Towards Comprehension of the ABCB1/P-Glycoprotein Role in Chronic Myeloid Leukemia


Abstract
:1. Introduction
2. Clinical Relevance of ABCB1/Pgp Expression and Activity in CML Patients
2.1. ABCB1/Pgp Expression/Activity in Different CML Phases/Stages
2.2. ABCB1/Pgp and CML Patient Response to TKI
3. ABCB1 Polymorphisms (SNPs) in Chronic Myeloid Leukemia Patients
3.1. SNPs at Position 1236
3.2. SNPs at Position 3435
3.3. SNPs at Position 2677
3.4. Haplotypes
3.5. Susceptibility to the Development of Leukemia and ABCB1 SNPs
3.6. Impact of ABCB1 SNPs on Pharmacokinetics
4. Interactions between ABCB1/Pgp and TKIs in CML Cells
5. Resistance Mediated by Pgp Associated with Cellular Microvesicles (MVs)
6. Associations between Pgp and ‘Onco-Molecules’: Exploring Multifactorial Resistance
6.1. Pgp and microRNAs (miRNAs) Interactions in CML Cells
6.2. Associations between Pgp and the Inhibitor of Apoptosis Proteins (IAPs)
7. Overcoming Drug Resistance in CML
7.1. Clinical Modulation of Pgp
7.2. Interaction of Second- and Third-Generation TKIs with Pgp
7.3. Efficacy of Non-TKI Drugs in CML
8. Conclusions and Future Prospects
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chereda, B.; Melo, J.V. Natural course and biology of CML. Ann. Hematol. 2015, 94, S107–S121. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Peng, C.; Li, D.; Li, S. Molecular and cellular bases of chronic myeloid leukemia. Protein Cell 2010, 1, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Cilloni, D.; Saglio, G. Molecular pathways: BCR-ABL. Clin. Cancer Res. 2012, 18, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Schwetz, B.A. From the Food and Drug Administration. JAMA 2001, 286, 2705. [Google Scholar] [CrossRef]
- Apperley, J.F. Part I: Mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol. 2007, 8, 1018–1029. [Google Scholar] [CrossRef]
- Santos, F.P.; Kantarjian, H.; Quintas-Cardama, A.; Cortes, J. Evolution of therapies for chronic myelogenous leukemia. Cancer J. 2011, 17, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Shah, N.P.; Hochhaus, A.; Cortes, J.; Shah, S.; Ayala, M.; Moiraghi, B.; Shen, Z.; Mayer, J.; Pasquini, R.; et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N. Engl. J. Med. 2010, 362, 2260–2270. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Shah, N.P.; Cortes, J.E.; Baccarani, M.; Agarwal, M.B.; Undurraga, M.S.; Wang, J.; Ipina, J.J.; Kim, D.W.; Ogura, M.; et al. Dasatinib or imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: 2-year follow-up from a randomized phase 3 trial (DASISION). Blood 2012, 119, 1123–1129. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Kim, D.W.; Pinilla-Ibarz, J.; le Coutre, P.; Paquette, R.; Chuah, C.; Nicolini, F.E.; Apperley, J.F.; Khoury, H.J.; Talpaz, M.; et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 2013, 369, 1783–1796. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Cortes, J.E.; Kim, D.W.; Khoury, H.J.; Brummendorf, T.H.; Porkka, K.; Martinelli, G.; Durrant, S.; Leip, E.; Kelly, V.; et al. Bosutinib safety and management of toxicity in leukemia patients with resistance or intolerance to imatinib and other tyrosine kinase inhibitors. Blood 2014, 123, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Balabanov, S.; Braig, M.; Brummendorf, T.H. Current aspects in resistance against tyrosine kinase inhibitors in chronic myelogenous leukemia. Drug Discov. Today Technol. 2014, 11, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Hochhaus, A.; Saglio, G.; Hughes, T.P.; Larson, R.A.; Kim, D.W.; Issaragrisil, S.; le Coutre, P.D.; Etienne, G.; Dorlhiac-Llacer, P.E.; Clark, R.E.; et al. Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia 2016, 30, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Guilhot, F.; O’Brien, S.G.; Gathmann, I.; Kantarjian, H.; Gattermann, N.; Deininger, M.W.; Silver, R.T.; Goldman, J.M.; Stone, R.M.; et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med. 2006, 355, 2408–2417. [Google Scholar] [CrossRef] [PubMed]
- Vigil, C.E.; Griffiths, E.A.; Wang, E.S.; Wetzler, M. Interpretation of cytogenetic and molecular results in patients treated for CML. Blood Rev. 2011, 25, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Baccarani, M.; Deininger, M.W.; Rosti, G.; Hochhaus, A.; Soverini, S.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Guilhot, F.; et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood 2013, 122, 872–884. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, S.G.; Guilhot, F.; Larson, R.A.; Gathmann, I.; Baccarani, M.; Cervantes, F.; Cornelissen, J.J.; Fischer, T.; Hochhaus, A.; Hughes, T.; et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N. Engl. J. Med. 2003, 348, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Castagnetti, F.; Gugliotta, G.; Breccia, M.; Stagno, F.; Iurlo, A.; Albano, F.; Abruzzese, E.; Martino, B.; Levato, L.; Intermesoli, T.; et al. Long-term outcome of chronic myeloid leukemia patients treated frontline with imatinib. Leukemia 2015, 29, 1823–1831. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.P.; Hochhaus, A.; Branford, S.; Muller, M.C.; Kaeda, J.S.; Foroni, L.; Druker, B.J.; Guilhot, F.; Larson, R.A.; O’Brien, S.G.; et al. Long-term prognostic significance of early molecular response to imatinib in newly diagnosed chronic myeloid leukemia: An analysis from the International Randomized Study of Interferon and STI571 (IRIS). Blood 2010, 116, 3758–3765. [Google Scholar] [CrossRef] [PubMed]
