Chace, D.F.; Rejniak, S.X.; Gordon, K.A.; DeBlanc, R.; Toki, B.E.; et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003, 102, 1458–1465. [Google Scholar] [CrossRef] [PubMed]
- Dubowchik, G.M.; Firestone, R.A.; Padilla, L.; Willner, D.; Hofstead, S.J.; Mosure, K.; Knipe, J.O.; Lasch, S.J.; Trail, P.A. Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: Model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconjug. Chem. 2002, 13, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Doronina, S.O.; Bovee, T.D.; Meyer, D.W.; Miyamoto, J.B.; Anderson, M.E.; Morris-Tilden, C.A.; Senter, P.D. Novel peptide linkers for highly potent antibody-auristatin conjugate. Bioconjug. Chem. 2008, 19, 1960–1963. [Google Scholar] [CrossRef] [PubMed]
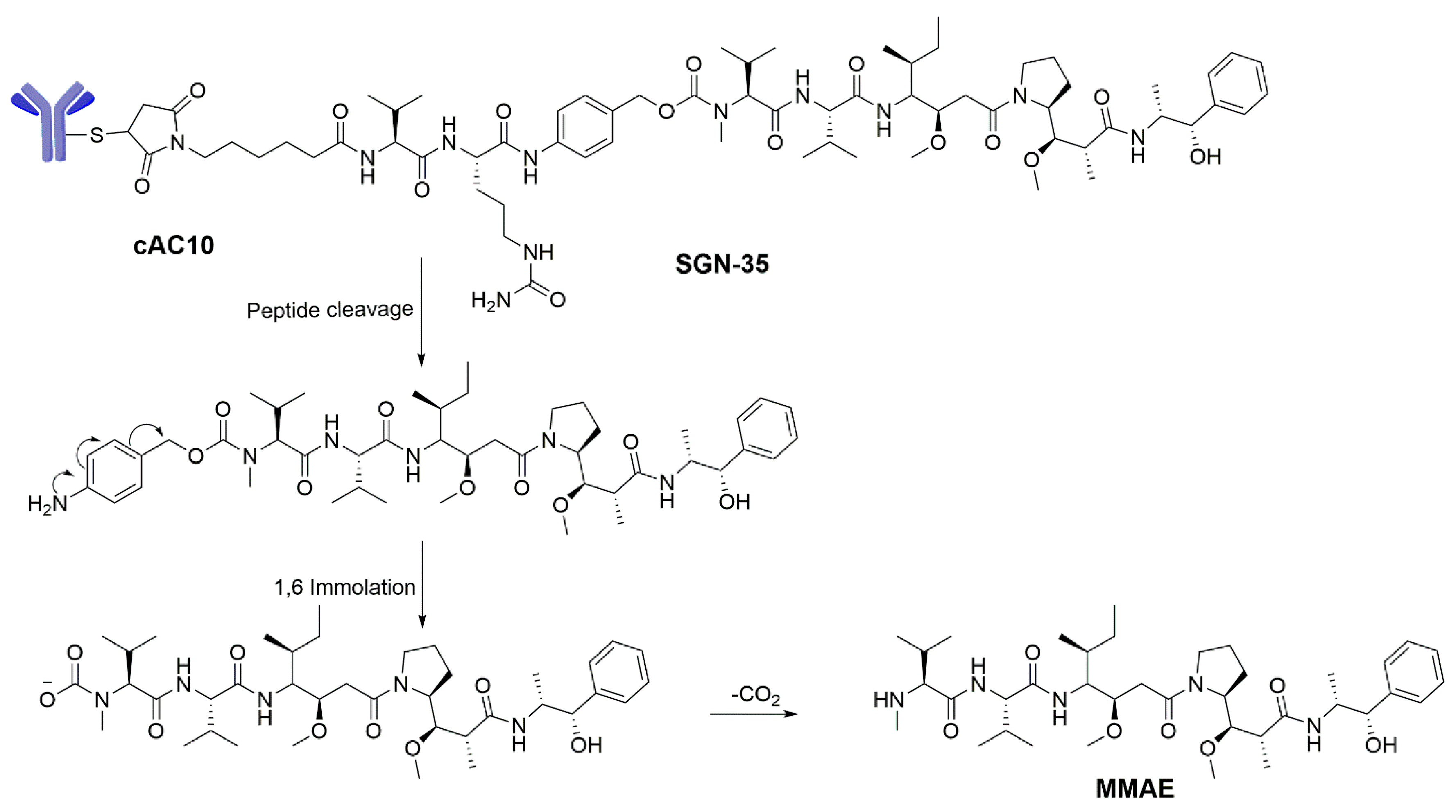
- Younes, A.; Bartlett, N.L.; Leonard, J.P.; Kennedy, D.A.; Lynch, C.M.; Sievers, E.L.; Forero-Torres, A. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N. Engl. J. Med. 2010, 363, 1812–1821. [Google Scholar] [CrossRef] [PubMed]
- Katz, J.; Janik, J.E.; Younes, A. Brentuximab Vedotin (SGN-35). Clin. Cancer Res. 2011, 17, 6428–6436. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Gopal, A.; Smith, S.; Ansell, S.; Rosenblatt, J.; Savage, K.; Connors, J.; Engert, A.; Larsen, E.; Kennedy, D. Results from a pivotal phase II study of brentuximab vedotin (SGN-35) in patients with relapsed or refractory hodgkin lymphoma (HL). J. Clin. Oncol. 2011, 29, 8031. [Google Scholar] [CrossRef]
- Singh, R.; Erickson, H.K. Antibody-cytotoxic agent conjugates: Preparation and characterization. Methods Mol. Biol. 2009, 525, 445–467. [Google Scholar] [PubMed]
- Doronina, S.; Alley, S.; Cerveney, C.; Francisco, J.; Law, C.-L.; Mendelsohn, B.; Okeley, N.; Sanderson, R.; Wahl, A.; Senter, P. Novel linkers for monoclonal antibody-mediated delivery of cell impermeable anticancer agents. Cancer Res. 2005, 65, 333. [Google Scholar]
- Jeffrey, S.C.; Andreyka, J.B.; Bernhardt, S.X.; Kissler, K.M.; Kline, T.; Lenox, J.S.; Moser, R.F.; Nguyen, M.T.; Okeley, N.M.; Stone, I.J.; et al. Development and properties of beta-glucuronide linkers for monoclonal antibody-drug conjugates. Bioconjug. Chem. 2006, 17, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Burke, P.J.; Hamilton, J.Z.; Pires, T.A.; Setter, J.R.; Hunter, J.H.; Cochran, J.H.; Waight, A.B.; Gordon, K.A.; Toki, B.E.; Emmerton, K.K.; et al. Development of novel quaternary ammonium linkers for antibody-drug conjugates. Mol. Cancer Ther. 2016, 15, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Bryant, P.; Pabst, M.; Badescu, G.; Bird, M.; McDowell, W.; Jamieson, E.; Swierkosz, J.; Jurlewicz, K.; Tommasi, R.; Henseleit, K.; et al. In vitro and in vivo evaluation of cysteine rebridged trastuzumab-MMAE antibody drug conjugates with defined drug-to-antibody ratios. Mol. Pharm. 2015, 12, 1872–1879. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.E.; Hirsch, C.F.; Sesin, D.F.; Flor, J.E.; Chartrain, M.; Fromtling, R.E.; Harris, G.H.; Salvatore, M.J.; Liesch, J.M.; Yudin, K. Pharmaceuticals from cultured algae. J. Ind. Microbiol. 1990, 5, 113–123. [Google Scholar] [CrossRef]
