Carbohydrate-Based Host-Guest Complexation of Hydrophobic Antibiotics for the Enhancement of Antibacterial Activity


Abstract
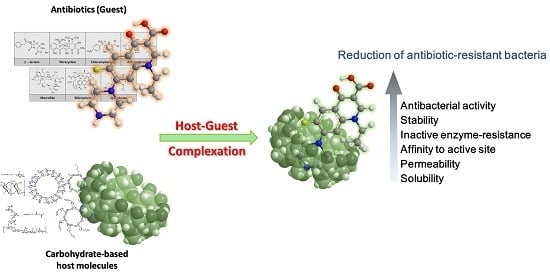
:
1. Introduction
2. Mechanisms of Host-Guest Complexation in Drug Delivery
3. Mechanisms of Antibacterial Resistance
4. β-Cyclodextrin and Its Derivatives
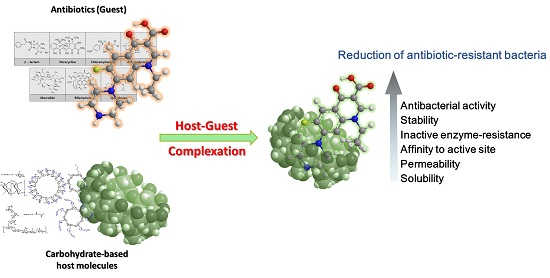
4.1. Methicillin/per-6(4-methoxylbenzyl)-amino-6-deoxy-β-cyclodextrin Complex
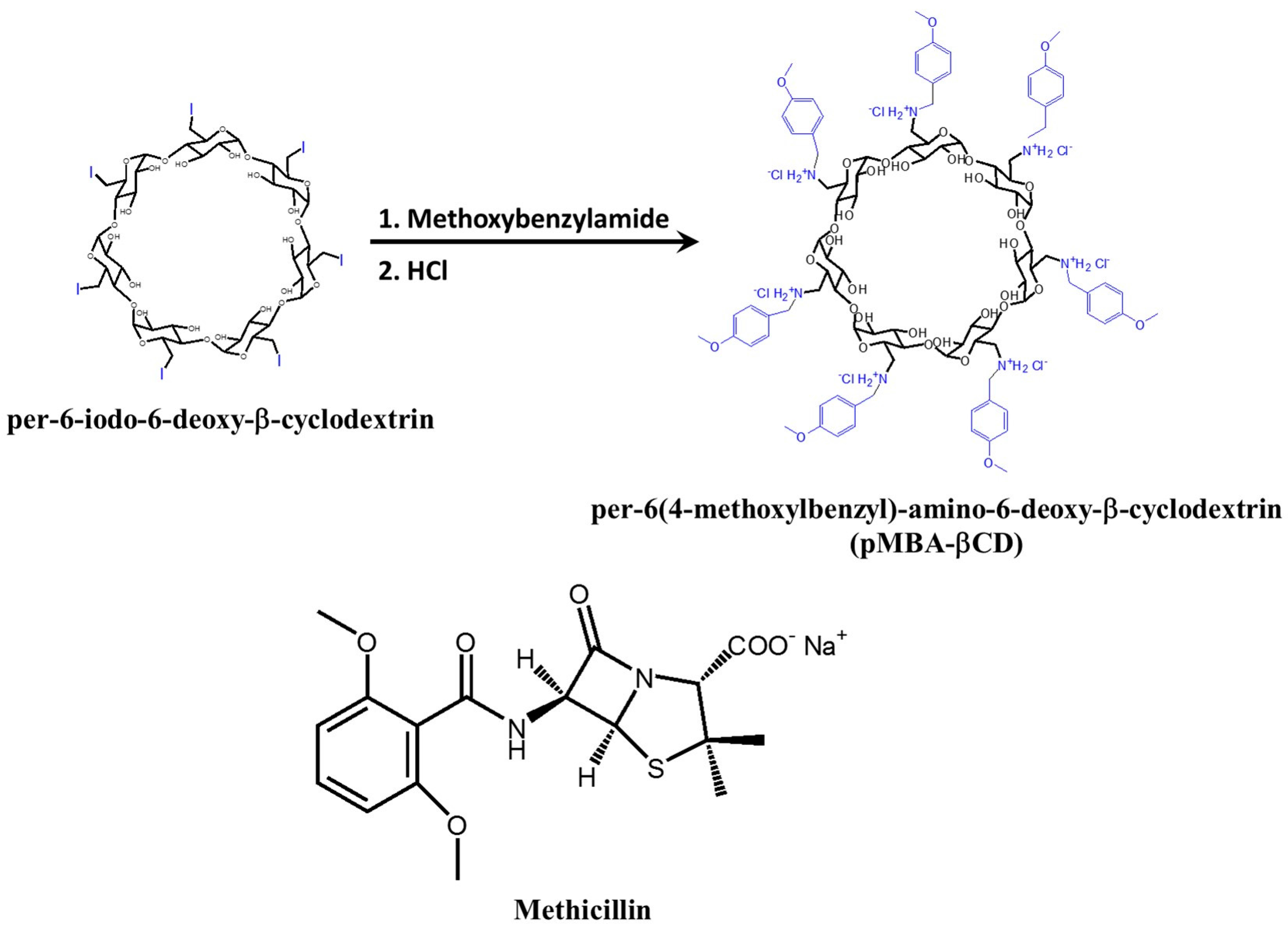
4.2. Ciprofloxacin/mono-6-Deoxy-6-aminoethylamino-β-cyclodextrin Complex
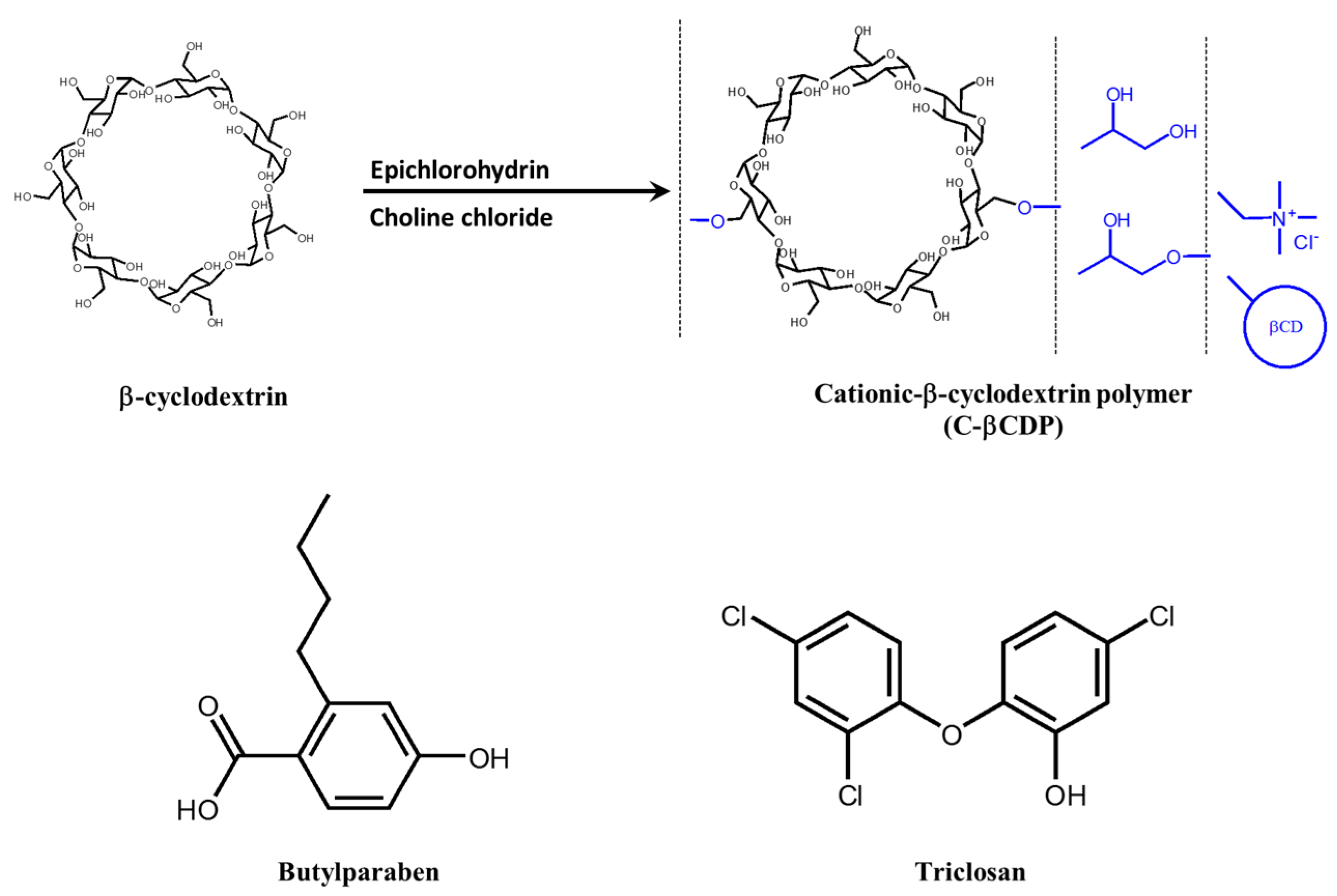
4.3. Butylparaben and Triclosan with Cationic β-Cyclodextrin Polymer
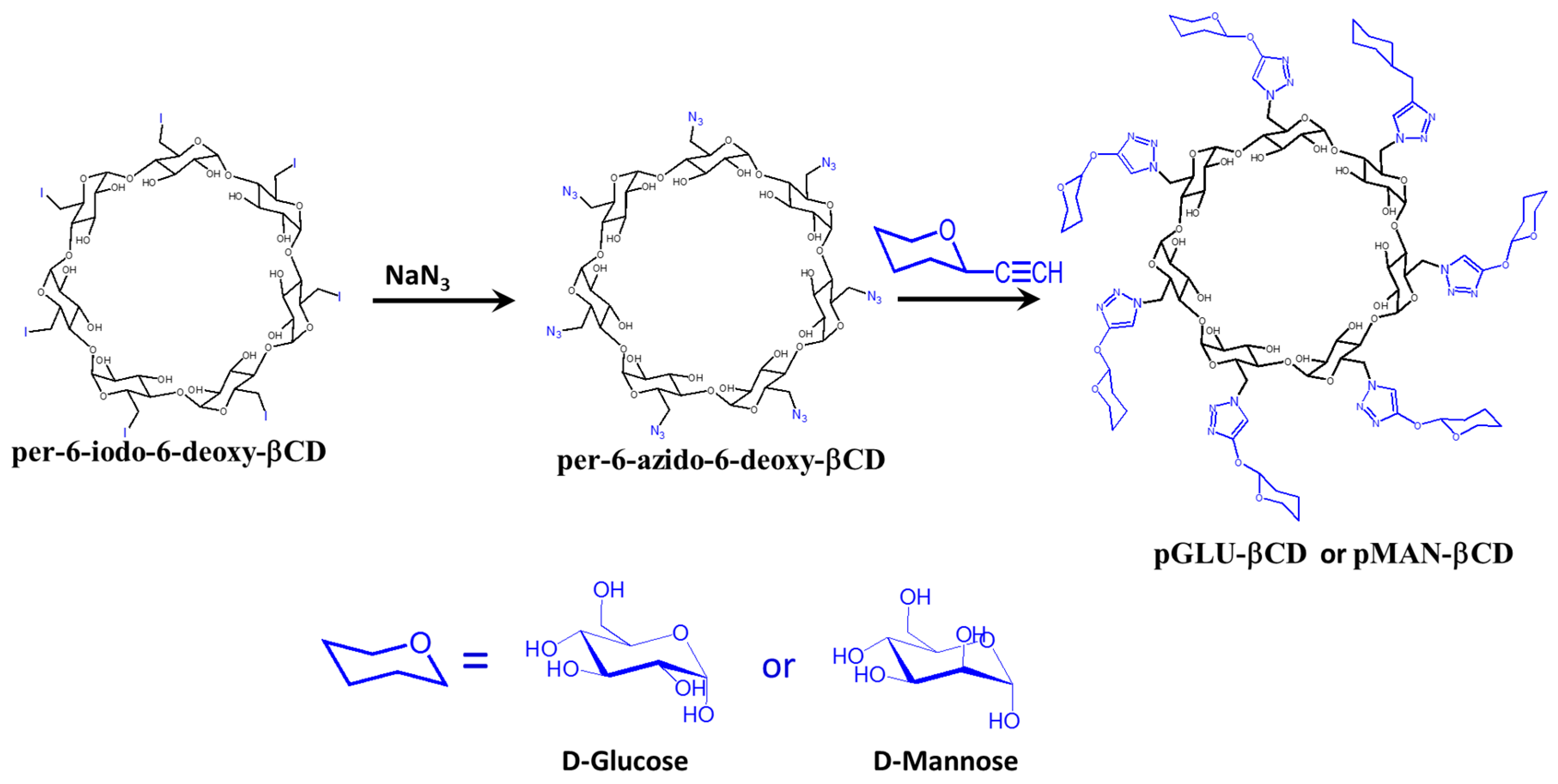
4.4. Sugar-Grafted β-Cyclodextrin for Delivering Antibiotics
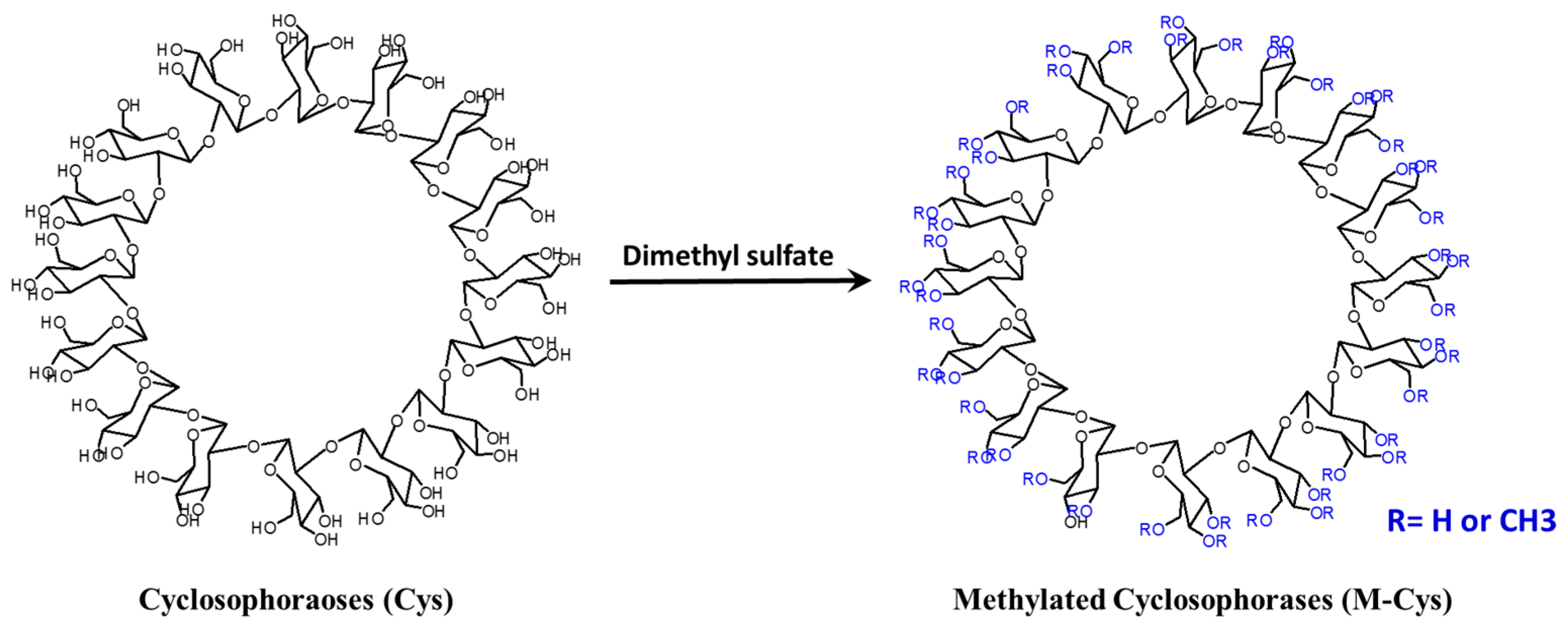
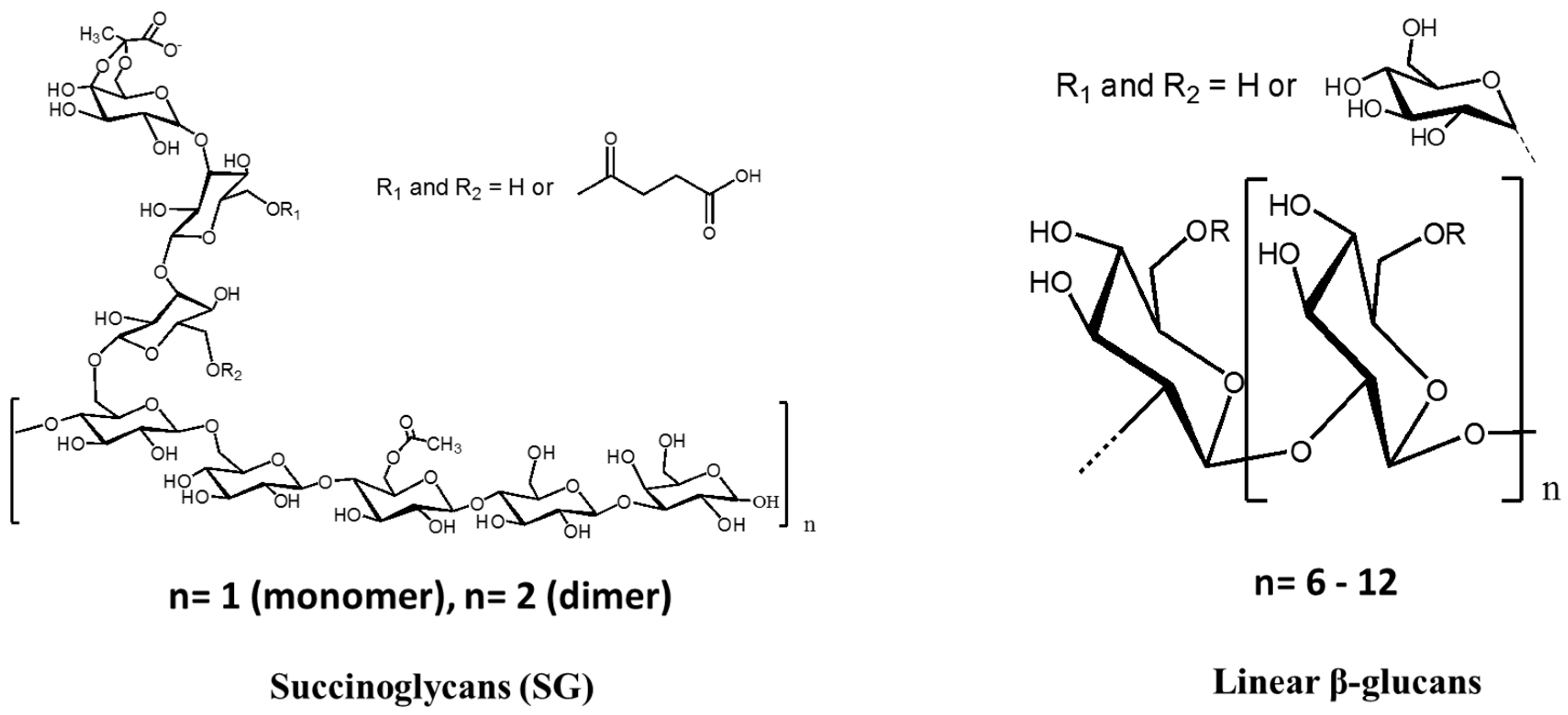
5. Cyclosophoroase and Its Derivatives
6. Linear Oligosaccharides
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Levy, S.B.; Marshall, B. Antibacterial resistance worldwide: Causes, challenges and responses. Nat. Med. 2004, 10, S122–S129. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.L.; Manna, A. Compositions and Methods for Affecting Virulence Determinants in Bacteria. U.S. Patent 7339043 B2, 4 March 2008. [Google Scholar]
- Nathan, C.; Cars, O. Antibiotic resistance—Problems, progress, and prospects. N. Engl. J. Med. 2014, 371, 1761–1763. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, M.A.; Walsh, C.T. Antibiotics for emerging pathogens. Science 2009, 325, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C. Where will new antibiotics come from? Nat. Rev. Microbiol. 2003, 1, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Neu, H.C. The crisis in antibiotic resistance. Science 1992, 257, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Jukes, T.H. Antibiotics in animal feeds and animal production. Bioscience 1972, 22, 526–534. [Google Scholar] [CrossRef]
- O’Neill, J. Antimicrobial Resistance: Tackling a crisis for the health and wealth of nations. Rev. Antimicrob. Resist. 2014, 1–16. [Google Scholar]
- Walsh, C.T.; Wencewicz, T.A. Prospects for new antibiotics: A molecule-centered perspective. J. Antibiot. 2014, 67, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Alanis, A.J. Resistance to antibiotics: Are we in the post-antibiotic era? Arch. Med. Res. 2005, 36, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Davies, J. Inactivation of antibiotics and the dissemination of resistance genes. Science 1994, 264, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.D. Bacterial resistance to antibiotics: Enzymatic degradation and modification. Adv. Drug Deliv. Rev. 2005, 57, 1451–1470. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.R.; Betts, M.J. Carbapenems: The pinnacle of the β-lactam antibiotics or room for improvement? J. Antimicrob. Chemother. 