- Kalmanti, L.; Saussele, S.; Lauseker, M.; Muller, M.C.; Dietz, C.T.; Heinrich, L.; Hanfstein, B.; Proetel, U.; Fabarius, A.; Krause, S.W.; et al. Safety and efficacy of imatinib in CML over a period of 10 years: Data from the randomized CML-study IV. Leukemia 2015, 29, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Khorashad, J.S.; Kelley, T.W.; Szankasi, P.; Mason, C.C.; Soverini, S.; Adrian, L.T.; Eide, C.A.; Zabriskie, M.S.; Lange, T.; Estrada, J.C.; et al. BCR-ABL1 compound mutations in tyrosine kinase inhibitor-resistant CML: Frequency and clonal relationships. Blood 2013, 121, 489–498. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, T.; Shakespeare, W.C.; Zhu, X.; Eide, C.A.; Rivera, V.M.; Wang, F.; Adrian, L.T.; Zhou, T.; Huang, W.S.; Xu, Q.; et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 2009, 16, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Pagnano, K.B.; Bendit, I.; Boquimpani, C.; De Souza, C.A.; Miranda, E.C.; Zalcberg, I.; Larripa, I.; Nardinelli, L.; Silveira, R.A.; Fogliatto, L.; et al. BCR-ABL mutations in chronic myeloid leukemia treated with tyrosine kinase inhibitors and impact on survival. Cancer Investig. 2015, 33, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Kantarjian, H.; Jones, D.; Talpaz, M.; Bekele, N.; O’Brien, S.; Zhou, X.; Luthra, R.; Garcia-Manero, G.; Giles, F.; et al. Frequency and clinical significance of BCR-ABL mutations in patients with chronic myeloid leukemia treated with imatinib mesylate. Leukemia 2006, 20, 1767–1773. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.N.; Haislip, S.; Sharpe, J.; Hess, T.; Gilmore, J.; Jackson, J.; Sail, K.R.; Ericson, S.G.; Chen, L. Assessment of treatment and monitoring patterns and subsequent outcomes among patients with chronic myeloid leukemia treated with imatinib in a community setting. Curr. Med. Res. Opin. 2014, 30, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Nestal de Moraes, G.; Souza, P.S.; Costas, F.C.; Vasconcelos, F.C.; Reis, F.R.; Maia, R.C. The Interface between BCR-ABL-Dependent and -Independent Resistance Signaling Pathways in Chronic Myeloid Leukemia. Leuk. Res. Treat. 2012, 2012, 671702. [Google Scholar] [CrossRef] [PubMed]
- Copland, M.; Hamilton, A.; Elrick, L.J.; Baird, J.W.; Allan, E.K.; Jordanides, N.; Barow, M.; Mountford, J.C.; Holyoake, T.L. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood 2006, 107, 4532–4539. [Google Scholar] [CrossRef] [PubMed]
- Corbin, A.S.; Agarwal, A.; Loriaux, M.; Cortes, J.; Deininger, M.W.; Druker, B.J. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J. Clin. Investig. 2011, 121, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Breier, A.; Gibalova, L.; Seres, M.; Barancik, M.; Sulova, Z. New insight into p-glycoprotein as a drug target. Anticancer Agents Med. Chem. 2013, 13, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Lavi, O.; Hall, M.D.; Gillet, J.P. Toward a Better Understanding of the Complexity of Cancer Drug Resistance. Ann. Rev. Pharmacol. Toxicol. 2016, 56, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Lage, H. An overview of cancer multidrug resistance: A still unsolved problem. Cell. Mol. life Sci. 2008, 65, 3145–3167. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.D.; Doyle, L.A. Mining our ABCs: Pharmacogenomic approach for evaluating transporter function in cancer drug resistance. Cancer Cell 2004, 6, 105–107. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Klukovits, A.; Krajcsi, P. Mechanisms and therapeutic potential of inhibiting drug efflux transporters. Expert Opin. Drug Metab. Toxicol. 2015, 11, 907–920. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.W.; Ruefli, A.A.; Tainton, K.M.; Smyth, M.J. A role for P-glycoprotein in regulating cell death. Leuk. Lymphoma 2000, 38, 1–11. [Google Scholar] [PubMed]
- Carter, A.; Dann, E.J.; Katz, T.; Shechter, Y.; Oliven, A.; Regev, R.; Eytan, E.; Rowe, J.M.; Eytan, G.D. Cells from chronic myelogenous leukaemia patients at presentation exhibit multidrug resistance not mediated by either MDR1 or MRP1. Br. J. Haematol. 2001, 114, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, F.C.; Cavalcanti, G.B., Jr.; Silva, K.L.; de Meis, E.; Kwee, J.K.; Rumjanek, V.M.; Maia, R.C. Contrasting features of MDR phenotype in leukemias by using two fluorochromes: Implications for clinical practice. Leuk. Res. 2007, 31, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, F.C.; Silva, K.L.; Souza, P.S.; Silva, L.F.; Moellmann-Coelho, A.; Klumb, C.E.; Maia, R.C. Variation of MDR proteins expression and activity levels according to clinical status and evolution of CML patients. Cytom. Part B Clin. Cytom. 2011, 80, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, F.C.; Nestal de Moraes, G.; Moellmann-Coelho, A.; Maia, R.C. Phosphorylated Crkl reduction levels are associated with the lowest P-glycoprotein activity levels in cells from chronic myeloid leukemia patients. Leuk. Res. 2013, 37, 1711–1718. [Google Scholar] [CrossRef] [PubMed]
- Vivona, D.; Lima, L.T.; Rodrigues, A.C.; Bueno, C.T.; Alcantara, G.K.; Barros, L.S.; De Moraes Hungria, V.T.; Chiattone, C.S.; De Lourdes Lopes Ferrari Chauffaille, M.; Guerra-Shinohara, E.M. ABCB1 haplotypes are associated with P-gp activity and affect a major molecular response in chronic myeloid leukemia patients treated with a standard dose of imatinib. Oncol. Lett. 2014. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Park, C.J.; Kim, D.Y.; Lee, B.R.; Kim, Y.J.; Cho, Y.U.; Jang, S. MRP1 and P-glycoprotein expression assays would be useful in the additional detection of treatment non-responders in CML patients without ABL1 mutation. Leuk. Res. 2015, 39, 1109–1116. [Google Scholar] [CrossRef] [PubMed]