- Trimurtulu, G.; Ohtani, I.; Patterson, G.M.L.; Moore, R.E.; Corbett, T.H.; Valeriote, F.A.; Demchik, L. Total structures of cryptophycins, potent antitumor depsipeptides from the Blue-green alga nostoc sp. strain GSV 224. J. Am. Chem. Soc. 1994, 116, 4729–4737. [Google Scholar] [CrossRef]
- Smith, C.D.; Zhang, X.; Mooberry, S.L.; Patterson, G.M.; Moore, R.E. Cryptophycin: A new antimicrotubule agent active against drug-resistant cells. Cancer Res. 1994, 54, 3779–3784. [Google Scholar] [PubMed]
- Smith, A.B.; Cho, Y.S.; Pettit, G.R.; Hirschmann, R. Design, synthesis, and evaluation of azepine-based cryptophycin mimetics. Tetrahedron 2003, 59, 6991–7009. [Google Scholar] [CrossRef]
- Patel, V.F.; Andis, S.L.; Kennedy, J.H.; Ray, J.E.; Schultz, R.M. Novel cryptophycin antitumor agents: Synthesis and cytotoxicity of fragment “B” analogues. J. Med. Chem. 1999, 42, 2588–2603. [Google Scholar] [CrossRef] [PubMed]
- Eggen, M.; Georg, G.I. The cryptophycins: their synthesis and anticancer activity. Med. Res. Rev. 2002, 22, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Georg, G.I.; Ali, S.M.; Stella, V.J.; Waugh, W.N.; Himes, R.H. Halohydrin analogues of cryptophycin 1: Synthesis and biological activity. Bioorg. Med. Chem. Lett. 1998, 8, 1959–1962. [Google Scholar] [CrossRef]
- Al-Awar, R.S.; Ray, J.E.; Schultz, R.M.; Andis, S.L.; Kennedy, J.H.; Moore, R.E.; Liang, J.; Golakoti, T.; Subbaraju, G.V.; Corbett, T.H. A convergent approach to cryptophycin 52 analogues: Synthesis and biological evaluation of a novel series of fragment a epoxides and chlorohydrins. J. Med. Chem. 2003, 46, 2985–3007. [Google Scholar] [CrossRef] [PubMed]
- Weiss, C.; Sammet, B.; Sewald, N. Recent approaches for the synthesis of modified cryptophycins. Nat. Prod. Rep. 2013, 30, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Weiss, C.; Bogner, T.; Sammet, B.; Sewald, N. Total synthesis and biological evaluation of fluorinated cryptophycins. Beilstein J. Org. Chem. 2012, 8, 2060–2066. [Google Scholar] [CrossRef] [PubMed]
- Varie, D.L.; Shih, C.; Hay, D.A.; Andis, S.L.; Corbett, T.H.; Gossett, L.S.; Janisse, S.K.; Martinelli, M.J.; Moher, E.D.; Schultz, R.M.; et al. Synthesis and biological evaluation of cryptophycin analogs with substitution at C-6 (fragment C region). Bioorg. Med. Chem. Lett. 1999, 9, 369–374. [Google Scholar] [CrossRef]
- Nahrwold, M.; Bogner, T.; Eissler, S.; Verma, S.; Sewald, N. “Clicktophycin-52”: A bioactive cryptophycin-52 triazole analogue. Org. Lett. 2010, 12, 1064–1067. [Google Scholar] [CrossRef] [PubMed]
- Nahrwold, M.; Weiss, C.; Bogner, T.; Mertink, F.; Conradi, J.; Sammet, B.; Palmisano, R.; Royo Gracia, S.; Preusse, T.; Sewald, N. Conjugates of modified cryptophycins and RGD-peptides enter target cells by endocytosis. J. Med. Chem. 2013, 56, 1853–1864. [Google Scholar] [CrossRef] [PubMed]
- Buck, S.B.; Huff, J.K.; Himes, R.H.; Georg, G.I. Total synthesis and anti-tubulin activity of epi-c3 analogues of cryptophycin-24. J. Med. Chem. 2004, 47, 3697–3699. [Google Scholar] [CrossRef] [PubMed]
