The Inhibitory Effects of Purple Sweet Potato Color on Hepatic Inflammation Is Associated with Restoration of NAD+ Levels and Attenuation of NLRP3 Inflammasome Activation in High-Fat-Diet-Treated Mice


Abstract
:1. Introduction
2. Results
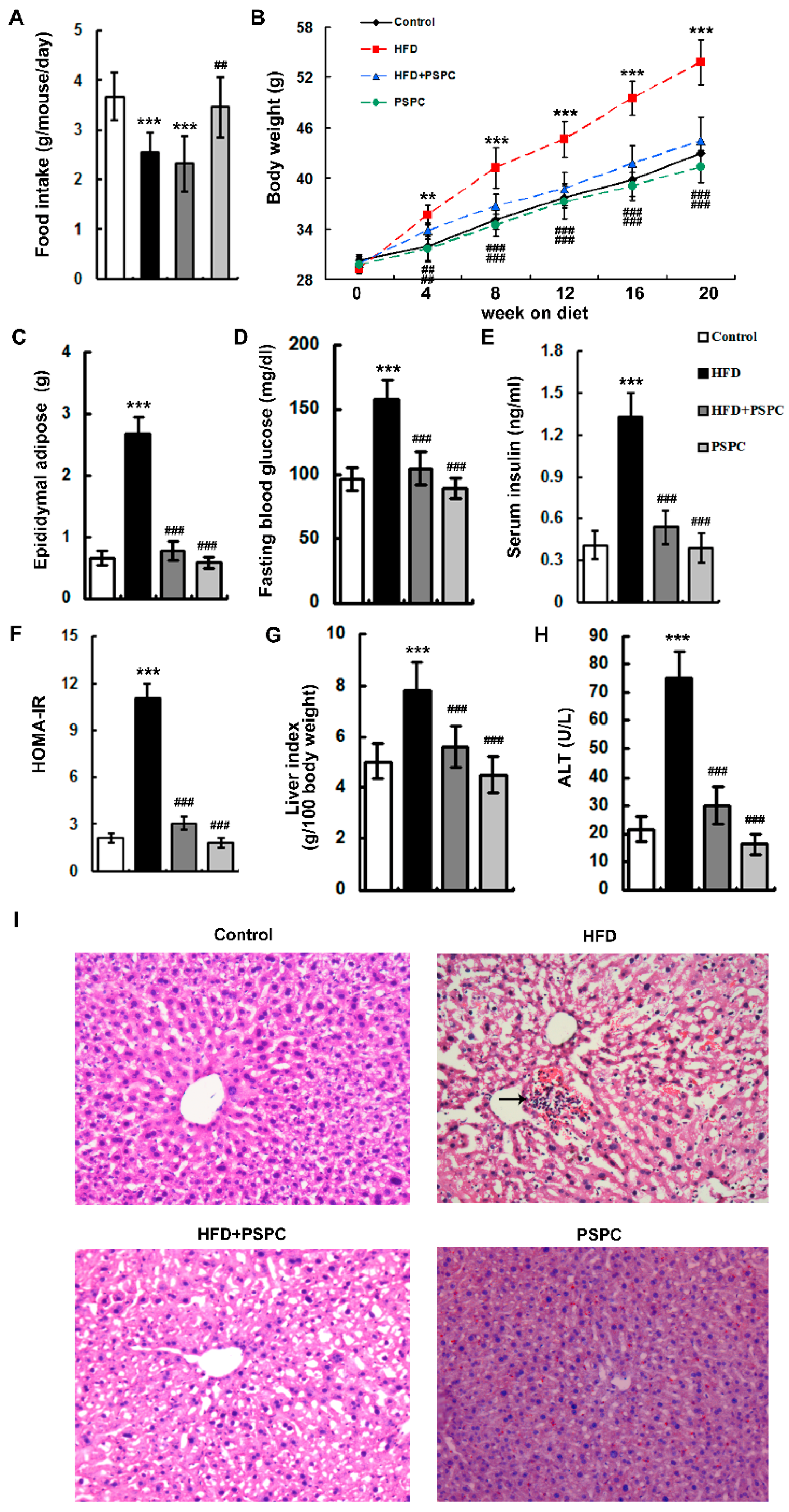
2.1. PSPC Ameliorates Obesity and Liver Injury in HFD-Treated Mice
2.2. PSPC Attenuates Oxidative Stress in HFD-Treated Mouse Livers
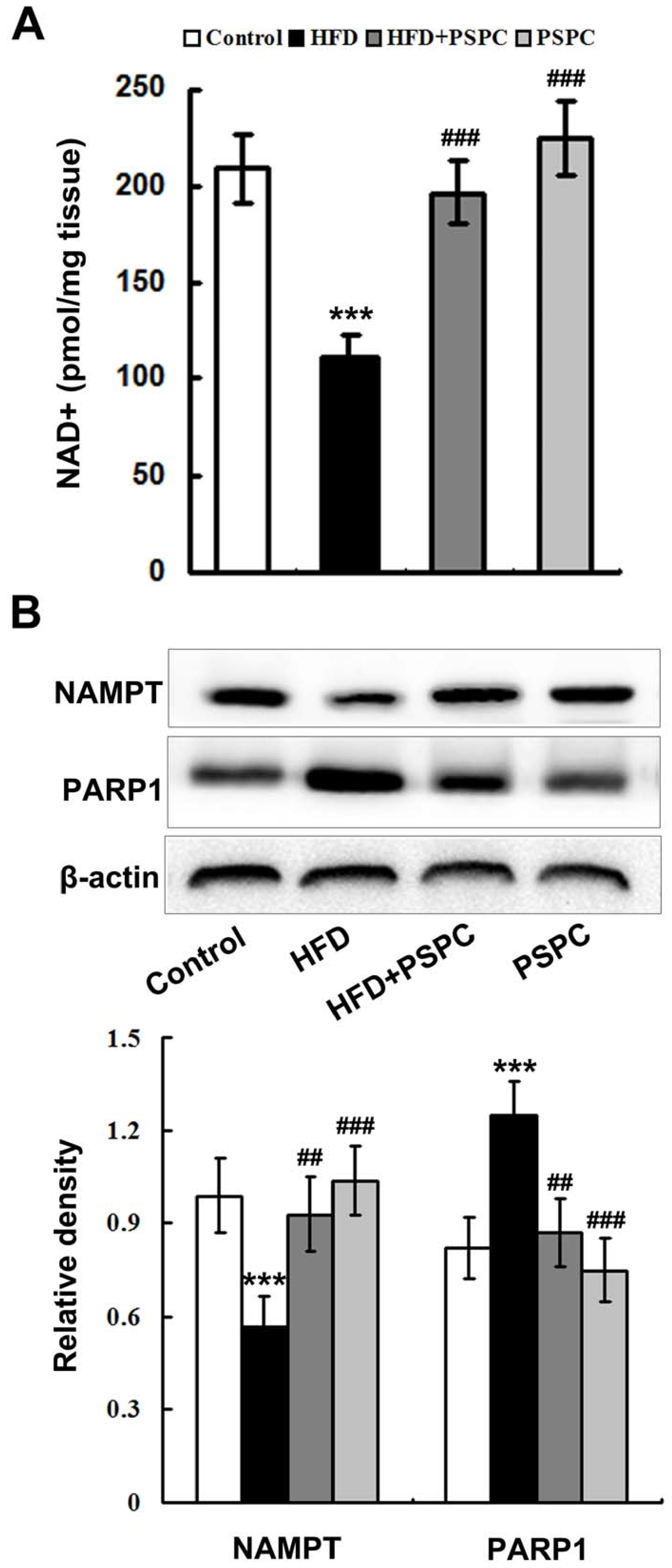
2.3. PSPC Restores NAD+ Level in HFD-Treated Mouse Livers
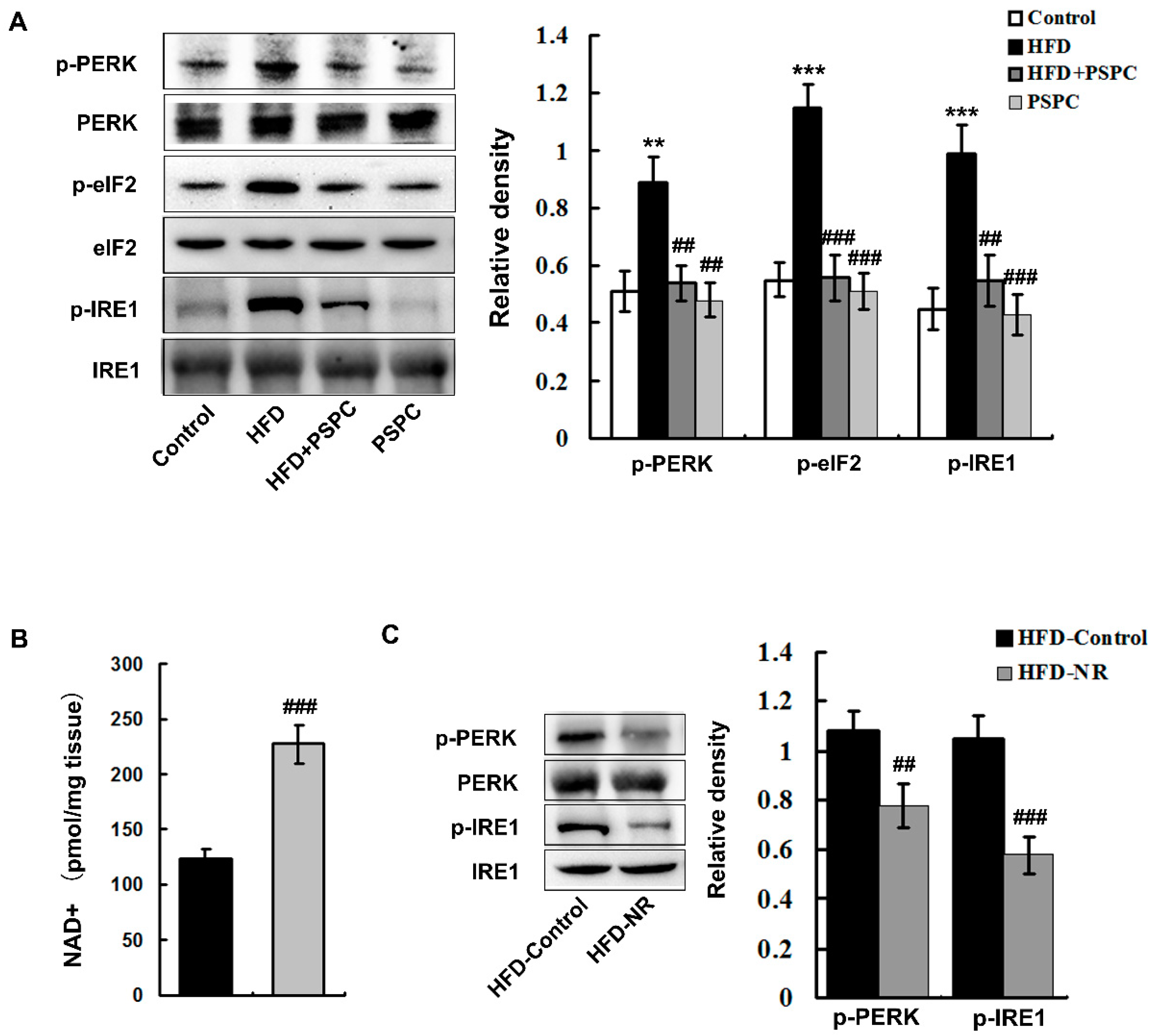
2.4. PSPC Depresses NAD+ Depletion-Mediated ER Stress in HFD-Treated Mouse Livers
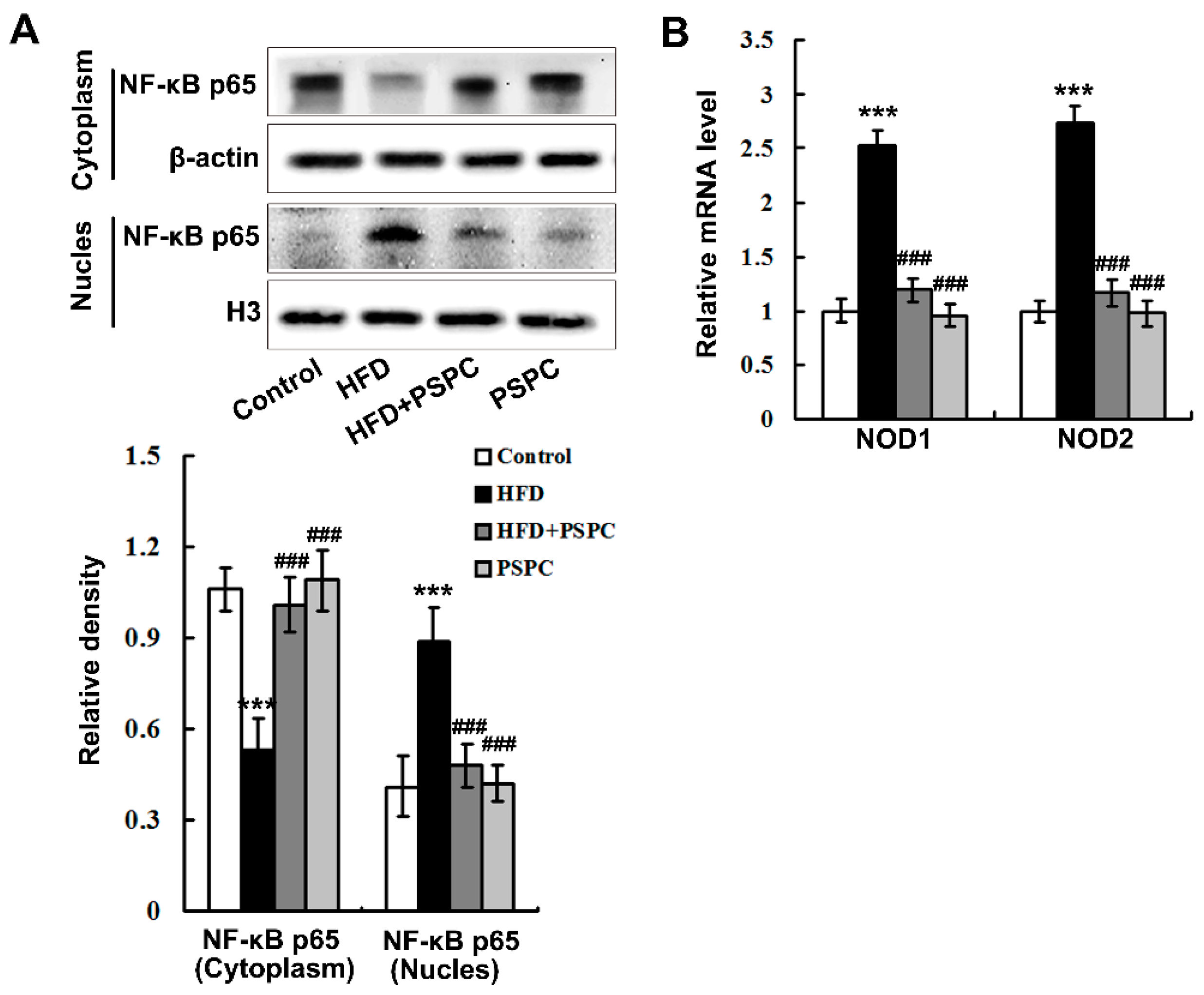
2.5. PSPC Abates Nuclear Factor-κB (NF-κB) p65 Nuclear Translocation and Nucleotide Oligomerization Domain Protein (NOD) Expression in HFD-Treated Mouse Livers
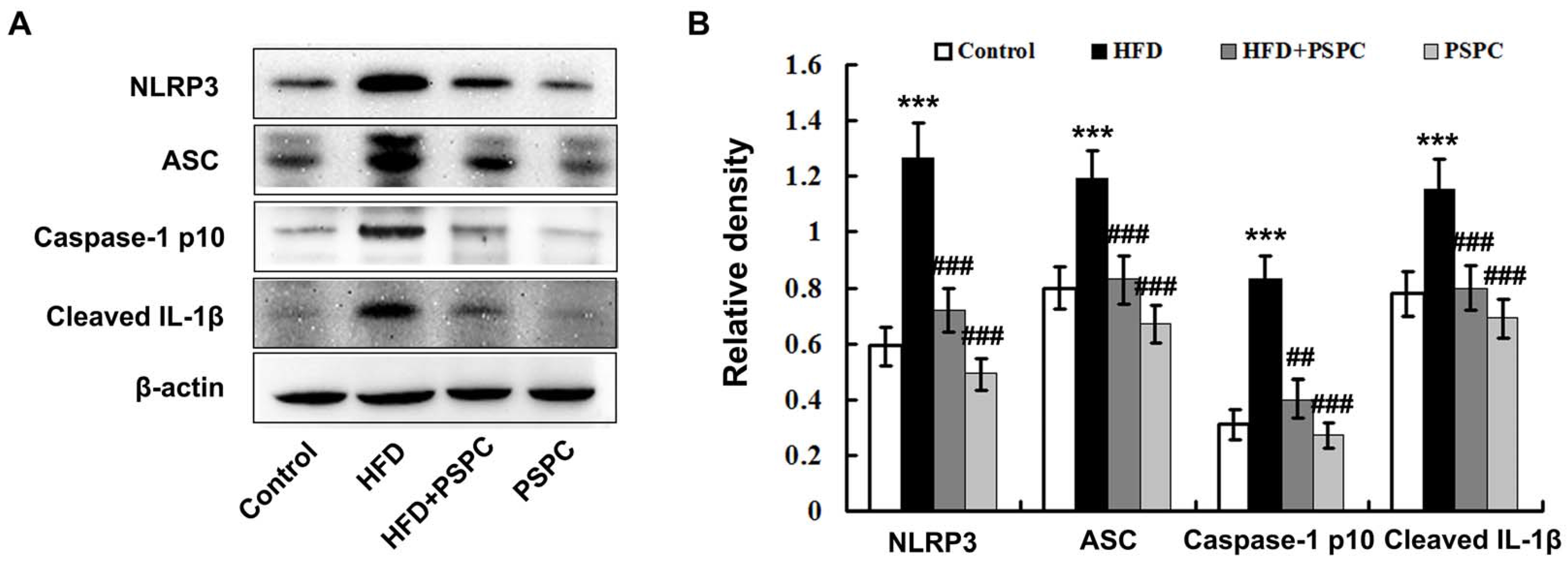
2.6. PSPC Prevents NLRP3 Inflammasome Activation in HFD-Treated Mouse Livers
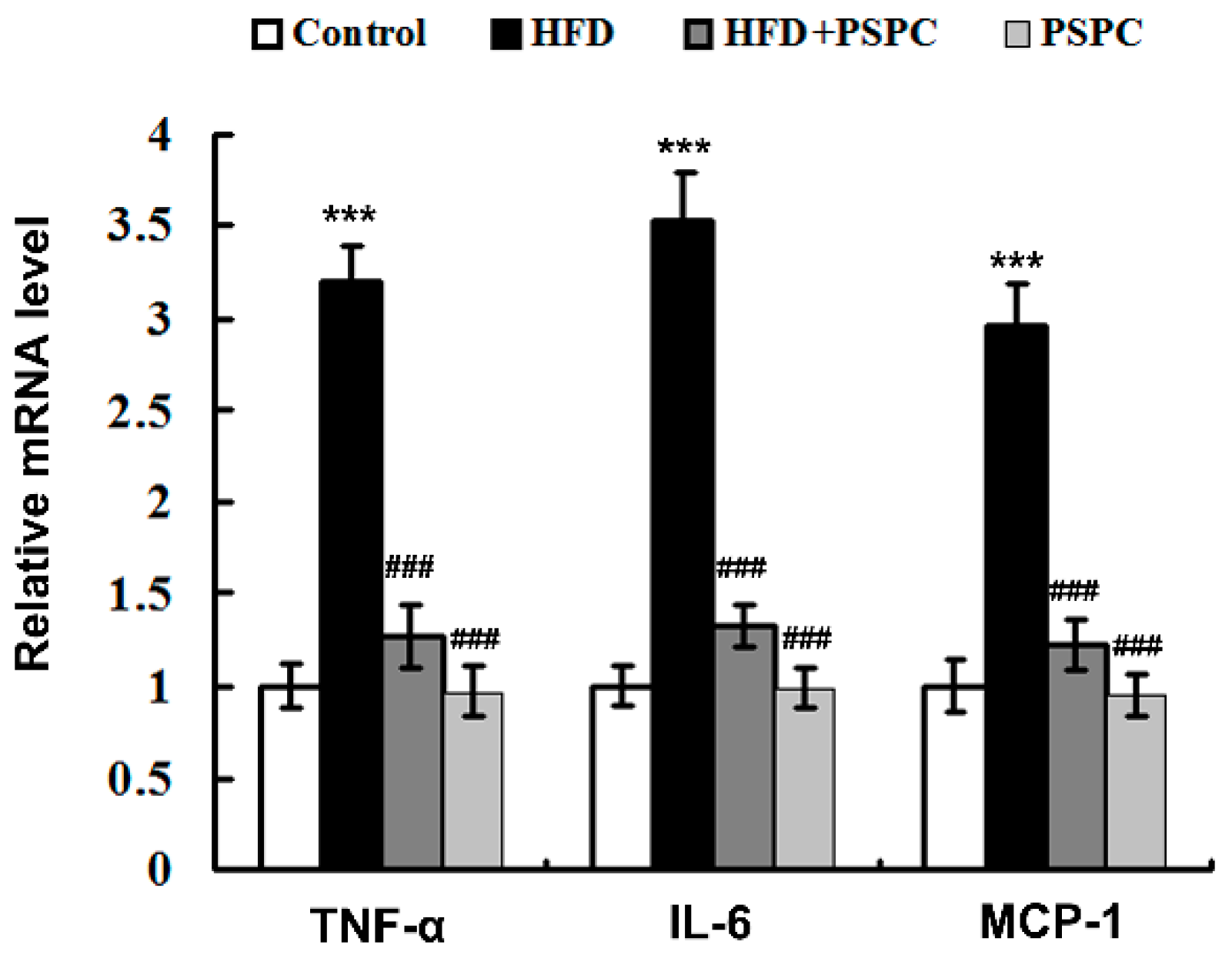
2.7. PSPC Suppresses Inflammation-Related Genes Expressions in HFD-Treated Mouse Livers
3. Discussion
4. Materials and Methods
4.1. Animals and Treatment
4.1.1. PSPC Treatment
4.1.2. NR Treatment
4.2. Tissue Homogenates
4.3. Biochemical Analyses
4.4. ROS Assay
4.5. Liver Slice Collection and Histopathological Analysis
4.6. Immunofluorescence Staining
4.7. NAD+ Assay
4.8. Quantitative Real Time Polymerase Chain Reaction
4.9. Western Blot Analysis
4.10. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Okin, D.; Medzhitov, R. The Effect of Sustained Inflammation on Hepatic Mevalonate Pathway Results in Hyperglycemia. Cell 2016, 165, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Duparc, T.; Plovier, H.; Marrachelli, V.G.; Van Hul, M.; Essaghir, A.; Ståhlman, M.; Matamoros, S.; Geurts, L.; Pardo-Tendero, M.M.; Druart, C.; et al. Hepatocyte MyD88 affects bile acids, gut microbiota and metabolome contributing to regulate glucose and lipid metabolism. Gut 2017, 66, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Ke, B.; Zhao, Z.; Ye, X.; Gao, Z.; Manganiello, V.; Wu, B.; Ye, J. Inactivation of NF-κB p65 (RelA) in Liver Improves Insulin Sensitivity and Inhibits cAMP/PKA Pathway. Diabetes 2015, 64, 3355–3362. [Google Scholar] [CrossRef] [PubMed]