- Silva, K.L.; de Souza, P.S.; Nestal de Moraes, G.; Moellmann-Coelho, A.; Vasconcelos Fda, C.; Maia, R.C. XIAP and P-glycoprotein co-expression is related to imatinib resistance in chronic myeloid leukemia cells. Leuk. Res. 2013, 37, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Razga, F.; Racil, Z.; Machova Polakova, K.; Buresova, L.; Klamova, H.; Zackova, D.; Dvorakova, D.; Polivkova, V.; Cetkovsky, P.; Mayer, J. The predictive value of human organic cation transporter 1 and ABCB1 expression levels in different cell populations of patients with de novo chronic myelogenous leukemia. Int. J. Hematol. 2011, 94, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, H.; Sharma, P.; Malhotra, B.; Bhargava, S.; Jasuja, S.; Kumar, M. Molecular response to imatinib & its correlation with mRNA expression levels of imatinib influx & efflux transporters in patients with chronic myeloid leukaemia in chronic phase. Indian J. Med. Res. 2015, 142, 175–182. [Google Scholar] [PubMed]
- Da Cunha Vasconcelos, F.; Mauricio Scheiner, M.A.; Moellman-Coelho, A.; Mencalha, A.L.; Renault, I.Z.; Rumjanek, V.M.; Maia, R.C. Low ABCB1 and high OCT1 levels play a favorable role in the molecular response to imatinib in CML patients in the community clinical practice. Leuk. Res. 2016, 51, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, M.; Hanfstein, B.; Erben, P.; Wolf, D.; Ernst, T.; Fabarius, A.; Saussele, S.; Purkayastha, D.; Woodman, R.C.; Hofmann, W.K.; et al. MDR1 expression predicts outcome of Ph+ chronic phase CML patients on second-line nilotinib therapy after imatinib failure. Leukemia 2014, 28, 1478–1485. [Google Scholar] [CrossRef] [PubMed]
- Eadie, L.N.; Dang, P.; Saunders, V.A.; Yeung, D.T.; Osborn, M.P.; Grigg, A.P.; Hughes, T.P.; White, D.L. The clinical significance of ABCB1 overexpression in predicting outcome of CML patients undergoing first-line imatinib treatment. Leukemia 2017, 31, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Stromskaya, T.P.; Rybalkina, E.Y.; Kruglov, S.S.; Zabotina, T.N.; Mechetner, E.B.; Turkina, A.G.; Stavrovskaya, A.A. Role of P-glycoprotein in evolution of populations of chronic myeloid leukemia cells treated with imatinib. Biochem. Biokhimiia 2008, 73, 29–37. [Google Scholar] [CrossRef]
- Yeung, D.T.; Osborn, M.P.; White, D.L.; Branford, S.; Braley, J.; Herschtal, A.; Kornhauser, M.; Issa, S.; Hiwase, D.K.; Hertzberg, M.; et al. TIDEL-II: First-line use of imatinib in CML with early switch to nilotinib for failure to achieve time-dependent molecular targets. Blood 2015, 125, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Mahon, F.X.; Hayette, S.; Lagarde, V.; Belloc, F.; Turcq, B.; Nicolini, F.; Belanger, C.; Manley, P.W.; Leroy, C.; Etienne, G.; et al. Evidence that resistance to nilotinib may be due to BCR-ABL, Pgp, or Src kinase overexpression. Cancer Res. 2008, 68, 9809–9816. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Sauna, Z.E.; Ambudkar, S.V. Evidence for the interaction of imatinib at the transport-substrate site(s) of the multidrug-resistance-linked ABC drug transporters ABCB1 (P-glycoprotein) and ABCG2. Leukemia 2008, 22, 445–447. [Google Scholar] [CrossRef] [PubMed]
- Fung, K.L.; Gottesman, M.M. A synonymous polymorphism in a common MDR1 (ABCB1) haplotype shapes protein function. Biochim. Biophys. 2009, 1794, 860–871. [Google Scholar] [CrossRef] [PubMed]
- Vivona, D.; Bueno, C.T.; Lima, L.T.; Hirata, R.D.; Hirata, M.H.; Luchessi, A.D.; Zanichelli, M.A.; Chiattone, C.S.; Guerra-Shinohara, E.M. ABCB1 haplotype is associated with major molecular response in chronic myeloid leukemia patients treated with standard-dose of imatinib. Blood Cells Mol. Dis. 2012, 48, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Dulucq, S.; Bouchet, S.; Turcq, B.; Lippert, E.; Etienne, G.; Reiffers, J.; Molimard, M.; Krajinovic, M.; Mahon, F.X. Multidrug resistance gene (MDR1) polymorphisms are associated with major molecular responses to standard-dose imatinib in chronic myeloid leukemia. Blood 2008, 112, 2024–2027. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Evans, S.; Rivory, L.P.; Hoskins, J.M.; Mann, G.J.; Farlow, D.; Clarke, C.L.; Balleine, R.L.; Gurney, H. Hepatic technetium Tc 99m-labeled sestamibi elimination rate and ABCB1 (MDR1) genotype as indicators of ABCB1 (P-glycoprotein) activity in patients with cancer. Clin. Pharmacol. Ther. 2005, 77, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Hamada, A.; Miyano, H.; Watanabe, H.; Saito, H. Interaction of imatinib mesilate with human P-glycoprotein. J. Pharmaco. Exp. Ther. 2003, 307, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Dulucq, S.; Krajinovic, M. The pharmacogenetics of imanitib. Genome Med. 2010, 2, 85. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.N.; Li, J.Y.; Miao, K.R.; Qiao, C.; Zhang, S.J.; Qiu, H.R.; Qian, S.X. Multidrug resistance gene (MDR1) polymorphisms correlate with imatinib response in chronic myeloid leukemia. Med. Oncol. 2011, 28, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Deenik, W.; van der Holt, B.; Janssen, J.J.; Chu, I.W.; Valk, P.J.; Ossenkoppele, G.J.; van der Heiden, I.P.; Sonneveld, P.; van Schaik, R.H.; Cornelissen, J.J. Polymorphisms in the multidrug resistance gene MDR1 (ABCB1) predict for molecular resistance in patients with newly diagnosed chronic myeloid leukemia receiving high-dose imatinib. Blood 2010, 116, 6144–6145; Author reply 6145–6146. [Google Scholar] [CrossRef] [PubMed]