- Golakoti, T.; Ogino, J.; Heltzel, C.E.; Le Husebo, T.; Jensen, C.M.; Larsen, L.K.; Patterson, G.M.L.; Moore, R.E.; Mooberry, S.L. Structure determination, conformational analysis, chemical stability studies, and antitumor evaluation of the cryptophycins. Isolation of 18 new analogs from Nostoc sp. strain GSV 224. J. Am. Chem. Soc. 1995, 117, 12030–12049. [Google Scholar] [CrossRef]
- Sammet, B.; Bogner, T.; Nahrwold, M.; Weiss, C.; Sewald, N. Approaches for the synthesis of functionalized cryptophycins. J. Org. Chem. 2010, 75, 6953–6960. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, J.P.; Sun, W.; Gallagher, M.; Johnson, R.; Vaughn, D.; Schuchter, L.; Algazy, K.; Hahn, S.; Enas, N.; Ellis, D.; et al. Phase I trial of the cryptophycin analogue LY355703 administered as an intravenous infusion on a day 1 and 8 schedule every 21 days. Clin. Cancer Res. 2002, 8, 2524–2529. [Google Scholar] [PubMed]
- Sessa, C.; Weigang-Kohler, K.; Pagani, O.; Greim, G.; Mora, O.; De Pas, T.; Burgess, M.; Weimer, I.; Johnson, R. Phase I and pharmacological studies of the cryptophycin analogue LY355703 administered on a single intermittent or weekly schedule. Eur. J. Cancer 2002, 38, 2388–2396. [Google Scholar] [CrossRef]
- Edelman, M.J.; Gandara, D.R.; Hausner, P.; Israel, V.; Thornton, D.; DeSanto, J.; Doyle, L.A. Phase 2 study of cryptophycin 52 (LY355703) in patients previously treated with platinum based chemotherapy for advanced non-small cell lung cancer. Lung Cancer 2003, 39, 197–199. [Google Scholar] [CrossRef]
- D’Agostino, G.; del Campo, J.; Mellado, B.; Izquierdo, M.A.; Minarik, T.; Cirri, L.; Marini, L.; Perez-Gracia, J.L.; Scambia, G. A multicenter phase II study of the cryptophycin analog LY355703 in patients with platinum-resistant ovarian cancer. Int. J. Gynecol. Cancer 2006, 16, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Verma, V.A.; Pillow, T.H.; DePalatis, L.; Li, G.; Phillips, G.L.; Polson, A.G.; Raab, H.E.; Spencer, S.; Zheng, B. The cryptophycins as potent payloads for antibody drug conjugates. Bioorg. Med. Chem. Lett. 2015, 25, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Sasse, F.; Steinmetz, H.; Heil, J.; Hofle, G.; Reichenbach, H. Tubulysins, new cytostatic peptides from myxobacteria acting on microtubuli. Production, isolation, physico-chemical and biological properties. J. Antibiot. 2000, 53, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.W.; Sasse, F.; Lunsdorf, H.; Elnakady, Y.A.; Reichenbach, H. Mechanism of action of tubulysin, an antimitotic peptide from myxobacteria. ChemBiolChem 2006, 7, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Leamon, C.; Reddy, J.; Wang, K.; Dorton, R.; Westrick, E.; Dawson, A.; Smith, T.; Vlahov, I.; Vetzel, M. Folate receptor specific anti-tumor activity of EC0305, a folate-tubulysin conjugate. Cancer Res. 2007, 67, 2261. [Google Scholar]