- Morrison, M.C.; Mulder, P.; Salic, K.; Verheij, J.; Liang, W.; van Duyvenvoorde, W.; Menke, A.; Kooistra, T.; Kleemann, R.; Wielinga, P.Y. Intervention with a caspase-1 inhibitor reduces obesity-associated hyperinsulinemia, non-alcoholic steatohepatitis and hepatic fibrosis in LDLR-/-.Leiden mice. Int. J. Obes. 2016, 40, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Garten, A.; Schuster, S.; Penke, M.; Gorski, T.; de Giorgis, T.; Kiess, W. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat. Rev. Endocrinol. 2015, 11, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Escande, C.; Nin, V.; Price, N.L.; Capellini, V.; Gomes, A.P.; Barbosa, M.T.; O’Neil, L.; White, T.A.; Sinclair, D.A.; Chini, E.N. Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes 2013, 62, 1084–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, D.; Yang, Q.; Kong, D.; Banks, A.S.; Zhang, L.; Rodgers, J.T.; Pirinen, E.; Pulinilkunnil, T.C.; Gong, F.; Wang, Y.; et al. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature 2014, 508, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Stromsdorfer, K.L.; Yamaguchi, S.; Yoon, M.J.; Moseley, A.C.; Franczyk, M.P.; Kelly, S.C.; Qi, N.; Imai, S.; Yoshino, J. NAMPT-Mediated NAD(+) Biosynthesis in Adipocytes Regulates Adipose Tissue Function and Multi-organ Insulin Sensitivity in Mice. Cell Rep. 2016, 16, 1851–1860. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Kim, J.J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 2013, 62, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, T.; Godlewski, G.; Cinar, R.; Bertola, A.; Szanda, G.; Liu, J.; Tam, J.; Han, T.; Mukhopadhyay, B.; Skarulis, M.C.; et al. Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates beta cell loss in type 2 diabetes. Nat. Med. 2013, 19, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, J.H.; Chen, X.Y.; Hu, Q.H.; Wang, M.X.; Jin, R.; Zhang, Q.Y.; Wang, W.; Wang, R.; Kang, L.L.; et al. Reactive oxygen species-induced TXNIP drives fructose-mediated hepatic inflammation and lipid accumulation through NLRP3 inflammasome activation. Antioxid. Redox Signal. 2015, 22, 848–870. