Structural Examination of Halogen-Bonded Co-Crystals of Tritopic Acceptors

Abstract
:1. Introduction
2. Results
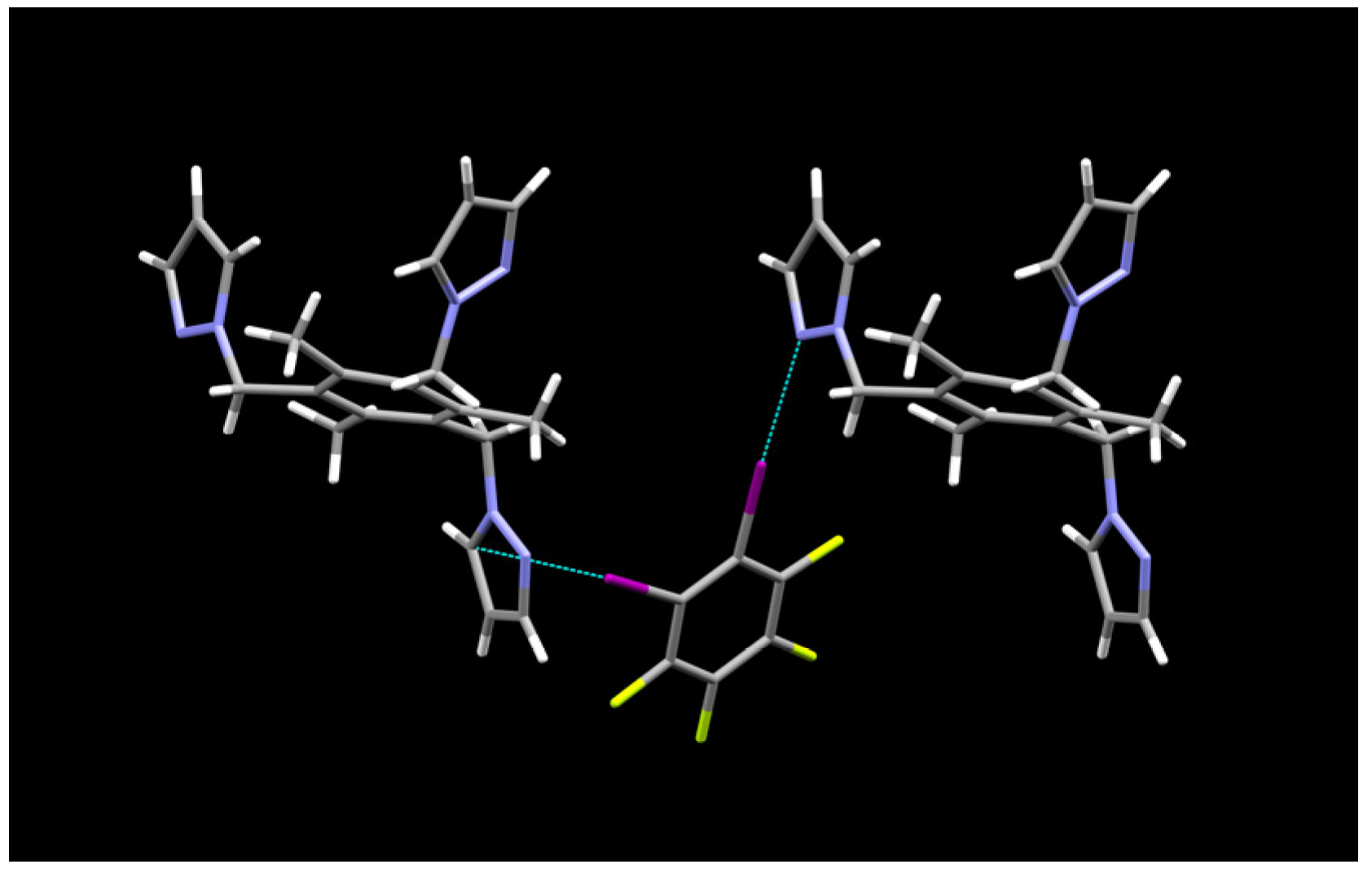
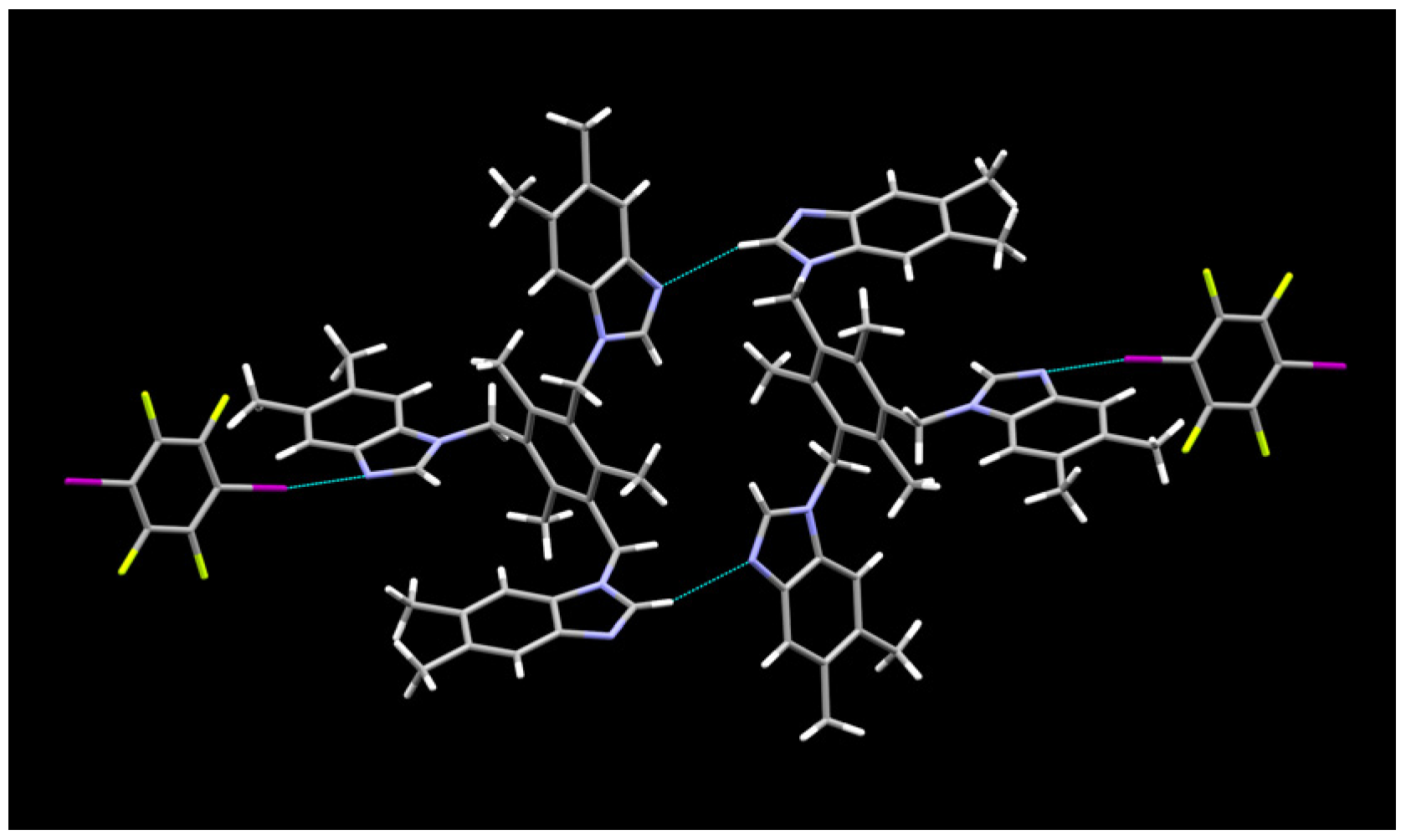
