Stringent Nucleotide Recognition by the Ribosome at the Middle Codon Position



Abstract
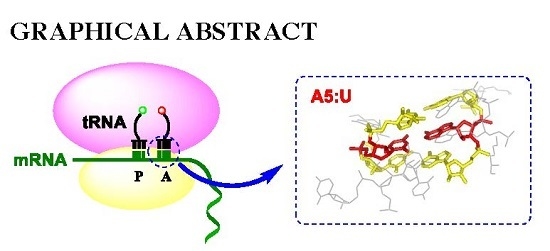
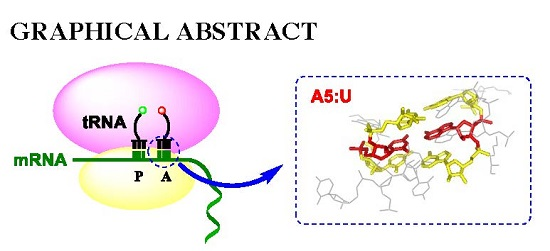
:
1. Introduction
2. Results
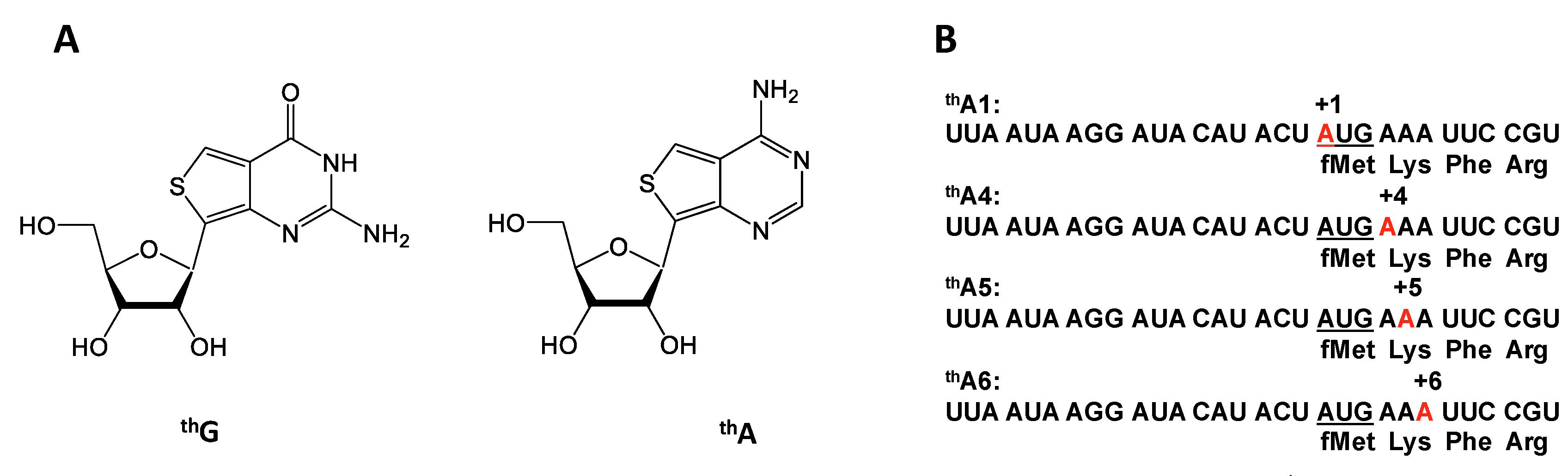
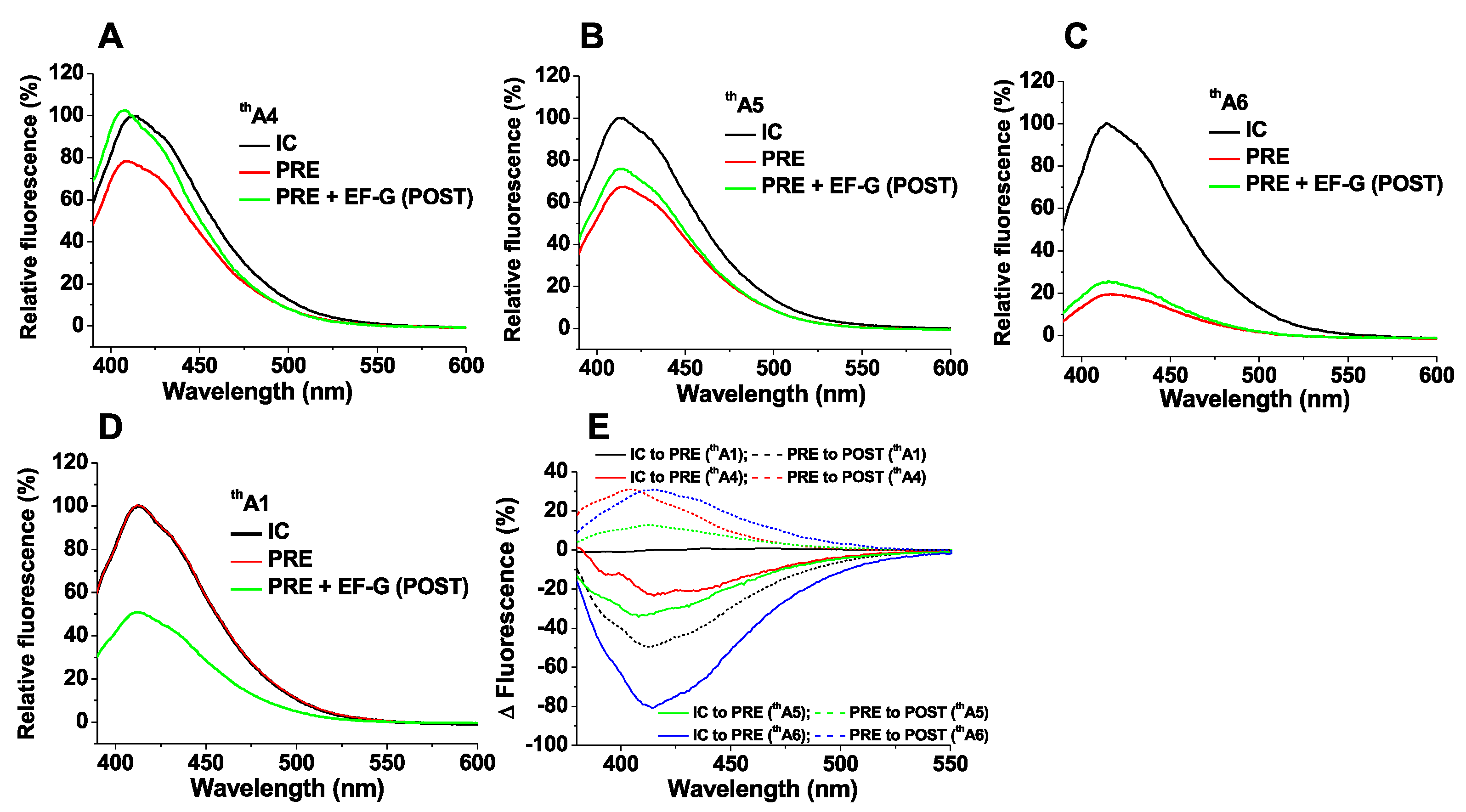
2.1. thA Fluorescence Intensity Changes Accompanying PRE and POST Complex Formation
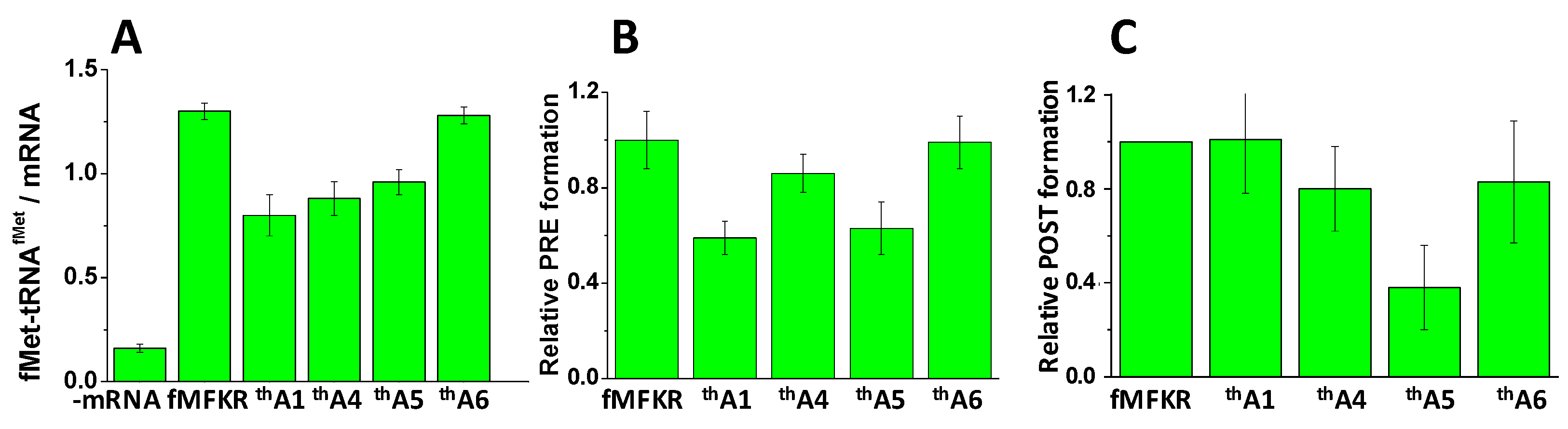
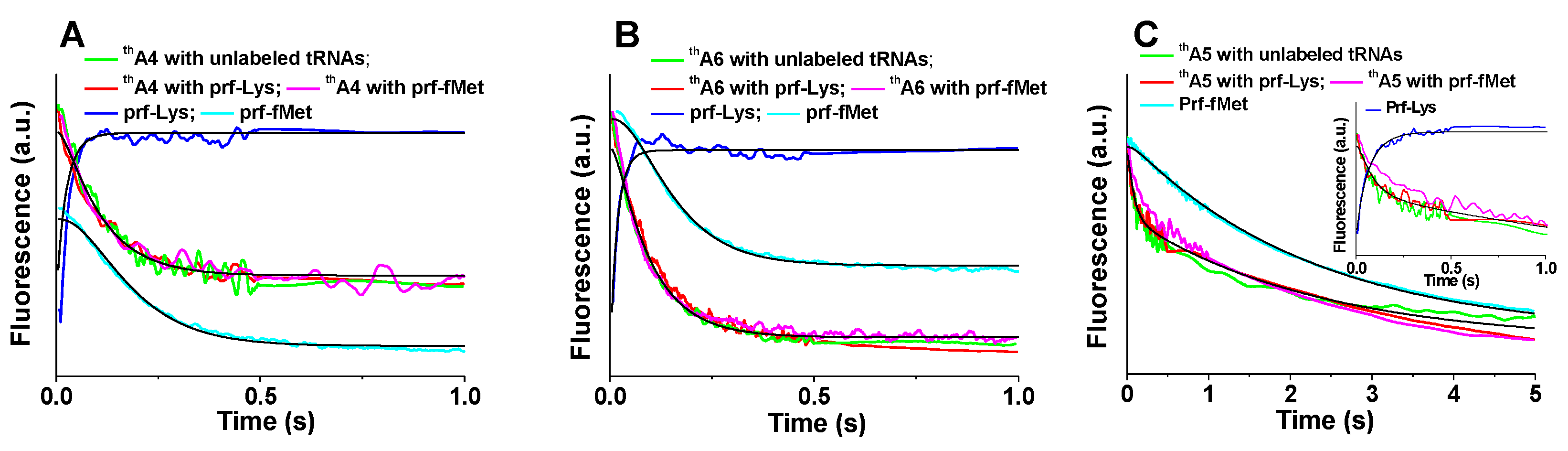
2.2. Kinetics of PRE Complex Formation
2.3. Kinetics of POST Complex Formation
3. Discussion
4. Materials and Methods
4.1. Synthesis of Modified mRNAs
4.2. Complex Preparation and Quantification
4.3. Fluorescence Experiments
4.4. Solvent Volume Calculations
4.5. Apparent Rate Constants
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Voorhees, R.M.; Ramakrishnan, V. Structural basis of the translational elongation cycle. Annu. Rev. Biochem. 2013, 82, 203–236. [Google Scholar] [CrossRef] [PubMed]
- Yusupova, G.; Yusupov, M. Crystal structure of eukaryotic ribosome and its complexes with inhibitors. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160184. [Google Scholar] [CrossRef] [PubMed]
- Dever, T.E.; Green, R. The elongation, termination, and recycling phases of translation in eukaryotes. Cold Spring Harb. Perspect. Biol. 2012, 4, a013706. [Google Scholar] [CrossRef] [PubMed]
- Frank, J.; Gonzalez, R.L., Jr. Structure and dynamics of a processive Brownian motor: The translating ribosome. Annu. Rev. Biochem. 2010, 79, 381–412. [Google Scholar] [CrossRef] [PubMed]
- Wohlgemuth, I.; Pohl, C.; Mittelstaet, J.; Konevega, A.L.; Rodnina, M.V. Evolutionary optimization of speed and accuracy of decoding on the ribosome. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 2979–2986. [Google Scholar] [CrossRef] [PubMed]