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.J.; Lim, Y. Resveratrol ameliorates hepatic metaflammation and inhibits NLRP3 inflammasome activation. Metabolism 2014, 63, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Esatbeyoglu, T.; Rodríguez-Werner, M.; Schlösser, A.; Winterhalter, P.; Rimbach, G. Fractionation, enzyme inhibitory and cellular antioxidant activity of bioactives from purple sweet potato (Ipomoea batatas). Food Chem. 2017, 221, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.J.; Zheng, Y.L.; Lu, J.; Chen, G.Q.; Wang, X.H.; Feng, J.; Ruan, J.; Sun, X.; Li, C.X.; Sun, Q.J. Purple sweet potato color suppresses lipopolysaccharide-induced acute inflammatory response in mouse brain. Neurochem. Int. 2010, 56, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Wu, D.M.; Zheng, Y.L.; Hu, B.; Cheng, W.; Zhang, Z.F. Purple sweet potato color attenuates domoic acid-induced cognitive deficits by promoting estrogen receptor-α-mediated mitochondrial biogenesis signaling in mice. Free Radic. Boil. Med. 2012, 52, 646–659. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.F.; Fan, S.H.; Zheng, Y.L.; Lu, J.; Wu, D.M.; Shan, Q.; Hu, B. Purple sweet potato color attenuates oxidative stress and inflammatory response induced by D-galactose in mouse liver. Food Chem. Toxicol. 2009, 47, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.F.; Lu, J.; Zheng, Y.L.; Hu, B.; Fan, S.H.; Wu, D.M.; Zheng, Z.H.; Shan, Q.; Liu, C.M. Purple sweet potato color protects mouse liver against d-galactose-induced apoptosis via inhibiting caspase-3 activation and enhancing PI3K/Akt pathway. Food Chem. Toxicol. 2010, 48, 2500–2507. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.F.; Lu, J.; Zheng, Y.L.; Wu, D.M.; Hu, B.; Shan, Q.; Cheng, W.; Li, M.Q.; Sun, Y.Y. Purple sweet potato color attenuates hepatic insulin resistance via blocking oxidative stress and endoplasmic reticulum stress in high-fat-diet-treated mice. J. Nutr. Biochem. 2013, 24, 1008–1018. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Jiang, W.; Spinetti, T.; Tardivel, A.; Castillo, R.; Bourquin, C.; Guarda, G.; Tian, Z.; Tschopp, J.; Zhou, R. Omega-3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome activation. Immunity 2013, 38, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Gerdts, J.; Brace, E.J.; Sasaki, Y.; DiAntonio, A.; Milbrandt, J. SARM1 activation triggers axon degeneration locally via NAD⁺ destruction. Science 2015, 348, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xu, M.; Lee, J.; He, C.; Xie, Z. Leucine supplementation increases SIRT1 expression and prevents mitochondrial dysfunction and metabolic disorders in high-fat diet-induced obese mice. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1234–E1244. [Google Scholar] [CrossRef] [PubMed]
- Pirinen, E.; Cantó, C.; Jo, Y.S.; Morato, L.; Zhang, H.; Menzies, K.J.; Williams, E.G.; Mouchiroud, L.; Moullan, N.; Hagberg, C.; et al. Pharmacological Inhibition of poly(ADP-ribose) polymerases improves fitness and mitochondrial function in skeletal muscle. Cell Metab. 2014, 19, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.F.; Fan, S.H.; Zheng, Y.L.; Lu, J.; Wu, D.M.; Shan, Q.; Hu, B. Troxerutin improves hepatic lipid homeostasis by restoring NAD+-depletion-mediated dysfunction of lipin 1 signaling in high-fat diet-treated mice. Biochem. Pharmacol. 2014, 91, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Aragonès, G.; Suárez, M.; Ardid-Ruiz, A.; Vinaixa, M.; Rodríguez, M.A.; Correig, X.; Arola, L.; Bladé, C. Dietary proanthocyanidins boost hepatic NAD(+) metabolism and SIRT1 expression and activity in a dose-dependent manner in healthy rats. Sci. Rep. 2016, 6, 24977. [Google Scholar] [CrossRef]
- Luccarini, I.; Pantano, D.; Nardiello, P.; Cavone, L.; Lapucci, A.; Miceli, C.; Nediani, C.; Berti, A.; Stefani, M.; Casamenti, F. The Polyphenol Oleuropein Aglycone Modulates the PARP1-SIRT1 Interplay: An In Vitro and In Vivo Study. J. Alzheimers Dis. 2016, 54, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Boesten, D.M.; von Ungern-Sternberg, S.N.; den Hartog, G.J.; Bast, A. Protective Pleiotropic Effect of Flavonoids on NAD⁺ Levels in Endothelial Cells Exposed to High Glucose. Oxid. Med. Cell Longev. 2015, 2015, 894597. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Tai, H.; Wang, X.; Wang, Z.; Zhou, J.; Wei, X.; Ding, Y.; Gong, H.; Mo, C.; Zhang, J.; et al. AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD(+) elevation. Aging Cell 2016, 15, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Gueguen, C.; Palmier, B.; Plotkine, M.; Marchand-Leroux, C.; Besson, V.C. Neurological and histological consequences induced by in vivo cerebral oxidative stress: Evidence for beneficial effects of SRT1720, a sirtuin 1 activator, and sirtuin 1-mediated neuroprotective effects of poly(ADP-ribose) polymerase inhibition. PLoS ONE 2014, 9, e87367. [Google Scholar] [CrossRef] [PubMed]