- Cohen, R.; Vugts, D.J.; Visser, G.W.; Stigter-van Walsum, M.; Bolijn, M.; Spiga, M.; Lazzari, P.; Shankar, S.; Sani, M.; Zanda, M.; et al. Development of novel ADCs: Conjugation of tubulysin analogues to trastuzumab monitored by dual radiolabeling. Cancer Res. 2014, 74, 5700–5710. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Perry, S.R.; Muniz-Medina, V.; Wang, X.; Wetzel, L.K.; Rebelatto, M.C.; Hinrichs, M.J.; Bezabeh, B.Z.; Fleming, R.L.; Dimasi, N.; et al. A biparatopic HER2-targeting antibody-drug conjugate induces tumor regression in primary models refractory to or ineligible for HER2-targeted therapy. Cancer Cell 2016, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Schluep, T.; Gunawan, P.; Ma, L.; Jensen, G.S.; Duringer, J.; Hinton, S.; Richter, W.; Hwang, J. Polymeric tubulysin-peptide nanoparticles with potent antitumor activity. Clin. Cancer Res. 2009, 15, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Patterson, A.W.; Peltier, H.M.; Sasse, F.; Ellman, J.A. Design, synthesis, and biological properties of highly potent tubulysin D analogues. Chemistry 2007, 13, 9534–9541. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.P.; Jagodzinska, M.; Malpezzi, L.; Lazzari, P.; Manca, I.; Greig, I.R.; Sani, M.; Zanda, M. Synthesis and structure-activity relationship studies of novel tubulysin U analogues—Effect on cytotoxicity of structural variations in the tubuvaline fragment. Org. Biomol. Chem. 2013, 11, 2273–2287. [Google Scholar] [CrossRef] [PubMed]
- Crews, P.; Farias, J.J.; Emrich, R.; Keifer, P.A. Milnamide A, an unusual cytotoxic tripeptide from the Marine sponge auletta cf. constricta. J. Org. Chem. 1994, 59, 2932–2934. [Google Scholar] [CrossRef]
- Loganzo, F.; Discafani, C.M.; Annable, T.; Beyer, C.; Musto, S.; Hari, M.; Tan, X.; Hardy, C.; Hernandez, R.; Baxter, M.; et al. HTI-286, a synthetic analogue of the tripeptide hemiasterlin, is a potent antimicrotubule agent that circumvents P-glycoprotein-mediated resistance in vitro and in vivo. Cancer Res. 2003, 63, 1838–1845. [Google Scholar] [PubMed]
- Bai, R.; Durso, N.A.; Sackett, D.L.; Hamel, E. Interactions of the sponge-derived antimitotic tripeptide hemiasterlin with tubulin: Comparison with dolastatin 10 and cryptophycin 1. Biochemistry 1999, 38, 14302–14310. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Benz, F.W.; Wu, Y.; Wang, Q.; Chen, Y.; Chen, X.; Li, H.; Zhang, Y.; Zhang, R.; Yang, J. Structural insights into the pharmacophore of vinca domain inhibitors of microtubules. Mol. Pharmacol. 2016, 89, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Lesma, G.; Sacchetti, A.; Bai, R.; Basso, G.; Bortolozzi, R.; Hamel, E.; Silvani, A.; Vaiana, N.; Viola, G. Hemiasterlin analogues incorporating an aromatic, and heterocyclic type C-terminus: Design, synthesis and biological evaluation. Mol. Divers. 2014, 18, 357–373. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, G.; Cheng, W.; Lo, M.C.; Aulabaugh, A.; Razinkov, V.; Ding, W.; Loganzo, F.; Zask, A.; Ellestad, G. Biophysical characterization of the interactions of HTI-286 with tubulin heterodimer and microtubules. Biochemistry 2003, 42, 13484–13495. [Google Scholar] [CrossRef] [PubMed]