- Rodnina, M.V.; Wintermeyer, W. The ribosome as a molecular machine: The mechanism of tRNA-mRNA movement in translocation. Biochem. Soc. Trans. 2011, 39, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Sanbonmatsu, K.Y. Computational studies of molecular machines: The ribosome. Curr. Opin. Struct. Biol. 2012, 22, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Choi, J.; O’Leary, S.E.; Prabhakar, A.; Petrov, A.; Grosely, R.; Puglisi, E.V.; Puglisi, J.D. The molecular choreography of protein synthesis: Translational control, regulation, and pathways. Q. Rev. Biophys. 2016, 49, e11. [Google Scholar] [CrossRef] [PubMed]
- Loveland, A.B.; Demo, G.; Grigorieff, N.; Korostelev, A.A. Ensemble cryo-EM elucidates the mechanism of translation fidelity. Nature 2017, 546, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Rodnina, M.V.; Fischer, N.; Maracci, C.; Stark, H. Ribosome dynamics during decoding. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160182. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhang, H.; Broitman, S.L.; Reiche, M.; Farrell, I.; Cooperman, B.S.; Goldman, Y.E. Dynamics of translation by single ribosomes through mRNA secondary structures. Nat. Struct. Mol. Biol. 2013, 20, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Pape, T.; Wintermeyer, W.; Rodnina, M.V. Complete kinetic mechanism of elongation factor Tu-dependent binding of aminoacyl-tRNA to the A site of the E. coli ribosome. EMBO J. 1998, 17, 7490–7497. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, S.C.; Gonzalez, R.L.; Kim, H.D.; Chu, S.; Puglisi, J.D. tRNA selection and kinetic proofreading in translation. Nat. Struct. Mol. Biol. 2004, 11, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Geggier, P.; Dace, R.; Feldman, M.B.; Terry, D.S.; Altman, R.B.; Munro, J.B.; Blanchard, S.C. Conformational sampling of aminoacyl-tRNA during selection on the bacterial ribosome. J. Biol. Chem. 2010, 399, 576–595. [Google Scholar] [CrossRef] [PubMed]