- Au, A.; Aziz Baba, A.; Goh, A.S.; Wahid Fadilah, S.A.; Teh, A.; Rosline, H.; Ankathil, R. Association of genotypes and haplotypes of multi-drug transporter genes ABCB1 and ABCG2 with clinical response to imatinib mesylate in chronic myeloid leukemia patients. Biomed. Pharmacother. 2014, 68, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Adeagbo, B.A.; Bolaji, O.O.; Olugbade, T.A.; Durosinmi, M.A.; Bolarinwa, R.A.; Masimirembwa, C. Influence of CYP3A5*3 and ABCB1 C3435T on clinical outcomes and trough plasma concentrations of imatinib in Nigerians with chronic myeloid leukaemia. J. Clin. Pharm. Ther. 2016, 41, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Maffioli, M.; Camos, M.; Gaya, A.; Hernandez-Boluda, J.C.; Alvarez-Larran, A.; Domingo, A.; Granell, M.; Guillem, V.; Vallansot, R.; Costa, D.; et al. Correlation between genetic polymorphisms of the hOCT1 and MDR1 genes and the response to imatinib in patients newly diagnosed with chronic-phase chronic myeloid leukemia. Leuk. Res. 2011, 35, 1014–1019. [Google Scholar] [CrossRef] [PubMed]
- Angelini, S.; Soverini, S.; Ravegnini, G.; Barnett, M.; Turrini, E.; Thornquist, M.; Pane, F.; Hughes, T.P.; White, D.L.; Radich, J.; et al. Association between imatinib transporters and metabolizing enzymes genotype and response in newly diagnosed chronic myeloid leukemia patients receiving imatinib therapy. Haematologica 2013, 98, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A.; Elsalakawy, W.A. ABCB1 haplotypes but not individual SNPs predict for optimal response/failure in Egyptian patients with chronic-phase chronic myeloid leukemia receiving imatinib mesylate. Med. Oncol. 2014, 31, 279. [Google Scholar] [CrossRef] [PubMed]
- Elghannam, D.M.; Ibrahim, L.; Ebrahim, M.A.; Azmy, E.; Hakem, H. Association of MDR1 gene polymorphism (G2677T) with imatinib response in Egyptian chronic myeloid leukemia patients. Hematology 2014, 19, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Sriharsha, L.; Xu, W.; Kamel-Reid, S.; Liu, X.; Siminovitch, K.; Messner, H.A.; Lipton, J.H. Clinical relevance of a pharmacogenetic approach using multiple candidate genes to predict response and resistance to imatinib therapy in chronic myeloid leukemia. Clin. Cancer Res. 2009, 15, 4750–4758. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Miura, M.; Scott, S.A.; Kagaya, H.; Kameoka, Y.; Tagawa, H.; Saitoh, H.; Fujishima, N.; Yoshioka, T.; Hirokawa, M.; et al. Influence of CYP3A5 and drug transporter polymorphisms on imatinib trough concentration and clinical response among patients with chronic phase chronic myeloid leukemia. J. Hum. Genet. 2010, 55, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Vine, J.; Cohen, S.B.; Ruchlemer, R.; Goldschmidt, N.; Levin, M.; Libster, D.; Gural, A.; Gatt, M.E.; Lavie, D.; Ben-Yehuda, D.; et al. Polymorphisms in the human organic cation transporter and the multidrug resistance gene: Correlation with imatinib levels and clinical course in patients with chronic myeloid leukemia. Leuk. Lymphoma 2014, 55, 2525–2531. [Google Scholar] [CrossRef] [PubMed]
- Kimchi-Sarfaty, C.; Oh, J.M.; Kim, I.W.; Sauna, Z.E.; Calcagno, A.M.; Ambudkar, S.V.; Gottesman, M.M. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science 2007, 315, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Hoffmeyer, S.; Burk, O.; von Richter, O.; Arnold, H.P.; Brockmoller, J.; Johne, A.; Cascorbi, I.; Gerloff, T.; Roots, I.; Eichelbaum, M.; et al. Functional polymorphisms of the human multidrug-resistance gene: Multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc. Natl. Acad. Sci. USA 2000, 97, 3473–3478. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Lamba, J.K.; Zhang, L.; Schuetz, E.; Shukla, N.; Meibohm, B.; Yates, C.R. G2677T and C3435T genotype and haplotype are associated with hepatic ABCB1 (MDR1) expression. J. Clin. Pharmacol. 2006, 46, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Penna, G.; Allegra, A.; Alonci, A.; Aguennouz, M.; Garufi, A.; Cannavo, A.; Gerace, D.; Alibrandi, A.; Musolino, C. MDR-1 polymorphisms (G2677T and C3435T) in B-chronic lymphocytic leukemia: An impact on susceptibility and prognosis. Med. Oncol. 2011, 28, 1549–1554. [Google Scholar] [CrossRef] [PubMed]
- Goreva, O.B.; Grishanova, A.Y.; Mukhin, O.V.; Domnikova, N.P.; Lyakhovich, V.V. Possible prediction of the efficiency of chemotherapy in patients with lymphoproliferative diseases based on MDR1 gene G2677T and C3435T polymorphisms. Bull. Exp. Biol. Med. 2003, 136, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Jamroziak, K.; Mlynarski, W.; Balcerczak, E.; Mistygacz, M.; Trelinska, J.; Mirowski, M.; Bodalski, J.; Robak, T. Functional C3435T polymorphism of MDR1 gene: An impact on genetic susceptibility and clinical outcome of childhood acute lymphoblastic leukemia. Eur. J. Haematol. 2004, 72, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Yaya, K.; Hind, D.; Meryem, Q.; Asma, Q.; Said, B.; Sellama, N. Single nucleotide polymorphisms of multidrug resistance gene 1 (MDR1) and risk of chronic myeloid leukemia. Tumour Biol. 2014, 35, 10969–10975. [Google Scholar] [CrossRef] [PubMed]
- Gurney, H.; Wong, M.; Balleine, R.L.; Rivory, L.P.; McLachlan, A.J.; Hoskins, J.M.; Wilcken, N.; Clarke, C.L.; Mann, G.J.; Collins, M.; et al. Imatinib disposition and ABCB1 (MDR1, P-glycoprotein) genotype. Clin. Pharmacol. Ther. 2007, 82, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Galeotti, L.; Ceccherini, F.; Domingo, D.; Laurino, M.; Polillo, M.; Di Paolo, A.; Barate, C.; Fava, C.; D’Avolio, A.; Cervetti, G.; et al. Association of the hOCT1/ABCB1 genotype with efficacy and tolerability of imatinib in patients affected by chronic myeloid leukemia. Cancer Chemother. Pharmacol. 2017, 79, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Ambudkar, S.V.; Sauna, Z.E.; Gottesman, M.M.; Szakacs, G. A novel way to spread drug resistance in tumor cells: Functional intercellular transfer of P-glycoprotein (ABCB1). Trends Pharmacol. Sci. 2005, 26, 385–387. [Google Scholar] [CrossRef] [PubMed]