2000, 45, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Dalhoff, A.; Nasu, T.; Okamoto, K. Beta-lactamase stability of faropenem. Chemotherapy 2003, 49, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Yuen, K.-Y.; Kumana, C.R. Clinical role of β-lactam/β-lactamase inhibitor combinations. Drugs 2003, 63, 1511–1524. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.E.; Blosser-Middleton, R.S.; Critchley, I.A.; Karlowsky, J.A.; Thornsberry, C.; Sahm, D.F. Activity of faropenem, a new furanem, against european respiratory pathogens collected during 2000–2001: A comparison with other β-lactam agents. J. Antimicrob. Chemother. 2003, 51, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Wiens, T.; Redelmeier, T.; Av-Gay, Y. Development of a liposome formulation of ethambutol. Antimicrob. Agents Chemother. 2004, 48, 1887–1888. [Google Scholar] [CrossRef] [PubMed]
- Khuller, G.; Kapur, M.; Sharma, S. Liposome technology for drug delivery against mycobacterial infections. Curr. Pharm. Des. 2004, 10, 3263–3274. [Google Scholar] [CrossRef] [PubMed]
- Engelsen, S.B.; Cros, S.; Mackie, W.; Perez, S. A molecular builder for carbohydrates: Application to polysaccharides and complex carbohydrates. Biopolymers 1996, 39, 417–433. [Google Scholar] [CrossRef]
- Sharon, N.; Lis, H. Carbohydrates in cell recognition. Sci. Am. 1993, 268, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Sewalt, V.; Shanahan, D.; Gregg, L.; La Marta, J.; Carrillo, R. The generally recognized as safe (gras) process for industrial microbial enzymes. Ind. Biotechnol. 2016, 12, 295–302. [Google Scholar] [CrossRef]
- Ramesh, H.P.; Tharanathan, R.N. Carbohydrates—The renewable raw materials of high biotechnological value. Crit. Rev. Biotechnol. 2003, 23, 149–173. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.; Jung, S. Supramolecular complexation of carbohydrates for the bioavailability enhancement of poorly soluble drugs. Molecules 2015, 20, 19620–19646. [Google Scholar] [CrossRef] [PubMed]
- Del Valle, E.M.M. Cyclodextrins and their uses: A review. Process Biochem. 2004, 39, 1033–1046. [Google Scholar] [CrossRef]
- Szejtli, J. Introduction and general overview of cyclodextrin chemistry. Chem. Rev. 1998, 98, 1743–1754. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Bharti, N.; Madan, J.; Hiremath, S. Characterization of cyclodextrin inclusion complexes—A review. J. Pharm. Sci. Technol. 2010, 2, 171–183. [Google Scholar]
- Hedges, A.R. Industrial applications of cyclodextrins. Chem. Rev. 1998, 98, 2035–2044. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Jarho, P.; Masson, M.; Järvinen, T. Cyclodextrins in drug delivery. Expert Opin. Drug Deliv. 2005, 2, 335–351. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, K.; Okada, Y.; Horiyama, S.; Utamura, T.; Higashiura, T.; Ikeda, M. Preparation of cyclosophoraose-A and its complex-forming ability. J. Incl. Phenom. 1984, 2, 891–899. [Google Scholar] [CrossRef]
- Breedveld, M.W.; Miller, K.J. Cyclic beta-glucans of members of the family rhizobiaceae. Microbiol. Rev. 1994, 58, 145–161. [Google Scholar] [PubMed]