- de Kreutzenberg, S.V.; Ceolotto, G.; Papparella, I.; Bortoluzzi, A.; Semplicini, A.; Dalla Man, C.; Cobelli, C.; Fadini, G.P.; Avogaro, A. Downregulation of the longevity-associated protein sirtuin 1 in insulin resistance and metabolic syndrome: Potential biochemical mechanisms. Diabetes 2010, 59, 1006–1015. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Gariani, K.; Ryu, D.; Menzies, K.J.; Yi, H.S.; Stein, S.; Zhang, H.; Perino, A.; Lemos, V.; Katsyuba, E.; Jha, P.; et al. Inhibiting poly ADP-ribosylation increases fatty acid oxidation and protects against fatty liver disease. J. Hepatol. 2017, 66, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Módis, K.; Gero, D.; Erdélyi, K.; Szoleczky, P.; DeWitt, D.; Szabo, C. Cellular bioenergetics is regulated by PARP1 under resting conditions and during oxidative stress. Biochem. Pharmacol. 2012, 83, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zalzala, M.; Jadhav, K.; Xu, Y.; Kasumov, T.; Yin, L.; Zhang, Y. Carboxylesterase 2 prevents liver steatosis by modulating lipolysis, endoplasmic reticulum stress, and lipogenesis and is regulated by hepatocyte nuclear factor 4 alpha in mice. Hepatology 2016, 63, 1860–1874. [Google Scholar] [CrossRef] [PubMed]
- Park, E.C.; Kim, S.I.; Hong, Y.; Hwang, J.W.; Cho, G.S.; Cha, H.N.; Han, J.K.; Yun, C.H.; Park, S.Y.; Jang, I.S.; et al. Inhibition of CYP4A reduces hepatic endoplasmic reticulum stress and features of diabetes in mice. Gastroenterology 2014, 147, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, B.; Huang, F.; Liu, B.; Xie, Y. Curcumin inhibits lipolysis via suppression of ER stress in adipose tissue and prevents hepatic insulin resistance. J. Lipid Res. 2016, 57, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dou, X.; Li, S.; Zhang, X.; Zeng, Y.; Song, Z. Nicotinamide ameliorates palmitate-induced ER stress in hepatocytes via cAMP/PKA/CREB pathway-dependent Sirt1 upregulation. Biochim. Biophys. Acta 2015, 1853, 2929–2936. [Google Scholar] [CrossRef] [PubMed]