- Bebawy, M.; Combes, V.; Lee, E.; Jaiswal, R.; Gong, J.; Bonhoure, A.; Grau, G.E. Membrane microparticles mediate transfer of P-glycoprotein to drug sensitive cancer cells. Leukemia 2009, 23, 1643–1649. [Google Scholar] [CrossRef] [PubMed]
- Souza, P.S.; Madigan, J.P.; Gillet, J.P.; Kapoor, K.; Ambudkar, S.V.; Maia, R.C.; Gottesman, M.M.; Fung, K.L. Expression of the multidrug transporter P-glycoprotein is inversely related to that of apoptosis-associated endogenous TRAIL. Exp. Cell Res. 2015, 336, 318–328. [Google Scholar] [CrossRef] [PubMed]
- McDermott, M.; Eustace, A.J.; Busschots, S.; Breen, L.; Crown, J.; Clynes, M.; O’Donovan, N.; Stordal, B. In vitro Development of Chemotherapy and Targeted Therapy Drug-Resistant Cancer Cell Lines: A Practical Guide with Case Studies. Front. Oncol. 2014, 4, 40. [Google Scholar] [CrossRef] [PubMed]
- Rumjanek, V.M.; Trindade, G.S.; Wagner-Souza, K.; Meletti-de-Oliveira, M.C.; Marques-Santos, L.F.; Maia, R.C.; Capella, M.A.M. Multidrug resistance in tumour cells: Characterisation of the multidrug resistant cell line K562-Lucena 1. An. Acad. Bras. Cienc. 2001, 73, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Daflon-Yunes, N.; Pinto-Silva, F.E.; Vidal, R.S.; Novis, B.F.; Berguetti, T.; Lopes, R.R.; Polycarpo, C.; Rumjanek, V.M. Characterization of a multidrug-resistant chronic myeloid leukemia cell line presenting multiple resistance mechanisms. Mol. Cell. Biochem. 2013, 383, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Moreira, M.A.; Bagni, C.; de Pinho, M.B.; Mac-Cormick, T.M.; dos Santos Mota, M.; Pinto-Silva, F.E.; Daflon-Yunes, N.; Rumjanek, V.M. Changes in gene expression profile in two multidrug resistant cell lines derived from a same drug sensitive cell line. Leuk. Res. 2014, 38, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Mahon, F.X.; Deininger, M.W.; Schultheis, B.; Chabrol, J.; Reiffers, J.; Goldman, J.M.; Melo, J.V. Selection and characterization of BCR-ABL positive cell lines with differential sensitivity to the tyrosine kinase inhibitor STI571: Diverse mechanisms of resistance. Blood 2000, 96, 1070–1079. [Google Scholar] [PubMed]
- Alves, R.; Fonseca, A.R.; Goncalves, A.C.; Ferreira-Teixeira, M.; Lima, J.; Abrantes, A.M.; Alves, V.; Rodrigues-Santos, P.; Jorge, L.; Matoso, E.; et al. Drug transporters play a key role in the complex process of Imatinib resistance in vitro. Leuk. Res. 2015, 39, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.K.; Sodani, K.; Wang, S.R.; Kuang, Y.H.; Ashby, C.R., Jr.; Chen, X.; Chen, Z.S. Nilotinib (AMN107, Tasigna) reverses multidrug resistance by inhibiting the activity of the ABCB1/Pgp and ABCG2/BCRP/MXR transporters. Biochem. Pharmacol. 2009, 78, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Hiwase, D.K.; Saunders, V.; Hewett, D.; Frede, A.; Zrim, S.; Dang, P.; Eadie, L.; To, L.B.; Melo, J.; Kumar, S.; et al. Dasatinib cellular uptake and efflux in chronic myeloid leukemia cells: Therapeutic implications. Clin. Cancer Res. 2008, 14, 3881–3888. [Google Scholar] [CrossRef] [PubMed]
- Redaelli, S.; Perini, P.; Ceccon, M.; Piazza, R.; Rigolio, R.; Mauri, M.; Boschelli, F.; Giannoudis, A.; Gambacorti-Passerini, C. In vitro and in vivo identification of ABCB1 as an efflux transporter of bosutinib. J. Hematol. Oncol. 2015, 8, 81. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.X.; Tiwari, A.K.; Wu, H.C.; Chen, Z.S. Overexpression of P-glycoprotein induces acquired resistance to imatinib in chronic myelogenous leukemia cells. Chin. J. Cancer 2012, 31, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Hiwase, D.K.; White, D.; Zrim, S.; Saunders, V.; Melo, J.V.; Hughes, T.P. Nilotinib-mediated inhibition of ABCB1 increases intracellular concentration of dasatinib in CML cells: Implications for combination TKI therapy. Leukemia 2010, 24, 658–660. [Google Scholar] [CrossRef] [PubMed]
- Dohse, M.; Scharenberg, C.; Shukla, S.; Robey, R.W.; Volkmann, T.; Deeken, J.F.; Brendel, C.; Ambudkar, S.V.; Neubauer, A.; Bates, S.E. Comparison of ATP-binding cassette transporter interactions with the tyrosine kinase inhibitors imatinib, nilotinib, and dasatinib. Drug Metab. Dispos. 2010, 38, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.F.; Slater, A.; Kantharidis, P.; Rischin, D.; Juneja, S.; Rossi, R.; Lee, G.; Parkin, J.D.; Zalcberg, J.R. Altered multidrug resistance phenotype caused by anthracycline analogues and cytosine arabinoside in myeloid leukemia. Blood 1999, 93, 4086–4095. [Google Scholar] [PubMed]
- Callaghan, R.; Crowley, E.; Potter, S.; Kerr, I.D. P-glycoprotein: So many ways to turn it on. J. Clin. Pharmacol. 2008, 48, 365–378. [Google Scholar] [CrossRef] [PubMed]