- Schmeing, T.M.; Voorhees, R.M.; Kelley, A.C.; Ramakrishnan, V. How mutations in tRNA distant from the anticodon effect the fidelity of decoding. Nat. Struct. Mol. Biol. 2011, 18, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Chen, C.; Kavaliauskas, D.; Knudsen, C.R.; Goldman, Y.E.; Cooperman, B.S. EF-Tu dynamics during pre-translocation complex formation: EF-Tu GDP exits the ribosome via two different pathways. Nucleic Acids Res. 2015, 43, 9519–9528. [Google Scholar] [CrossRef] [PubMed]
- Savelsbergh, A.; Katunin, V.I.; Mohr, D.; Peske, F.; Rodnina, M.V.; Wintermeyer, W. An elongation factor G-induced ribosome rearrangement precedes tRNA-mRNA translocation. Mol. Cell 2003, 11, 1517–1523. [Google Scholar] [CrossRef]
- Pan, D.; Kirillov, S.V.; Cooperman, B.S. Kinetically competent intermediates in the translocation step of protein synthesis. Mol. Cell 2007, 25, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Munro, J.B.; Wasserman, M.R.; Altman, R.B.; Wang, L.; Blanchard, S.C. Correlated conformational events in EF-G and the ribosome regulate translocation. Nat. Struct. Mol. Biol. 2010, 17, 1470–1477. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Stevens, B.; Kaur, J.; Cabral, D.; Liu, H.; Wang, Y.; Zhang, H.; Rosenblum, G.; Smilansky, Z.; Goldman, Y.E.; et al. Single-molecule fluorescence measurements of ribosomal translocation dynamics. Mol. Cell 2011, 42, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Cui, X.; Beausang, J.F.; Zhang, H.; Farrell, I.; Cooperman, B.S.; Goldman, Y.E. Elongation factor G initiates translocation through a power stroke. Proc. Natl. Acad. Sci. USA 2016, 113, 7515–7520. [Google Scholar] [CrossRef] [PubMed]