- Gariani, K.; Menzies, K.J.; Ryu, D.; Wegner, C.J.; Wang, X.; Ropelle, E.R.; Moullan, N.; Zhang, H.; Perino, A.; et al. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology 2016, 63, 1190–1204. [Google Scholar] [CrossRef] [PubMed]
- Ocampo, A.; Liu, J.; Barrientos, A. NAD+ salvage pathway proteins suppress proteotoxicity in yeast models of neurodegeneration by promoting the clearance of misfolded/oligomerized proteins. Hum. Mol. Genet. 2013, 22, 1699–1708. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.G.; Upton, J.P.; Praveen, P.V.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef] [PubMed]
- Menu, P.; Mayor, A.; Zhou, R.; Tardivel, A.; Ichijo, H.; Mori, K.; Tschopp, J. ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death Dis. 2012, 3, e261. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.M.; Ka, S.M.; Wu, H.L.; Yeh, Y.C.; Kuo, C.H.; Hua, K.F.; Shi, G.Y.; Hung, Y.J.; Hsiao, F.C.; Yang, S.S.; et al. Thrombomodulin domain 1 ameliorates diabetic nephropathy in mice via anti-NF-κB/NLRP3 inflammasome-mediated inflammation, enhancement of NRF2 antioxidant activity and inhibition of apoptosis. Diabetologia 2014, 57, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Keestra-Gounder, A.M.; Byndloss, M.X.; Seyffert, N.; Young, B.M.; Chávez-Arroyo, A.; Tsai, A.Y.; Cevallos, S.A.; Winter, M.G.; Pham, O.H.; Tiffany, C.R.; et al. NOD1 and NOD2 signalling links ER stress with inflammation. Nature 2016, 532, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Moreira, L.O.; Zamboni, D.S. NOD1 and NOD2 Signaling in Infection and Inflammation. Front. Immunol. 2012, 3, 328. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.Y.; Ka, S.M.; Chang, J.M.; Chen, H.C.; Shui, H.A.; Li, C.Y.; Hua, K.F.; Chang, W.L.; Huang, J.J.; Yang, S.S.; et al. Epigallocatechin-3-gallate prevents lupus nephritis development in mice via enhancing the Nrf2 antioxidant pathway and inhibiting NLRP3 inflammasome activation. Free Radic. Boil. Med. 2011, 51, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, J.; Li, S.; Li, Y.; Wang, X.; Liu, B.; Fu, Q.; Ma, S. Curcumin attenuates glutamate neurotoxicity in the hippocampus by suppression of ER stress-associated TXNIP/NLRP3 inflammasome activation in a manner dependent on AMPK. Toxicol. Appl. Pharmacol. 