- Jeong, D.; Ki Kim, H.; Jeong, J.-P.; Dindulkar, S.D.; Cho, E.; Yang, Y.-H.; Jung, S. Cyclosophoraose/cellulose hydrogels as an efficient delivery system for galangin, a hydrophobic antibacterial drug. Cellulose 2016, 23, 2609–2625. [Google Scholar] [CrossRef]
- Siemoneit, U.; Schmitt, C.; Alvarez-Lorenzo, C.; Luzardo, A.; Otero-Espinar, F.; Concheiro, A.; Blanco-Méndez, J. Acrylic/cyclodextrin hydrogels with enhanced drug loading and sustained release capability. Int. J. Pharm. 2006, 312, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-H.; Kim, K.-T.; Choi, J.-M.; Tahir, M.N.; Cho, E.; Choi, Y.-J.; Lee, I.-S.; Jung, S.-H. Solubilization of pyrimethamine, antibacterial drug, by low-molecular-weight succinoglycan dimers isolated from shinorhizobium meliloti. Bull. Korean Chem. Soc. 2012, 33, 2731–2736. [Google Scholar] [CrossRef]
- Balasubramanian, D.; Raman, B.; Sundari, C.S. Polysaccharides as amphiphiles. J. Am. Chem. Soc. 1993, 115, 74–77. [Google Scholar] [CrossRef]
- Wang, L.-X.; Wang, Y.; Pellock, B.; Walker, G.C. Structural characterization of the symbiotically important low-molecular-weight succinoglycan ofsinorhizobium meliloti. J. Bacteriol. 1999, 181, 6788–6796. [Google Scholar] [PubMed]
- Cho, E.; Choi, J.M.; Kim, H.; Lee, I.-S.; Jung, S. Hydrophobic interactions of succinoglycan dimers isolated from sinorhizobium meliloti with hydrophobic fluorescence probes, 8-anilino-1-naphthalenesulfonate and 6-p-toluidino-2-naphthalenesulfonate. Bull. Korean Chem. Soc. 2011, 32, 4071–4074. [Google Scholar] [CrossRef]
- Cho, E.; Choi, J.-M.; Jung, S.-H. Analysis on the molecular interactions between tobramycin, an aminoglycoside antibiotic and periplasmic glucans isolated from pseudomonas syringae. Bull. Korean Chem. Soc. 2011, 32, 347–349. [Google Scholar] [CrossRef]
- Athanassiou, G.; Michaleas, S.; Lada-Chitiroglou, E.; Tsitsa, T.; Antoniadou-Vyza, E. Antimicrobial activity of β-lactam antibiotics against clinical pathogens after molecular inclusion in several cyclodextrins. A novel approach to bacterial resistance. J. Pharm. Pharmacol. 2003, 55, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Stella, V.J.; Rajewski, R.A. Cyclodextrins: Their future in drug formulation and delivery. Pharm. Res. 1997, 14, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Magnúsdóttir, A.; Másson, M.; Sigurjónsdóttir, J.F. Self-association and cyclodextrin solubilization of drugs. J. Pharm. Sci. 2002, 91, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Zhang, L.; Guo, S. Mechanisms of lead biosorption on cellulose/chitin beads. Water Res. 2005, 39, 3755–3762. [Google Scholar] [CrossRef] [PubMed]
- Cramer, F.; Saenger, W.; Spatz, H.-C. Inclusion compounds. Xix. 1a the formation of inclusion compounds of α-cyclodextrin in aqueous solutions. Thermodynamics and kinetics. J. Am. Chem. Soc. 1967, 89, 14–20. [Google Scholar] [CrossRef]