- Loganzo, F.; Hari, M.; Annable, T.; Tan, X.; Morilla, D.B.; Musto, S.; Zask, A.; Kaplan, J.; Minnick, A.A.; May, M.K. Cells resistant to HTI-286 do not overexpress P-glycoprotein but have reduced drug accumulation and a point mutation in α-tubulin. Mol. Cancer Ther. 2004, 3, 1319–1327. [Google Scholar] [PubMed]
- Ratain, M.; Undevia, S.; Janisch, L.; Roman, S.; Mayer, P.; Buckwalter, M.; Foss, D.; Hamilton, B.; Fischer, J.; Bukowski, R. Phase 1 and pharmacological study of HTI-286, a novel antimicrotubule agent: Correlation of neutropenia with time above a threshold serum concentration. Proc. Am. Soc. Clin. Oncol. 2003, 22, 516. [Google Scholar]
- Harada, M.; Tsuchiya, M.; Miyazaki, R.; Inoue, T.; Tanaka, R.; Yanagisawa, Y.; Ito, M.; Ito, Y.; Naito, K. Preclinical evaluation of NC-6201, an antibody/drug-conjugated micelle incorporating novel hemiasterlin analogue E7974. In Proceedings of the AACR 107th Annual Meeting, New Orleans, LA, USA, 16–20 April 2016. [Google Scholar]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; McPhail, A.T. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef] [PubMed]
- Holmes, F.A.; Walters, R.S.; Theriault, R.L.; Forman, A.D.; Newton, L.K.; Raber, M.N.; Buzdar, A.U.; Frye, D.K.; Hortobagyi, G.N. Phase II trial of taxol, an active drug in the treatment of metastatic breast cancer. J. Natl. Cancer Inst. 1991, 83, 1797–1805. [Google Scholar] [CrossRef] [PubMed]
- Guillemard, V.; Saragovi, H.U. Taxane-antibody conjugates afford potent cytotoxicity, enhanced solubility, and tumor target selectivity. Cancer Res. 2001, 61, 694–699. [Google Scholar] [PubMed]
- Ojima, I.; Geng, X.; Wu, X.; Qu, C.; Borella, C.P.; Xie, H.; Wilhelm, S.D.; Leece, B.A.; Bartle, L.M.; Goldmacher, V.S.; et al. Tumor-specific novel taxoid-monoclonal antibody conjugates. J. Med. Chem. 2002, 45, 5620–5623. [Google Scholar] [CrossRef] [PubMed]
- Quiles, S.; Raisch, K.P.; Sanford, L.L.; Bonner, J.A.; Safavy, A. Synthesis and preliminary biological evaluation of high-drug-load paclitaxel-antibody conjugates for tumor-targeted chemotherapy. J. Med. Chem. 2010, 53, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Ojima, I.; Kuduk, S.D.; Chakravarty, S. Recent advances in the medicinal chemistry of taxoid anticancer agents. Adv. Med. Chem. 1999, 4, 69–124. [Google Scholar]
- Singh, R.; Sharma, M.; Joshi, P.; Rawat, D.S. Clinical status of anti-cancer agents derived from marine sources. Anticancer Agents Med. Chem. 2008, 8, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Ter Haar, E.; Kowalski, R.J.; Hamel, E.; Lin, C.M.; Longley, R.E.; Gunasekera, S.P.; Rosenkranz, H.S.; Day, B.W. Discodermolide, a cytotoxic marine agent that stabilizes microtubules more potently than taxol. Biochemistry 1996, 35, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Risinger, A.L.; Peng, J.; Chen, Z.; Hu, L.; Mooberry, S.L. Potent taccalonolides, AF and AJ, inform significant structure-activity relationships and tubulin as the binding site of these microtubule stabilizers. J. Am. Chem. Soc. 2011, 133, 19064–19067. [Google Scholar] [CrossRef] [PubMed]