- De Souza, P.S.; Faccion, R.S.; Bernardo, P.S.; Maia, R.C. Membrane microparticles: Shedding new light into cancer cell communication. J. Cancer Res. Clin. Oncol. 2016, 142, 1395–1406. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, R.; Gong, J.; Sambasivam, S.; Combes, V.; Mathys, J.M.; Davey, R.; Grau, G.E.; Bebawy, M. Microparticle-associated nucleic acids mediate trait dominance in cancer. FASEB J. 2012, 26, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.F.; Luk, F.; Gong, J.; Jaiswal, R.; Grau, G.E.; Bebawy, M. Microparticles mediate MRP1 intercellular transfer and the re-templating of intrinsic resistance pathways. Pharmacol. Res. 2013, 76, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Rodrigues, V.; Di Luca, A.; Sousa, D.; Seca, H.; Meleady, P.; Henry, M.; Lima, R.T.; O’Connor, R.; Vasconcelos, M.H. Multidrug resistant tumour cells shed more microvesicle-like EVs and less exosomes than their drug-sensitive counterpart cells. Biochim. Biophys. Acta 2016, 1860, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Milani, G.; Lana, T.; Bresolin, S.; Aveic, S.; Pasto, A.; Frasson, C.; Te Kronnie, G. Expression Profiling of Circulating Microvesicles Reveals Intercellular Transmission of Oncogenic Pathways. Mol. Cancer Res. 2017, 15, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Corrado, C.; Raimondo, S.; Saieva, L.; Flugy, A.M.; De Leo, G.; Alessandro, R. Exosome-mediated crosstalk between chronic myelogenous leukemia cells and human bone marrow stromal cells triggers an interleukin 8-dependent survival of leukemia cells. Cancer Lett. 2014, 348, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Raimondo, S.; Saieva, L.; Corrado, C.; Fontana, S.; Flugy, A.; Rizzo, A.; De Leo, G.; Alessandro, R. Chronic myeloid leukemia-derived exosomes promote tumor growth through an autocrine mechanism. Cell Commun. Signal. 2015, 13, 8. [Google Scholar] [CrossRef] [PubMed]
- Corrado, C.; Saieva, L.; Raimondo, S.; Santoro, A.; De Leo, G.; Alessandro, R. Chronic myelogenous leukaemia exosomes modulate bone marrow microenvironment through activation of epidermal growth factor receptor. J. Cell. Mol. Med. 2016, 20, 1829–1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caivano, A.; Laurenzana, I.; De Luca, L.; La Rocca, F.; Simeon, V.; Trino, S.; D’Auria, F.; Traficante, A.; Maietti, M.; Izzo, T.; et al. High serum levels of extracellular vesicles expressing malignancy-related markers are released in patients with various types of hematological neoplastic disorders. Tumour Biol. 2015, 36, 9739–9752. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Hu, S.; Wang, J.; Cai, J.; Xiao, L.; Yu, L.; Wang, Z. MiR-27a modulates MDR1/P-glycoprotein expression by targeting HIPK2 in human ovarian cancer cells. Gynecol. Oncol. 2010, 119, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, B.B. Friend or foe: The role of microRNA in chemotherapy resistance. Acta Pharmacol. Sin. 2013, 34, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, S.; Tong, Y.; Steitz, J.A. Switching from repression to activation: MicroRNAs can up-regulate translation. Science 2007, 318, 1931–1934. [Google Scholar] [CrossRef] [PubMed]
- Haenisch, S.; Werk, A.N.; Cascorbi, I. MicroRNAs and their relevance to ABC transporters. Br. J. Clin. Pharmacol. 2014, 77, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Rodrigues, V.; Seca, H.; Sousa, D.; Sousa, E.; Lima, R.T.; Vasconcelos, M.H. The network of P-glycoprotein and microRNAs interactions. Int. J. Cancer 2014, 135, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wu, H.; Liu, X.; Evans, B.R.; Medina, D.J.; Liu, C.G.; Yang, J.M. Role of MicroRNA miR-27a and miR-451 in the regulation of MDR1/P-glycoprotein expression in human cancer cells. Biochem. Pharmacol. 2008, 76, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Toscano-Garibay, J.D.; Aquino-Jarquin, G. Regulation exerted by miRNAs in the promoter and UTR sequences: MDR1/P-gp expression as a particular case. DNA Cell Biol. 2012, 31, 1358–1364. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.D.; Zhang, H.; Zhang, P.; Zheng, Y.S.; Zhang, X.J.; Han, B.W.; Luo, X.Q.; Xu, L.; Zhou, H.; Qu, L.H.; et al. Down-regulated miR-331-5p and miR-27a are associated with chemotherapy resistance and relapse in leukaemia. J. Cell. Mol. Med. 2011, 15, 2164–2175. [Google Scholar] [CrossRef] [PubMed]
- Fei, J.; Li, Y.; Zhu, X.; Luo, X. miR-181a post-transcriptionally downregulates oncogenic RalA and contributes to growth inhibition and apoptosis in chronic myelogenous leukemia (CML). PLoS ONE 2012, 7, e32834. [Google Scholar] [CrossRef] [PubMed]
- Mosakhani, N.; Mustjoki, S.; Knuutila, S. Down-regulation of miR-181c in imatinib-resistant chronic myeloid leukemia. Mol. Cytogenet. 2013, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Fallah, P.; Amirizadeh, N.; Poopak, B.; Toogeh, G.; Arefian, E.; Kohram, F.; Hosseini Rad, S.M.; Kohram, M.; Teimori Naghadeh, H.; Soleimani, M. Expression pattern of key microRNAs in patients with newly diagnosed chronic myeloid leukemia in chronic phase. Int. J. Lab. Hematol. 2015, 37, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Flamant, S.; Ritchie, W.; Guilhot, J.; Holst, J.; Bonnet, M.L.; Chomel, J.C.; Guilhot, F.; Turhan, A.G.; Rasko, J.E. Micro-RNA response to imatinib mesylate in patients with chronic myeloid leukemia. Haematologica 2010, 95, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, L.; Li, N.; Miao, Y.; Zhou, H.; Jia, L. miR-9 regulates the multidrug resistance of chronic myelogenous leukemia by targeting ABCB1. Oncol. Rep. 2017, 37, 2193–2200. [Google Scholar] [CrossRef] [PubMed]
- Ohyashiki, K.; Umezu, T.; Katagiri, S.; Kobayashi, C.; Azuma, K.; Tauchi, T.; Okabe, S.; Fukuoka, Y.; Ohyashiki, J.H. Downregulation of Plasma miR-215 in Chronic Myeloid Leukemia Patients with Successful Discontinuation of Imatinib. Int. J. Mol. Sci. 2016, 17, 570. [Google Scholar] [CrossRef] [PubMed]