- Belardinelli, R.; Sharma, H.; Peske, F.; Wintermeyer, W.; Rodnina, M.V. Translocation as continuous movement through the ribosome. RNA Biol. 2016, 13, 1197–1203. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, M.R.; Alejo, J.L.; Altman, R.B.; Blanchard, S.C. Multiperspective smFRET reveals rate-determining late intermediates of ribosomal translocation. Nat. Struct. Mol. Biol. 2016, 23, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Qin, H.; Cooperman, B.S. Synthesis and functional activity of tRNAs labeled with fluorescent hydrazides in the D-loop. RNA 2009, 15, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Sinkeldam, R.W.; Tor, Y. Emissive RNA alphabet. J. Am. Chem. Soc. 2011, 133, 14912–14915. [Google Scholar] [CrossRef] [PubMed]
- Samanta, P.K.; Manna, A.K.; Pati, S.K. Thieno analogues of RNA nucleosides: A detailed theoretical study. J. Phys. Chem. B 2012, 116, 7618–7626. [Google Scholar] [CrossRef] [PubMed]
- Gedik, M.; Brown, A. Computational study of the excited state properties of modified RNA nucleobases. J. Photochem. Photobiol. A 2013, 259, 25–32. [Google Scholar] [CrossRef]
- Liu, W.; Shin, D.; Tor, Y.; Cooperman, B.S. Monitoring translation with modified mRNAs strategically labeled with isomorphic fluorescent guanosine mimetic. ACS Chem. Biol. 2013, 8, 2017–2023. [Google Scholar] [CrossRef] [PubMed]
- Erlacher, M.D.; Chirkova, A.; Voegele, P.; Polacek, N. Generation of chemically engineered ribosomes for atomic mutagenesis studies on protein biosynthesis. Nat. Protoc. 2011, 6, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Forconi, M.; Benz-Moy, T.; Gleitsman, K.R.; Ruben, E.; Metz, C.; Herschlag, D. Exploring purine N7 interactions via atomic mutagenesis: The group I ribozyme as a case study. RNA 2012, 18, 1222–1229. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Zhang, C.M.; Kirillov, S.; Hou, Y.M.; Cooperman, B.S. Perturbation of the tRNA tertiary core differentially affects specific steps of the elongation cycle. J. Biol. Chem. 2008, 283, 18431–18440. [Google Scholar] [CrossRef] [PubMed]
- Daviter, T.; Gromadski, K.B.; Rodnina, M.V. The ribosome’s response to codon-anticodon mismatches. Biochimie 2006, 88, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Gromadski, K.B.; Rodnina, M.V. Kinetic determinants of high-fidelity tRNA discrimination on the ribosome. Mol. Cell 2004, 13, 191–200. [Google Scholar] [CrossRef]
- Demeshkina, N.; Jenner, L.; Westhof, E.; Yusupov, M.; Yusupova, G. A new understanding of the decoding principle on the ribosome. Nature 2012, 484, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Khade, P.K.; Shi, X.; Joseph, S. Steric complementarity in the decoding center isimportant for tRNA selection by the ribosome. J. Mol. Biol. 2013, 425, 3778–3789. [Google Scholar] [CrossRef] [PubMed]
- Ogle, J.M.; Brodersen, D.E.; Clemons, W.M.; Tarry, M.J.; Carter, A.P.; Ramakrishnan, V. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science 2001, 292, 897–902. [Google Scholar] [CrossRef] [PubMed]
- Demeshkina, N.; Jenner, L.; Westhof, E.; Yusupov, M.; Yusupova, G. New structural insights into the decoding mechanism: Translation infidelity via a G·U pair with Watson-Crick geometry. FEBS Lett. 2013, 587, 1848–1857. [Google Scholar] [CrossRef] [PubMed]
- Sanbonmatsu, K.Y.; Joseph, S. Understanding discrimination by the ribosome: Stability testing and groove measurement of codon-anticodon pairs. J. Mol. Biol. 2003, 328, 33–47. [Google Scholar] [CrossRef]