- Stella, V.J.; Rao, V.M.; Zannou, E.A.; Zia, V. Mechanisms of drug release from cyclodextrin complexes. Adv. Drug Deliv. Rev. 1999, 36, 3–16. [Google Scholar] [CrossRef]
- Loftsson, T.; Moya-Ortega, M.D.; Alvarez-Lorenzo, C.; Concheiro, A. Pharmacokinetics of cyclodextrins and drugs after oral and parenteral administration of drug/cyclodextrin complexes. J. Pharm. Pharmacol. 2016, 68, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Vogensen, S.B.; Brewster, M.E.; Konráðsdóttir, F. Effects of cyclodextrins on drug delivery through biological membranes. J. Pharm. Sci. 2007, 96, 2532–2546. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Chang, C.-E.; Gilson, M.K. Calculation of cyclodextrin binding affinities: Energy, entropy, and implications for drug design. Biophys. J. 2004, 87, 3035–3049. [Google Scholar] [CrossRef] [PubMed]
- Uekama, K. Design and evaluation of cyclodextrin-based drug formulation. Chem. Pharm. Bull. 2004, 52, 900–915. [Google Scholar] [CrossRef] [PubMed]
- Pistolis, G.; Balomenou, I. Cyclodextrin cavity size effect on the complexation and rotational dynamics of the laser dye 2,5-diphenyl-1,3,4-oxadiazole: From singly occupied complexes to their nanotubular self-assemblies. J. Phys. Chem. B 2006, 110, 16428–16438. [Google Scholar] [CrossRef] [PubMed]
- Frijlink, H.W.; Visser, J.; Hefting, N.R.; Oosting, R.; Meijer, D.K.; Lerk, C.F. The pharmacokinetics of β-cyclodextrin and hydroxypropyl-β-cyclodextrin in the rat. Pharm. Res. 1990, 7, 1248–1252. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, F.; Kurihara, M.; Horiuchi, Y.; Utsuki, T.; Uekama, K.; Yamasaki, M. Preparation of heptakis(2,6-di-O-ethyl)-β-cyclodextrin and its nuclear magnetic resonance spectroscopic characterization. Pharm. Res. 1993, 10, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Uekama, K.; Arima, H.; Irie, T.; Matsubara, K.; Kuriki, T. Sustained release of buserelin acetate, a luteinizing hormone-releasing hormone agonist, from an injectable oily preparation utilizing ethylated β-cyclodextrin. J. Pharm. Pharmacol. 1989, 41, 874–876. [Google Scholar] [CrossRef] [PubMed]
- Shiotani, K.; Uehata, K.; Irie, T.; Hirayama, F.; Uekama, K. Characterization of the inclusion mode of beta-cyclodextrin sulfate and its effect on the chlorpromazine-induced hemolysis of rabbit erighrocytes. Chem. Pharm. Bull. 1994, 42, 2332–2337. [Google Scholar] [CrossRef] [PubMed]
- Shiotani, K.; Irie, T.; Uekama, K.; Ishimaru, Y. Cyclodextrin sulfates in parenteral use: Protection against gentamicin nephrotoxicity in the rat. Eur. J. Pharm. Sci. 1995, 3, 139–151. [Google Scholar] [CrossRef]
- Cram, D.J. The design of molecular hosts, guests, and their complexes (nobel lecture). Angew. Chem. Int. Ed. 1988, 27, 1009–1020. [Google Scholar] [CrossRef]
- Connors, K.A. Binding Constants: The Measurement of Molecular Complex Stability 1987; J. Wiley & Sons: New York, NY, USA, 1991. [Google Scholar]