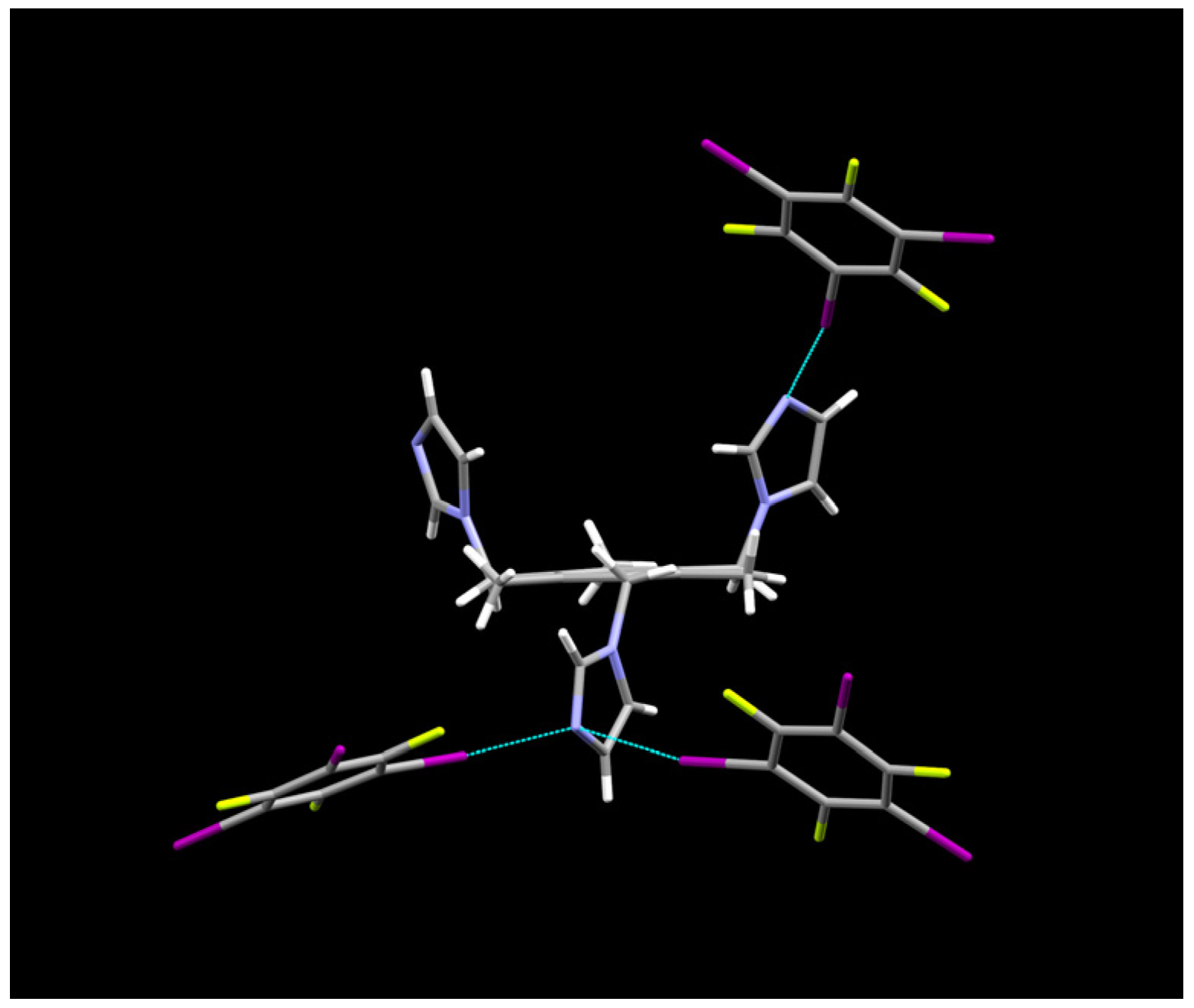