- Risinger, A.L.; Li, J.; Bennett, M.J.; Rohena, C.C.; Peng, J.; Schriemer, D.C.; Mooberry, S.L. Taccalonolide binding to tubulin imparts microtubule stability and potent in vivo activity. Cancer Res. 2013, 73, 6780–6792. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Risinger, A.L.; Li, J.; Mooberry, S.L. Synthetic reactions with rare taccalonolides reveal the value of C-22,23 epoxidation for microtubule stabilizing potency. J. Med. Chem. 2014, 57, 6141–6149. [Google Scholar] [CrossRef] [PubMed]
- Ferrandina, G.; Mariani, M.; Andreoli, M.; Shahabi, S.; Scambia, G.; Ferlini, C. Novel drugs targeting microtubules: The role of epothilones. Curr. Pharm. Des. 2012, 18, 2793–2803. [Google Scholar] [CrossRef] [PubMed]
- Prota, A.E.; Bargsten, K.; Northcote, P.T.; Marsh, M.; Altmann, K.H.; Miller, J.H.; Diaz, J.F.; Steinmetz, M.O. Structural basis of microtubule stabilization by laulimalide and peloruside A. Angew. Chem. Int. Ed. Engl. 2014, 53, 1621–1625. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, G. Colchicine semisynthetics: Chemotherapeutics for cancer? Curr. Med. Chem. 2013, 20, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Rajak, H.; Dewangan, P.K.; Patel, V.; Jain, D.K.; Singh, A.; Veerasamy, R.; Sharma, P.C.; Dixit, A. Design of combretastatin A-4 analogs as tubulin targeted vascular disrupting agent with special emphasis on their cis-restricted isomers. Curr. Pharm. Des. 2013, 19, 1923–1955. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Zhang, J.; Liu, Z.; Wang, M.; Dong, Y. Developments of combretastatin A-4 derivatives as anticancer agents. Curr. Med. Chem. 2011, 18, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wang, Y.; Wang, T.; Jiang, J.; Botting, C.H.; Liu, H.; Chen, Q.; Yang, J.; Naismith, J.H.; Zhu, X.; Chen, L. Pironetin reacts covalently with cysteine-316 of alpha-tubulin to destabilize microtubule. Nat. Commun. 2016, 7, 12103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, G.D.; Fu, Y.X. Design of next generation antibody drug conjugates. Acta Pharm. Sin. 2013, 48, 1053–1070. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Chemical Structure | Linker Type |
|---|---|---|
| MHH |  | Chemical labile (acid labile) linker |
| DSDM |  | Disulfide-containing reducible linker |
| Sulfo-SPDB |  | Disulfide-containing reducible linker |
| MC-VC-PABC |  | Enzymatically cleavable linker |
| SMCC |  | Non-cleavable bifunctional linker |
| Mal-PEG-NHS |  | Non-cleavable spacer linker |
| GBC |  | Enzyme-labile ß-Glucuronide linker |
| ADC | Status | Therapeutic Area | Target | mAb | Linker | Payload |
|---|---|---|---|---|---|---|
| Trastuzumab emtansine (T-DM1) | Approved | Breast cancer | HER2 | Trastuzumab | SMCC | DM1 a |
| Lorvotuzumab mertansine (IMGN901) | Phase II | Hematologic-blood cancer, sarcoma, neuroblastoma, SCLC, MM | CD56 | huN901 | SPP | DM1 |
| IMGN529 | Phase II | NHL | CD37 | K7153A | SMCC | DM1 |
| IMGN289 | Phase I | Squamous Cell carcinoma, NSCLC, solid tumors | EGFR | J2898A | SMCC | DM1 |
| AMG 172 | Phase I | RCC | CD27L | Anti-CD27L | MCC | DM1 |