- Bouchemal, K.; Mazzaferro, S. How to conduct and interpret itc experiments accurately for cyclodextrin-guest interactions. Drug Discov. Today 2012, 17, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Cram, D.J.; Cram, J.M. Design of complexes between synthetic hosts and organic guests. Acc. Chem. Res. 1978, 11, 8–14. [Google Scholar] [CrossRef]
- Palamini, M.; Canciani, A.; Forneris, F. Identifying and visualizing macromolecular flexibility in structural biology. Front. Mol. Biosci. 2016, 3, 47. [Google Scholar] [CrossRef] [PubMed]
- Landers, T.F.; Cohen, B.; Wittum, T.E.; Larson, E.L. A review of antibiotic use in food animals: Perspective, policy, and potential. Public Health Rep. 2012, 127, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Chambers, H. Methicillin-resistant staphylococcus aureus. Mechanisms of resistance and implications for treatment. Postgrad. Med. 2001, 109, 43–50. [Google Scholar] [PubMed]
- Reynolds, P.E. Structure, biochemistry and mechanism of action of glycopeptide antibiotics. Eur. J. Clin. Microbiol. Infect. Dis. 1989, 8, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Pootoolal, J.; Neu, J.; Wright, G.D. Glycopeptide antibiotic resistance. Ann. Rev. Pharmacol. Toxicol. 2002, 42, 381–408. [Google Scholar] [CrossRef] [PubMed]
- Sieradzki, K.; Tomasz, A. Suppression of glycopeptide resistance in a highly teicoplanin-resistant mutant of Staphylococcus aureus by transposon inactivation of genes involved in cell wall synthesis. Microb. Drug Resist. 1998, 4, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Halling-Sørensen, B.; Sengeløv, G.; Tjørnelund, J. Toxicity of tetracyclines and tetracycline degradation products to environmentally relevant bacteria, including selected tetracycline-resistant bacteria. Arch. Environ. Contam. Toxicol. 2002, 42, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Chopra, I.; Roberts, M. Tetracycline antibiotics: Mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232–260. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Nishino, K.; Roberts, M.C.; Tolmasky, M.; Aminov, R.I.; Zhang, L. Mechanisms of antibiotic resistance. Front. Microbiol. 2015, 6, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, J.M.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Jevons, M.P. “Celbenin”-resistant staphylococci. Br. Med. J. 1961, 1, 124–125. [Google Scholar] [CrossRef]
- Philippon, A.; Arlet, G.; Jacoby, G.A. Plasmid-determined ampc-type β-lactamases. Antimicrob. Agents Chemother. 2002, 46, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.F.; Meroueh, S.O.; Mobashery, S. Bacterial resistance to β-lactam antibiotics: Compelling opportunism, compelling opportunity. Chem. Rev. 2005, 105, 395–424. [Google Scholar] [CrossRef] [PubMed]