- Szaflarski, W.; Vesper, O.; Teraoka, Y.; Plitta, B.; Wilson, D.N.; Nierhaus, K.H. New features of the ribosome and ribosomal inhibiters: Non-enzymatic recycling, misreading and back-translocation. J. Mol. Biol. 2008, 380, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Voss, N.R.; Gerstein, M. 3V: Cavity, channel and cleft volume calculator and extractor. Nucleic Acids Res. 2010, 38, W555–W562. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.H. Available online: http://rna.urmc.rochester.edu/NNDB/turner04/stack.txt (accessed on 15 August 2017).
- Dima, R.I.; Hyeon, C.; Thirumalai, D. Extracting stacking interaction parameters for RNA from the data set of native structures. J. Mol. Biol. 2005, 347, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Pörschke, D.; Eigen, M. Co-operative non-enzymic base recognition. 3. Kinetics of the helix-coil transition of the oligoribouridylic—Oligoriboadenylic acid system and of oligoriboadenylic acid alone at acidic pH. J. Mol. Biol. 1971, 62, 361–381. [Google Scholar] [CrossRef]
- Mohan, S.; Hsiao, C.; VanDeusen, H.; Gallagher, R.; Krohn, E.; Kalahar, B.; Wartell, R.M.; Williams, L.D. Mechanism of RNA double helix-propagation at atomic resolution. J. Phys. Chem. B 2009, 113, 2614–2623. [Google Scholar] [CrossRef] [PubMed]
- Colizzi, F.; Bussi, G. RNA unwinding from reweighted pulling simulations. J. Am. Chem. Soc. 2012, 134, 5173–5179. [Google Scholar] [CrossRef] [PubMed]
- Rovira, A.R.; Fin, A.; Tor, Y. Chemical mutagenesis of an emissive RNA alphabet. J. Am. Chem. Soc. 2015, 137, 14602–14605. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
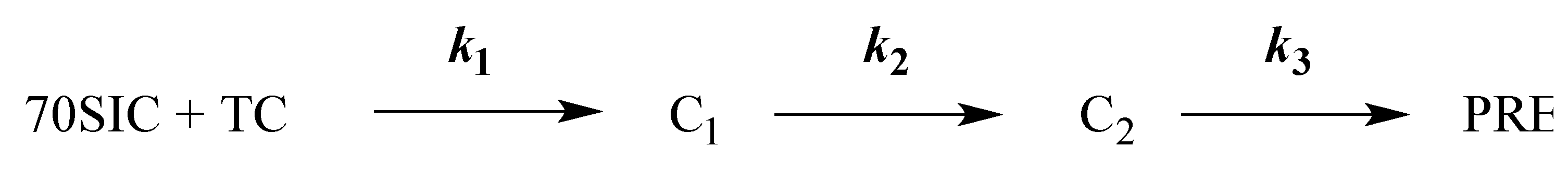{kind=link}
| mRNA | kTC 2 | kthA | kfMet 3 |
|---|---|---|---|
| thA4 | 53 ± 1 | 9 ± 1 | 5.2 ± 0.6 |
| thA5 | 18 ± 5 | 13 ± 3, minor 0.5 ± 0.2, major | 0.4 ± 0.1 |