- Yap, E.; Norziha, Z.A.; Simbun, A.; Tumian, N.R.; Cheong, S.K.; Leong, C.F.; Wong, C.L. MicroRNAs that affect the Fanconi Anemia/BRCA pathway are downregulated in imatinib-resistant chronic myeloid leukemia patients without detectable BCR-ABL kinase domain mutations. Leuk. Res. 2017, 59, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Nestal de Moraes, G.; Silva, K.L.; Vasconcelos, F.C.; Maia, R.C. Survivin overexpression correlates with an apoptosis-resistant phenotype in chronic myeloid leukemia cells. Oncol. Rep. 2011, 25, 1613–1619. [Google Scholar] [PubMed]
- Reis, F.R.; Vasconcelos, F.C.; Pereira, D.L.; Moellman-Coelho, A.; Silva, K.L.; Maia, R.C. Survivin and P-glycoprotein are associated and highly expressed in late phase chronic myeloid leukemia. Oncol. Rep. 2011, 26, 471–478. [Google Scholar] [PubMed]
- Souza, P.S.; Vasconcelos, F.C.; De Souza Reis, F.R.; Nestal De Moraes, G.; Maia, R.C. P-glycoprotein and survivin simultaneously regulate vincristine-induced apoptosis in chronic myeloid leukemia cells. Int. J. Oncol. 2011, 39, 925–933. [Google Scholar] [PubMed]
- Bernardo, P.S.; Reis, F.R.; Maia, R.C. Imatinib increases apoptosis index through modulation of survivin subcellular localization in the blast phase of CML cells. Leuk. Res. 2012, 36, 1510–1516. [Google Scholar] [CrossRef] [PubMed]
- Stenner, M.; Weinell, A.; Ponert, T.; Hardt, A.; Hahn, M.; Preuss, S.F.; Guntinas-Lichius, O.; Klussmann, J.P. Cytoplasmic expression of survivin is an independent predictor of poor prognosis in patients with salivary gland cancer. Histopathology 2010, 57, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Sokal, J.E.; Cox, E.B.; Baccarani, M.; Tura, S.; Gomez, G.A.; Robertson, J.E.; Tso, C.Y.; Braun, T.J.; Clarkson, B.D.; Cervantes, F.; et al. Prognostic discrimination in “good-risk” chronic granulocytic leukemia. Blood 1984, 63, 789–799. [Google Scholar] [PubMed]
- Seca, H.; Lima, R.T.; Guimaraes, J.E.; Helena Vasconcelos, M. Simultaneous targeting of P-gp and XIAP with siRNAs increases sensitivity of P-gp overexpressing CML cells to imatinib. Hematology 2011, 16, 100–108. [Google Scholar] [CrossRef] [PubMed]
- List, A.F.; Kopecky, K.J.; Willman, C.L.; Head, D.R.; Slovak, M.L.; Douer, D.; Dakhil, S.R.; Appelbaum, F.R. Cyclosporine inhibition of P-glycoprotein in chronic myeloid leukemia blast phase. Blood 2002, 100, 1910–1912. [Google Scholar] [PubMed]
- Maia, R.C.; Carrico, M.K.; Klumb, C.E.; Noronha, H.; Coelho, A.M.; Vasconcelos, F.C.; Ruimanek, V.M. Clinical approach to circumvention of multidrug resistance in refractory leukemic patients: Association of cyclosporin A with etoposide. J. Exp. Clin. Cancer Res. 1997, 16, 419–424. [Google Scholar] [PubMed]
- Illmer, T.; Schaich, M.; Platzbecker, U.; Freiberg-Richter, J.; Oelschlagel, U.; von Bonin, M.; Pursche, S.; Bergemann, T.; Ehninger, G.; Schleyer, E. P-glycoprotein-mediated drug efflux is a resistance mechanism of chronic myelogenous leukemia cells to treatment with imatinib mesylate. Leukemia 2004, 18, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Mahon, F.X.; Belloc, F.; Lagarde, V.; Chollet, C.; Moreau-Gaudry, F.; Reiffers, J.; Goldman, J.M.; Melo, J.V. MDR1 gene overexpression confers resistance to imatinib mesylate in leukemia cell line models. Blood 2003, 101, 2368–2373. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Wang, L.; Clark, R.E.; Pirmohamed, M. Active transport of imatinib into and out of cells: Implications for drug resistance. Blood 2004, 104, 3739–3745. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, M.H. ATP-binding-cassette transporters in hematopoietic stem cells and their utility as therapeutical targets in acute and chronic myeloid leukemia. Leukemia 2007, 21, 2094–2102. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Turnquist, H.; Jackson, J.; Sgagias, M.; Yan, Y.; Gong, M.; Dean, M.; Sharp, J.G.; Cowan, K. The multidrug resistance transporter ABCG2 (breast cancer resistance protein 1) effluxes Hoechst 33342 and is overexpressed in hematopoietic stem cells. Clin. Cancer Res. 2002, 8, 22–28. [Google Scholar] [PubMed]
- Wang, F.; Wang, X.K.; Shi, C.J.; Zhang, H.; Hu, Y.P.; Chen, Y.F.; Fu, L.W. Nilotinib enhances the efficacy of conventional chemotherapeutic drugs in CD34(+)CD38(−) stem cells and ABC transporter overexpressing leukemia cells. Molecules 2014, 19, 3356–3375. [Google Scholar] [CrossRef] [PubMed]
- Sen, R.; Natarajan, K.; Bhullar, J.; Shukla, S.; Fang, H.B.; Cai, L.; Chen, Z.S.; Ambudkar, S.V.; Baer, M.R. The novel BCR-ABL and FLT3 inhibitor ponatinib is a potent inhibitor of the MDR-associated ATP-binding cassette transporter ABCG2. Mol. Cancer Ther. 2012, 11, 2033–2044. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Saunders, V.A.; Leclercq, T.M.; Hughes, T.P.; White, D.L. Ponatinib is not transported by ABCB1, ABCG2 or OCT-1 in CML cells. Leukemia 2015, 29, 1792–1794. [Google Scholar] [CrossRef] [PubMed]
- Rai, D.; Singh, J.K.; Roy, N.; Panda, D. Curcumin inhibits FtsZ assembly: An attractive mechanism for its antibacterial activity. Biochem. J. 2008, 410, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.S.; Huang, H.P.; Yang, J.S.; Wu, J.Y.; Hsia, T.C.; Lin, C.C.; Lin, C.W.; Kuo, C.L.; Gibson Wood, W.; Chung, J.G. DNA damage and endoplasmic reticulum stress mediated curcumin-induced cell cycle arrest and apoptosis in human lung carcinoma A-549 cells through the activation caspases cascade- and mitochondrial-dependent pathway. Cancer Lett. 2008, 272, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Rodrigues, V.; Oliveira, A.; Correia-da-Silva, M.; Pinto, M.; Lima, R.T.; Sousa, E.; Vasconcelos, M.H. A novel curcumin derivative which inhibits P-glycoprotein, arrests cell cycle and induces apoptosis in multidrug resistance cells. Bioorg. Med. Chem. 2017, 25, 581–596. [Google Scholar] [CrossRef] [PubMed]