- Tenover, F.C. Mechanisms of antimicrobial resistance in bacteria. Am. J. Med. 2006, 119, S3–S10. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.-Z. Methicillin/per-6-(4-methoxylbenzyl)-amino-6-deoxy-β-cyclodextrin 1:1 complex and its potentiation in vitro against methicillin-resistant staphylococcus aureus. J. Antibiot. 2013, 66, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Melo, M.; Varanda, F.; Dohrn, R.; Marrucho, I. Solubility of ciprofloxacin and moxifloxacin in different solvents: The effect of the hcl group. In Proceedings of the EMPROMER, 2nd Mercosur Congress on Chemical Engineering and 4th Mercosur Congress on Process Systems Engineering, Rio de Janeiro, Brazil, 14–18 August 2005. [Google Scholar]
- Jianbin, C.; Liang, C.; Hao, X.; Dongpin, M. Preparation and study on the solid inclusion complex of ciprofloxacin with β-cyclodextrin. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2002, 58, 2809–2815. [Google Scholar] [CrossRef]
- Chao, J.; Meng, D.; Li, J.; Xu, H.; Huang, S. Preparation and study on the novel solid inclusion complex of ciprofloxacin with hp-β-cyclodextrin. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2004, 60, 729–734. [Google Scholar] [CrossRef]
- Choi, J.M.; Park, K.; Lee, B.; Jeong, D.; Dindulkar, S.D.; Choi, Y.; Cho, E.; Park, S.; Yu, J.-H.; Jung, S. Solubility and bioavailability enhancement of ciprofloxacin by induced oval-shaped mono-6-deoxy-6-aminoethylamino-β-cyclodextrin. Carbohydr. Polym. 2017, 163, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Aithal, K.S.; Udupa, N. Physicochemical study of ciprofloxacin with β-cyclodextrin. Pharm. Pharmacol. Commun. 1996, 2, 451–455. [Google Scholar]
- Liu, T.; Liu, H.; Wu, Z.; Chen, T.; Zhou, L.; Liang, Y.; Ke, B.; Huang, H.; Jiang, Z.; Xie, M. The use of poly (methacrylic acid) nanogel to control the release of amoxycillin with lower cytotoxicity. Mater. Sci. Eng. C 2014, 43, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Soni, M.; Burdock, G.; Taylor, S.; Greenberg, N. Safety assessment of propyl paraben: A review of the published literature. Food Chem. Toxicol. 2001, 39, 513–532. [Google Scholar] [CrossRef]
- McMurry, L.M.; Oethinger, M.; Levy, S.B. Triclosan targets lipid synthesis. Nature 1998, 394, 531–532. [Google Scholar] [CrossRef] [PubMed]
- Heath, R.J.; Li, J.; Roland, G.E.; Rock, C.O. Inhibition of the Staphylococcus aureus NADPH-dependent enoyl-acyl carrier protein reductase by triclosan and hexachlorophene. J. Biol. Chem. 2000, 275, 4654–4659. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Furue, M.; Nozakura, S.I. Optical resolution of mandelic acid derivatives by column chromatography on crosslinked cyclodextrin gels. J. Polym. Sci. Part A Polym. Chem. 1978, 16, 189–196. [Google Scholar] [CrossRef]
- Li, J.; Xiao, H.; Li, J.; Zhong, Y. Drug carrier systems based on water-soluble cationic β-cyclodextrin polymers. Int. J. Pharm. 2004, 278, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Guan, Y.; Xiao, H. Preparation and characterization of inclusion complexes of a cationic β-cyclodextrin polymer with butylparaben or triclosan. Int. J. Pharm. 2008, 357, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.P. Rapid test for distinguishing membrane-active antibacterial agents. J. Microbiol. Methods 2006, 67, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lee, J.; Castillo, R.; Hixon, M.; Pujol, C.; Doppalapudi, V.; Shepard, H.; Wahl, G.; Lobl, T.; Chan, M. A novel antibacterial agent with broad-spectrum activity and enhanced potency against beta-lactamase producing strains. J. Antimicrob. Chemother. 2002, 46, 1262–1268. [Google Scholar] [CrossRef]