2015, 286, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Mulder, P.; Morrison, M.C.; Wielinga, P.Y.; van Duyvenvoorde, W.; Kooistra, T.; Kleemann, R. Surgical removal of inflamed epididymal white adipose tissue attenuates the development of non-alcoholic steatohepatitis in obesity. Int. J. Obes. 2016, 40, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Burrington, C.M.; Graff, E.C.; Zhang, J.; Judd, R.L.; Suksaranjit, P.; Kaewpoowat, Q.; Davenport, S.K.; O’Neill, A.M.; Greene, M.W. Metabolic phenotype and adipose and liver features in a high-fat Western diet-induced mouse model of obesity-linked NAFLD. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E418–E439. [Google Scholar] [CrossRef] [PubMed]
- Handa, P.; Maliken, B.D.; Nelson, J.E.; Morgan-Stevenson, V.; Messner, D.J.; Dhillon, B.K.; Klintworth, H.M.; Beauchamp, M.; Yeh, M.M.; Elfers, C.T.; et al. Reduced adiponectin signaling due to weight gain results in nonalcoholic steatohepatitis through impaired mitochondrial biogenesis. Hepatology 2014, 60, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.; Rivera, L.; Furness, J.B.; Angus, P.W. The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Brun, P.; Castagliuolo, I.; Di Leo, V.; Buda, A.; Pinzani, M.; Palù, G.; Martines, D. Increased intestinal permeability in obese mice: New evidence in the pathogenesis of nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G518–G525. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Yin, J.; Zhang, G.; Long, H.; Zheng, X. Mulberry and cherry anthocyanin consumption prevents oxidative stress and inflammation in diet-induced obese mice. Mol. Nutr. Food Res. 2016, 60, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Jamar, G.; Estadella, D.; Pisani, L.P. Contribution of anthocyanin-rich foods in obesity control through gut microbiota interactions. Biofactors 2017. [Google Scholar] [CrossRef] [PubMed]
- Trammell, S.A.; Schmidt, M.S.; Weidemann, B.J.; Redpath, P.; Jaksch, F.; Dellinger, R.W.; Li, Z.; Abel, E.D.; Migaud, M.E.; Brenner, C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 2016, 7, 12948. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.F.; Zhang, Y.Q.; Fan, S.H.; Zhuang, J.; Zheng, Y.L.; Lu, J.; Wu, D.M.; Shan, Q.; Hu, B. Troxerutin protects against 2,2′,4,4′-tetrabromodiphenyl ether (BDE-47)-induced liver inflammation by attenuating oxidative stress-mediated NAD+-depletion. J. Hazard. Mater. 2015, 283, 98–109. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diet | Treatment (Gavage) | ||||