- Mapoung, S.; Pitchakarn, P.; Yodkeeree, S.; Ovatlarnporn, C.; Sakorn, N.; Limtrakul, P. Chemosensitizing effects of synthetic curcumin analogs on human multi-drug resistance leukemic cells. Chem. Biol. Interact. 2016, 244, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Lin, M.T.; Yi, T.; Tang, Y.N.; Fan, L.L.; He, X.C.; Zhao, Z.Z.; Chen, H.B. Apoptosis sensitization by Euphorbia factor L1 in ABCB1-mediated multidrug resistant K562/ADR cells. Molecules 2013, 18, 12793–12808. [Google Scholar] [CrossRef] [PubMed]
- Netto, C.D.; da Silva, A.J.; Salustiano, E.J.; Bacelar, T.S.; Rica, I.G.; Cavalcante, M.C.; Rumjanek, V.M.; Costa, P.R. New pterocarpanquinones: Synthesis, antineoplasic activity on cultured human malignant cell lines and TNF-alpha modulation in human PBMC cells. Bioorg. Med. Chem. 2010, 18, 1610–1616. [Google Scholar] [CrossRef] [PubMed]
- Maia, R.C.; Vasconcelos, F.C.; de Sa Bacelar, T.; Salustiano, E.J.; da Silva, L.F.; Pereira, D.L.; Moellman-Coelho, A.; Netto, C.D.; da Silva, A.J.; Rumjanek, V.M.; et al. LQB-118, a pterocarpanquinone structurally related to lapachol [2-hydroxy-3-(3-methyl-2-butenyl)-1,4-naphthoquinone]: A novel class of agent with high apoptotic effect in chronic myeloid leukemia cells. Investig. New Drugs 2011, 29, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- De Sa Bacelar, T.; da Silva, A.J.; Costa, P.R.; Rumjanek, V.M. The pterocarpanquinone LQB 118 induces apoptosis in tumor cells through the intrinsic pathway and the endoplasmic reticulum stress pathway. Anticancer Drugs 2013, 24, 73–83. [Google Scholar] [CrossRef] [PubMed]
- De Faria, F.C.; Leal, M.E.; Bernardo, P.S.; Costa, P.R.; Maia, R.C. NFkappaB pathway and microRNA-9 and -21 are involved in sensitivity to the pterocarpanquinone LQB-118 in different CML cell lines. Anticancer Agents Med. Chem. 2015, 15, 345–352. [Google Scholar] [CrossRef] [PubMed]


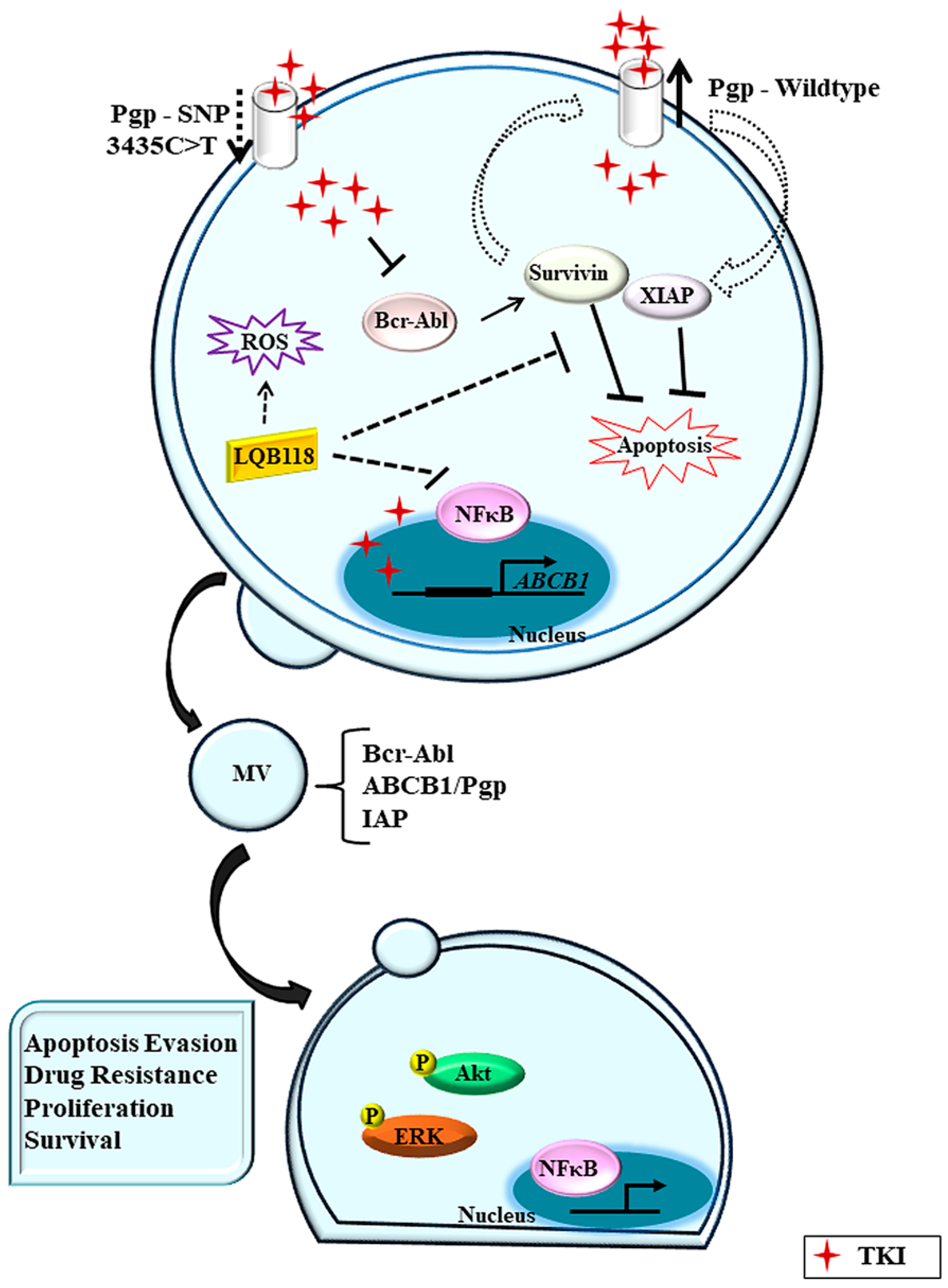{kind=link}
| Method | Number of Samples | Clinical Relevance | Reference |
|---|---|---|---|
| Tetramethylrosamine assay | 34 | Samples from patients with newly diagnosed CML showed resistance in comparison with cells from healthy donors. In later CML phases, samples exhibited more resistant profiles than samples on diagnosis of the disease. | [36] |
| Rho-123 assay and Pgp expression | 62 | Samples of patients in different CML phases, before or after receiving treatment, were analyzed. Pgp activity was present in 80% and Pgp expression in 84% of samples. The MDR phenotype was independent of the CML phase | [37] |
| Rho-123 assay and Pgp expression | 245 | Pgp expression and activity were present in all CML phases. The blast phase had the highest activity levels compared to chronic phase. | [38] |
| Rho-123 + CSA assay PhA + FTC assay | 55 | IM-resistant samples exhibited higher Pgp activity levels than the IM-sensitive ones. Greater Pgp activity (65.95%) was detected compared to BCRP activity (41.81%). There was no difference among the CML phases in relation to the frequency of Pgp positivity. | [39] |
| Rho-123 assay and Pgp expression | 224 | Pgp expression, but not Rho-123 efflux assay, showed differences between responder and non-responder IM-treated chronic phase patients. | [41] |
| Rho-123 assay and Pgp expression | 38 | Seventeen patients (44.7%) presented higher median Pgp expression levels in the advanced phased compared to the chronic phase. Pgp activity was found in 47.4% patients but this was not related to the CML phase. | [42] |
| ABCB1 mRNA levels | 30 | In samples analyzed on diagnosis of CML, there was no difference between ABCB1 mRNA levels and response to IM after 6 and 12 months of treatment. | [43] |
| ABCB1 mRNA levels | 63 | There was no difference between ABCB1 mRNA levels and response to IM treatment in patients in the chronic phase of CML. | [44] |
| ABCB1 mRNA levels | 68 | In most samples on diagnosis of CML (84%), low ABCB1 mRNA levels correlated with MMR, and high mRNA levels correlated with non-responders after IM treatment | [45] |
| ABCB1 mRNA levels | 83 | In the chronic phase of CML, high ABCB1 gene expression levels at the time of IM resistance were indicative of a favorable response to subsequent nilotinib treatment | [46] |
| ABCB1 mRNA levels | 155 | Patients in the chronic phase with increased levels of ABCB1 mRNA showed lower MMR rates at 12 and 24 months after IM treatment. | [47] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maia, R.C.; Vasconcelos, F.C.; Souza, P.S.; Rumjanek, V.M. Towards Comprehension of the ABCB1/P-Glycoprotein Role in Chronic Myeloid Leukemia. Molecules 2018, 23, 119. https://doi.org/10.3390/molecules23010119
Maia RC, Vasconcelos FC, Souza PS, Rumjanek VM. Towards Comprehension of the ABCB1/P-Glycoprotein Role in Chronic Myeloid Leukemia. Molecules. 2018; 23(1):119. https://doi.org/10.3390/molecules23010119
Chicago/Turabian StyleMaia, Raquel C., Flavia C. Vasconcelos, Paloma S. Souza, and Vivian M. Rumjanek. 2018. "Towards Comprehension of the ABCB1/P-Glycoprotein Role in Chronic Myeloid Leukemia" Molecules 23, no. 1: 119. https://doi.org/10.3390/molecules23010119
APA StyleMaia, R. C., Vasconcelos, F. C., Souza, P. S., & Rumjanek, V. M. (2018). Towards Comprehension of the ABCB1/P-Glycoprotein Role in Chronic Myeloid Leukemia. Molecules, 23(1), 119. https://doi.org/10.3390/molecules23010119