- Pauli, A.; Schilcher, H. Specific selection of essential oil compounds for treatment of children’s infection diseases. Pharmaceuticals 2004, 1, 1–30. [Google Scholar] [CrossRef]
- Mizuba, S.; Sheikh, W. Antimicrobial efficacy of potassium salts of four parabens. J. Ind. Microbiol. Biotechnol. 1987, 1, 363–369. [Google Scholar] [CrossRef]
- Li, M.; Neoh, K.G.; Xu, L.; Yuan, L.; Leong, D.T.; Kang, E.-T.; Chua, K.L.; Hsu, L.Y. Sugar-grafted cyclodextrin nanocarrier as a “trojan horse” for potentiating antibiotic activity. Pharm. Res. 2016, 33, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Postma, P.; Lengeler, J. Phosphoenolpyruvate: Carbohydrate phosphotransferase system of bacteria. Microbiol. Rev. 1985, 49, 232. [Google Scholar] [PubMed]
- Park, S. Cyclic glucans enhance solubility of bioavailable flavonoids. Molecules 2016, 21, 1556. [Google Scholar] [CrossRef] [PubMed]
- Markham, K.R. Techniques of Flavonoid Identification; Academic Press London: London, UK, 1982; Volume 31. [Google Scholar]
- Cushnie, T.T.; Lamb, A.J. Antimicrobial activity of flavonoids. Int. J. Antimicrob. Agents 2005, 26, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Eumkeb, G.; Sakdarat, S.; Siriwong, S. Reversing β-lactam antibiotic resistance of Staphylococcus aureus with galangin from alpinia officinarum hance and synergism with ceftazidime. Phytomedicine 2010, 18, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Betts, J.W.; Sharili, A.S.; Phee, L.M.; Wareham, D.W. In vitro activity of epigallocatechin gallate and quercetin alone and in combination versus clinical isolates of methicillin-resistant Staphylococcus aureus. J. Nat. Prod. 2015, 78, 2145–2148. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.-X.; Lee, S.F. Activity of plant flavonoids against antibiotic-resistant bacteria. Phytother. Res. 2001, 15, 39–43. [Google Scholar] [CrossRef]
- Chebil, L.; Humeau, C.; Anthoni, J.; Dehez, F.; Engasser, J.-M.; Ghoul, M. Solubility of flavonoids in organic solvents. J. Chem. Eng. Data 2007, 52, 1552–1556. [Google Scholar] [CrossRef]
- Kumar, S.; Pandey, A.K. Chemistry and biological activities of flavonoids: An overview. Sci. World J. 2013, 2013, 162750. [Google Scholar] [CrossRef] [PubMed]
- Denny, B.J.; Lambert, P.A.; West, P.W. The flavonoid galangin inhibits the L1 metallo-β-lactamase from Stenotrophomonas maltophilia. FEMS Microbiol. Lett. 2002, 208, 21–24. [Google Scholar] [CrossRef]