| AMG 595 | Phase I | Glioma, AA | EFGRvIII | Anti-EGFRvIII | SMCC | DM1 |
| MLN-2704 | Discontinued | Prostate cancer | PSMA | MLN-591 | SPP | DM1 |
| Cantuzumab mertansine (HuC242-DM1) | Discontinued | NSCLC, pancreatic cancer, colorectal cancer, solid tumors | CanAg | huC242 | SPP | DM1 |
| Mirvetuximab soravtansine (IMGN853) | Phase II | Solid tumors, NSCLC, ovarian cancer | Folate receptor 1 | M9346A | sSPDB | DM4 a |
| Anetumab ravtansine (BAY94-9343) | Phase II | NSCLC, Mesothelioma, solid tumors | Mesothelin | Anti-mesothelin | SPDB | DM4 |
| Coltuximab ravtansine (SAR3419) | Phase II | LBCL | CD19 | huB4 | SPDB | DM4 |
| BT-062 | Phase I/II | MM | CD138 | nBT062 | SPDB | DM4 |
| SAR-428926 | Phase I | Solid tumors | LAMP-1 | 853K3 | SPDB | DM4 |
| SAR-566658 | Phase I | Solid tumors | CA6 | huDS6 | SPDB | DM4 |
| IMGN388 | Discontinued | Solid tumors | Integrin αv | Anti-integrin αv | SPDB | DM4 |
| BIIB015 | Discontinued | Solid tumors | Crypto | Anti-cripto | SPDB | DM4 |
| AVE-9633 | Discontinued | AML | CD33 | huMy9-6 | Undisclosed | DM4 |
| Cantuzumab ravtansine (huC242-DM4) | Discontinued | Gastric cancer, pancreatic cancer | CanAg | huC242 | SPDB | DM4 |
| Adcetris (SGN-35) | Approved | HL, ALCL | CD30 | cAC10 (SGN-30) | VC | MMAE b |
| Glembatumumabvedotin (CDX-011) | Phase II | Breast cancer, melanoma, osteosarcoma, NSCLC | GPNMB | CR-011 | VC | MMAE |
| Pinatuzumab vedotin (DCDT2980S) | Phase II | NHL, DLBCL | CD22 | Anti-CD22 | VC | MMAE |
| Polatuzumab vedotin (DCDS4501A) | Phase II | NHL, DLBCL, CLL | CD79b | Anti-CD79b | VC | MMAE |
| Lifastuzumab vedotin (DNIB0600A) | Phase II | Ovarian cancer, NSCLC | NaPi2b | Anti-NaPi2b | VC | MMAE |
| PSMA ADC | Phase II | Prostate cancer | PSMA | Anti-PSMA | VC | MMAE |
| DMOT-4039A | Phase I | Pancreatic cancer, ovarian cancer | MSLN | MMOT-0530A | VC | MMAE |
| AGS-22M6E (ASG-22CE) | Phase I | Genitourinary cancer, solid tumors | Nectin-4 | Anti-Nectin-4 | VC | MMAE |
| SGN-LIV1A | Phase I | Breast cancer | LIV1 | Anti-LIV1 | VC | MMAE |
| HuMax-TF-ADC | Phase I | Solid tumors | tissue factor | TF-011 | VC | MMAE |
| AGS-15E | Phase I | urothelial | SLITRK6 | AGS15 | VC | MMAE |
| DLYE-5953A | Phase I | breast cancer, NSCLC, solid tumors | LY6E | Anti-Ly6E | VC | MMAE |
| DEDN-6526A | Phase I | Melanoma | EDNRB | Anti-EDNRB | VC | MMAE |
| AGS-67E | Phase I | Hematologic-blood cancer, AML | CD37 | Anti-CD37 | VC | MMAE |
| MLN-0264 | Discontinued | Pancreatic cancer, gastric cancer | GCC | 5F9 | VC | MMAE |
| ASG-5ME | Discontinued | Pancreas cancer, Prostate cancer, gastric cancer | AGS-5 | Anti-AGS-5 | VC | MMAE |
| Bay 79-4620 | Discontinued | Solid tumors | CA9 | 3ee9 | VC | MMAE |
| Sofituzumab vedotin (RG-7458) | Discontinued | Ovarian cancer, pancreatic cancer | MUC16 | Anti-MUC16 | VC | MMAE |
| Vandortuzumab vedotin (DSTP-3086S) | Discontinued | Prostate cancer | STEAP1 | Anti-STEAP1 | VC | MMAE |
| Denintuzumab mafodotin (SGN-CD19A) | Phase II | DLBL, follicular lymphoma | CD19 | Anti-CD19 | MC | MMAF b |
| AGS-16M8F | Phase II | RCC | ENPP3 | Anti-AGS-16 | MC | MMAF |