| thA6 | 62 ± 11 | 11 ± 1 | 6.6 ± 0.8 |
| fMKFR | 44 ± 3 | - | 8.1 ± 0.3 |
| mRNA | thA4 | thA6 | thA5 |
|---|---|---|---|
| Rate Constants | |||
| k1 (µM−1·s−1) | 123 ± 13 | 170 ± 9 | 42 ± 1 |
| k2 (s−1) | 10.6 ± 0.3 | 11.3 ± 0.1 | 40 ± 16 |
| k3 (s−1) | 12.0 ± 0.4 | 15.0 ± 0.5 | 0.44 ± 0.07 |
| Relative Intensity Change from 70SIC 1 | |||
| C1 | 0.14 | 0.16 | 0.05 |
| C2 | 1.00 | 1.00 | 0.35 |
| PRE | 1.00 | 1.00 | 1.00 |
| mRNA | kA (s−1) | kprf (s−1) |
|---|---|---|
| thA4 | 4.4 ± 0.3 | 4.4 ± 0.3 |
| thA6 | n.d. 2 | 2.6 ± 0.2 |
| thA1 | 1.3 ± 0.1 | 4.2 ± 0.2 |
| thA5 | 2.3 ± 0.2 | 13 ± 1, major 3 0.94 ± 0.16, minor 3 |
| Codon | Codon Position | Volume (Å3) |
|---|---|---|
| AAA | 5′-A4 | 424 |
| middle-A5 | 271 | |
| 3′-A6 | 396 | |
| GUG | 5′-G4 | 419 |
| CGU | middle-G5 | 252 |
| GUG | 3′-G6 | 406 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, W.; Shin, D.; Ng, M.; Sanbonmatsu, K.Y.; Tor, Y.; Cooperman, B.S. Stringent Nucleotide Recognition by the Ribosome at the Middle Codon Position. Molecules 2017, 22, 1427. https://doi.org/10.3390/molecules22091427
Liu W, Shin D, Ng M, Sanbonmatsu KY, Tor Y, Cooperman BS. Stringent Nucleotide Recognition by the Ribosome at the Middle Codon Position. Molecules. 2017; 22(9):1427. https://doi.org/10.3390/molecules22091427
Chicago/Turabian StyleLiu, Wei, Dongwon Shin, Martin Ng, Karissa Y. Sanbonmatsu, Yitzhak Tor, and Barry S. Cooperman. 2017. "Stringent Nucleotide Recognition by the Ribosome at the Middle Codon Position" Molecules 22, no. 9: 1427. https://doi.org/10.3390/molecules22091427
APA StyleLiu, W., Shin, D., Ng, M., Sanbonmatsu, K. Y., Tor, Y., & Cooperman, B. S. (2017). Stringent Nucleotide Recognition by the Ribosome at the Middle Codon Position. Molecules, 22(9), 1427. https://doi.org/10.3390/molecules22091427