- Kim, H.; Choi, J.; Choi, Y.; Tahir, M.; Yang, Y.-H.; Cho, E.; Jung, S. Enhanced solubility of galangin based on the complexation with methylated microbial cyclosophoraoses. J. Incl. Phenom. Macrocycl. Chem. 2014, 79, 291–300. [Google Scholar] [CrossRef]
- Kang, S.; Lee, S.; Kwon, C.; Jung, S. Solubility enhancement of flavonoids by cyclosophoraose isolated from rhizobium meliloti 2011. J. Microbiol. Biotechnol. 2006, 16, 791–794. [Google Scholar]
- Higuchi, T.; Sato, Y.; Murasugi, S. Use of Flavone Derivatives for Induction of β-Lactam-Sensitivity of Mrsa. U.S. Patent 6294526 B1, 25 September 2001. [Google Scholar]
- Wang, Q.; Xie, M. Antibacterial activity and mechanism of luteolin on Staphylococcus aureus. Wei Sheng Wu Xue Bao 2010, 50, 1180–1184. [Google Scholar] [PubMed]
- Lee, S.; Seo, D.-H.; Park, H.-L.; Choi, Y.; Jung, S. Solubility enhancement of a hydrophobic flavonoid, luteolin by the complexation with cyclosophoraoses isolated from rhizobium meliloti. Antonie Van Leeuwenhoek 2003, 84, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Felt, O.; Buri, P.; Gurny, R. Chitosan: A unique polysaccharide for drug delivery. Drug Dev. Ind. Pharm. 1998, 24, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Tønnesen, H.H.; Karlsen, J. Alginate in drug delivery systems. Drug Dev. Ind. Pharm. 2002, 28, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Brogden, R.; Pinder, R.; Sawyer, P.R.; Speight, T.; Avery, G. Tobramycin: A review of its antibacterial and pharmacokinetic properties and therapeutic use. Drugs 1976, 12, 166–200. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Sample | MRSA COL | MRSA USA300 |
|---|---|---|
| Methicillin | >128 | >128 |
| HP-βCD/methicillin | >64 | >64 |
| pMBA-βCD/methicillin | 2.0–4.0 | 2.0–4.0 |
| Test Sample | Stability Constant (M−1) | Antibacterial Activity 1 |
|---|---|---|
| Ciprofloxacin | - | 5.78 [76] |
| Β CD | 29.84 [76] | 5.58 [76] |
| 29.1 [77] | ||
| HP-βCD | 278 [75] | |
| mET-βCD | 627.3 [76] | 8.825 [76] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, D.; Joo, S.-W.; Shinde, V.V.; Cho, E.; Jung, S. Carbohydrate-Based Host-Guest Complexation of Hydrophobic Antibiotics for the Enhancement of Antibacterial Activity. Molecules 2017, 22, 1311. https://doi.org/10.3390/molecules22081311
Jeong D, Joo S-W, Shinde VV, Cho E, Jung S. Carbohydrate-Based Host-Guest Complexation of Hydrophobic Antibiotics for the Enhancement of Antibacterial Activity. Molecules. 2017; 22(8):1311. https://doi.org/10.3390/molecules22081311
Chicago/Turabian StyleJeong, Daham, Sang-Woo Joo, Vijay Vilas Shinde, Eunae Cho, and Seunho Jung. 2017. "Carbohydrate-Based Host-Guest Complexation of Hydrophobic Antibiotics for the Enhancement of Antibacterial Activity" Molecules 22, no. 8: 1311. https://doi.org/10.3390/molecules22081311
APA StyleJeong, D., Joo, S. -W., Shinde, V. V., Cho, E., & Jung, S. (2017). Carbohydrate-Based Host-Guest Complexation of Hydrophobic Antibiotics for the Enhancement of Antibacterial Activity. Molecules, 22(8), 1311. https://doi.org/10.3390/molecules22081311