| Depatuxizumab mafodotin (ABT-414) | Phase II | Glioblastoma multiforme, NSCLC, brain cancer, solid tumors | EGFRvIII | ABT-806 | MC | MMAF |
| ARX-788 | Phase I | Breast cancer | Her2 | Anti-Her2 | PEG4 | MMAF |
| J6M0-mcMMAF (GSK-2857916) | Phase I | MM | BCMA | J6M0 | MC | MMAF |
| Vorsetuzumab mafodotin (SGN-75) | Discontinued | NHL, RCC | CD70 | HIF6 | MC | MMAF |
| PF-06263507 | Discontinued | Solid tumors | 5T4 | Anti-5T4 | MC | MMAF |
| MEDI-547 | Discontinued | Ovarian cancer, solid tumors | EphA2 | 1C1 | MC | MMAF |
| Hu2H11-SPDB-Ex1 | Preclinical | Breast cancer, colon cancer, | EphA2 | hu2H11 | SPDB | Cryptophycin analogue c |
| Hu2H11-SPDB-Ex2 | Preclinical | Breast cancer, colon cancer, | EphA2 | hu2H11 | SPDB | Cryptophycin analogue |
| Hu2H11-SPDB-Ex5 | Preclinical | Breast cancer, colon cancer, | EphA2 | hu2H11 | SPDB | Cryptophycin analogue |
| Hu2H11-SPDB-Ex6 | Preclinical | Breast cancer, colon cancer, | EphA2 | hu2H11 | SPDB | Cryptophycin analogue |
| Hu2H11-Ex17 | Preclinical | Breast cancer, colon cancer, | EphA2 | hu2H11 | Undisclosed | Cryptophycin analogue |
| Hu2H11-Ex18 | Preclinical | Breast cancer, colon cancer, | EphA2 | hu2H11 | Undisclosed | Cryptophycin analogue |
| Hu2H11-Ex19 | Preclinical | Breast cancer, colon cancer, | EphA2 | hu2H11 | Undisclosed | Cryptophycin analogue |
| VDC-886 | Preclinical | Prostate cancer, breast cancer, NHL | pl-CS | DBL1-ID2a | Undisclosed | KT886 d |
| NC-6201 | Preclinical | Breast cancer, pancreatic cancer, colon cancer | EGFR | NCAB001 | PEG-poly(glutamic acid benzyl ester) | E7974 d |
| MEDI-4276 (AZ13599185-Trastuzumab) | Phase I | Breast cancer, gastric cancer | HER2 | 39S | MC | AZ13599185 e |
| 131I-TUB-OH-Trastuzumab | Preclinical | Breast cancer | HER2 | Trastuzumab | - | TUB-OH e |
| 131I-TUB-OMOM-Trastuzumab | Preclinical | Breast cancer | HER2 | Trastuzumab | - | TUB-OMOM e |
| DX-109 | Preclinical | Breast cancer | HER2 | anti-HER2 | Thioether | Tub-001 e |
| αCD30–glucQ-Tub | Preclinical | HL, ALCL | CD30 | AC10 | Quaternary Ammonium | Tubulysin M e |
| α-CEA-680-PTX | Preclinical | Pancreatic cancer | CEA | Anti-CEA | Hemisuccinamide-DyLight™ 680 − 4 × PEG | Paclitaxel f |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Lin, Z.; Arnst, K.E.; Miller, D.D.; Li, W. Tubulin Inhibitor-Based Antibody-Drug Conjugates for Cancer Therapy. Molecules 2017, 22, 1281. https://doi.org/10.3390/molecules22081281
Chen H, Lin Z, Arnst KE, Miller DD, Li W. Tubulin Inhibitor-Based Antibody-Drug Conjugates for Cancer Therapy. Molecules. 2017; 22(8):1281. https://doi.org/10.3390/molecules22081281
Chicago/Turabian StyleChen, Hao, Zongtao Lin, Kinsie E. Arnst, Duane D. Miller, and Wei Li. 2017. "Tubulin Inhibitor-Based Antibody-Drug Conjugates for Cancer Therapy" Molecules 22, no. 8: 1281. https://doi.org/10.3390/molecules22081281
APA StyleChen, H., Lin, Z., Arnst, K. E., Miller, D. D., & Li, W. (2017). Tubulin Inhibitor-Based Antibody-Drug Conjugates for Cancer Therapy. Molecules, 22(8), 1281. https://doi.org/10.3390/molecules22081281