|---|---|---|---|---|---|
| Group | HFD | ND | PSPC (700 mg/kg/day) | Distilled Water Containing 0.1% Tween 80 | Exposure Time |
| Control | + | + | 0–20 weeks | ||
| HFD | + | + | 0–20 weeks | ||
| HFD + PSPC | + | + | 0–20 weeks | ||
| PSPC | + | + | 0–20 weeks | ||
| Diet | Treatment (Gavage) | ||||
|---|---|---|---|---|---|
| Group | HFD | ND | NR (300 mg/kg/day) | PBS | Exposure Time |
| HFD-Control | + | + | 19–20 weeks | ||
| HFD-NR | + | + | 19–20 weeks | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Zhang, Z.-F.; Zheng, G.-H.; Wang, A.-M.; Sun, C.-H.; Qin, S.-P.; Zhuang, J.; Lu, J.; Ma, D.-F.; Zheng, Y.-L. The Inhibitory Effects of Purple Sweet Potato Color on Hepatic Inflammation Is Associated with Restoration of NAD+ Levels and Attenuation of NLRP3 Inflammasome Activation in High-Fat-Diet-Treated Mice. Molecules 2017, 22, 1315. https://doi.org/10.3390/molecules22081315
Wang X, Zhang Z-F, Zheng G-H, Wang A-M, Sun C-H, Qin S-P, Zhuang J, Lu J, Ma D-F, Zheng Y-L. The Inhibitory Effects of Purple Sweet Potato Color on Hepatic Inflammation Is Associated with Restoration of NAD+ Levels and Attenuation of NLRP3 Inflammasome Activation in High-Fat-Diet-Treated Mice. Molecules. 2017; 22(8):1315. https://doi.org/10.3390/molecules22081315
Chicago/Turabian StyleWang, Xin, Zi-Feng Zhang, Gui-Hong Zheng, Ai-Min Wang, Chun-Hui Sun, Su-Ping Qin, Juan Zhuang, Jun Lu, Dai-Fu Ma, and Yuan-Lin Zheng. 2017. "The Inhibitory Effects of Purple Sweet Potato Color on Hepatic Inflammation Is Associated with Restoration of NAD+ Levels and Attenuation of NLRP3 Inflammasome Activation in High-Fat-Diet-Treated Mice" Molecules 22, no. 8: 1315. https://doi.org/10.3390/molecules22081315
APA StyleWang, X., Zhang, Z. -F., Zheng, G. -H., Wang, A. -M., Sun, C. -H., Qin, S. -P., Zhuang, J., Lu, J., Ma, D. -F., & Zheng, Y. -L. (2017). The Inhibitory Effects of Purple Sweet Potato Color on Hepatic Inflammation Is Associated with Restoration of NAD+ Levels and Attenuation of NLRP3 Inflammasome Activation in High-Fat-Diet-Treated Mice. Molecules, 22(8), 1315. https://doi.org/10.3390/molecules22081315