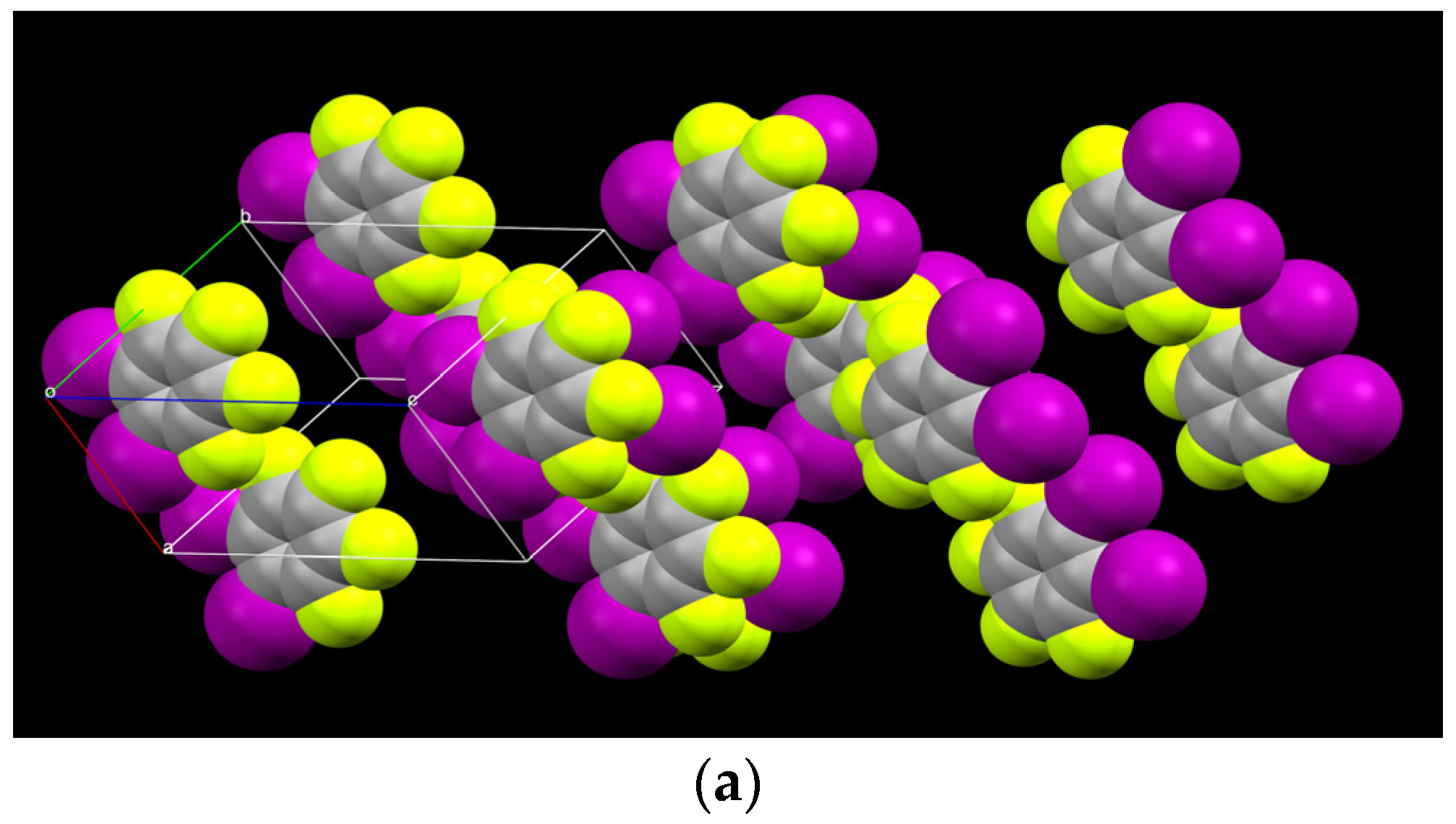
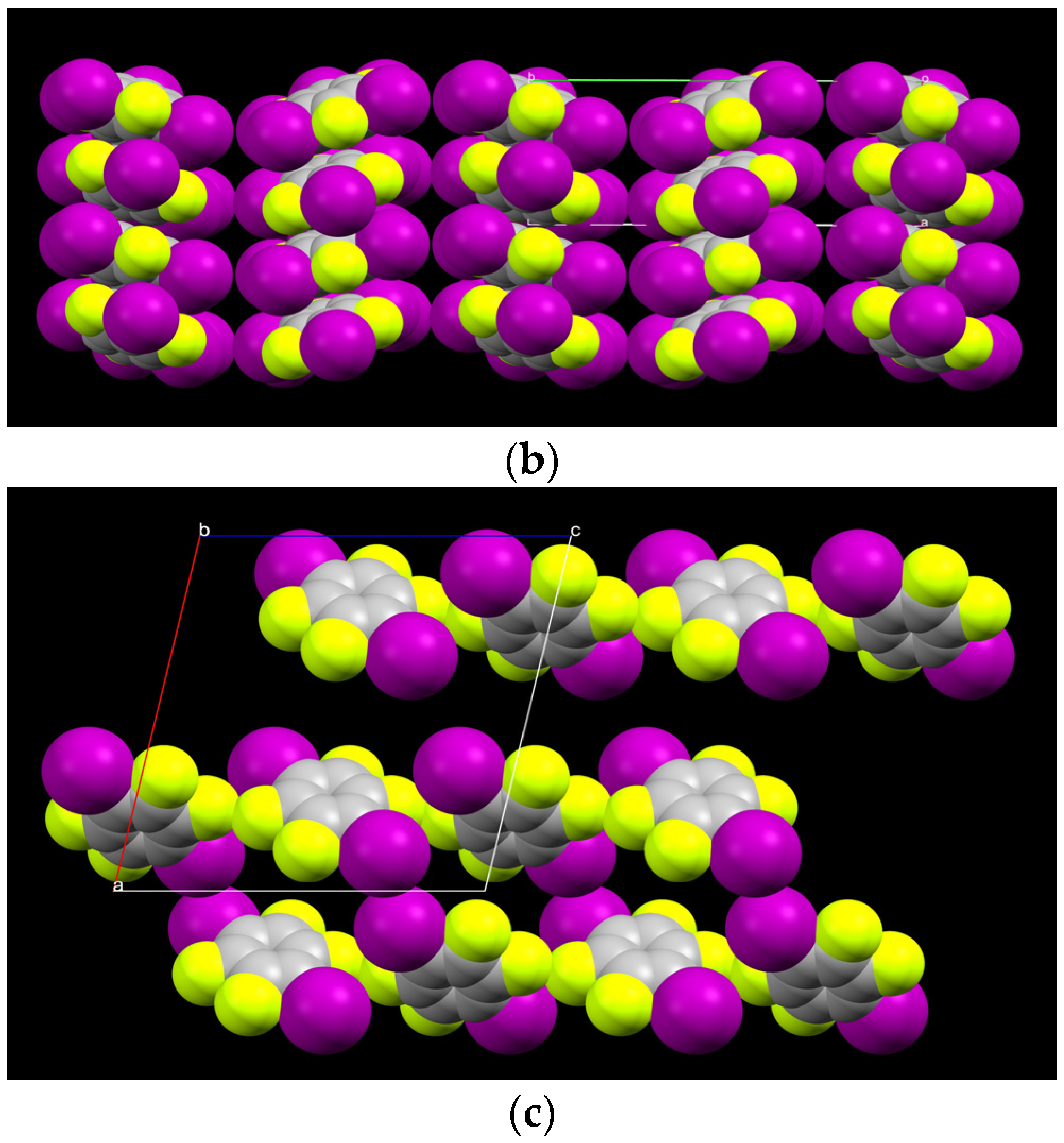
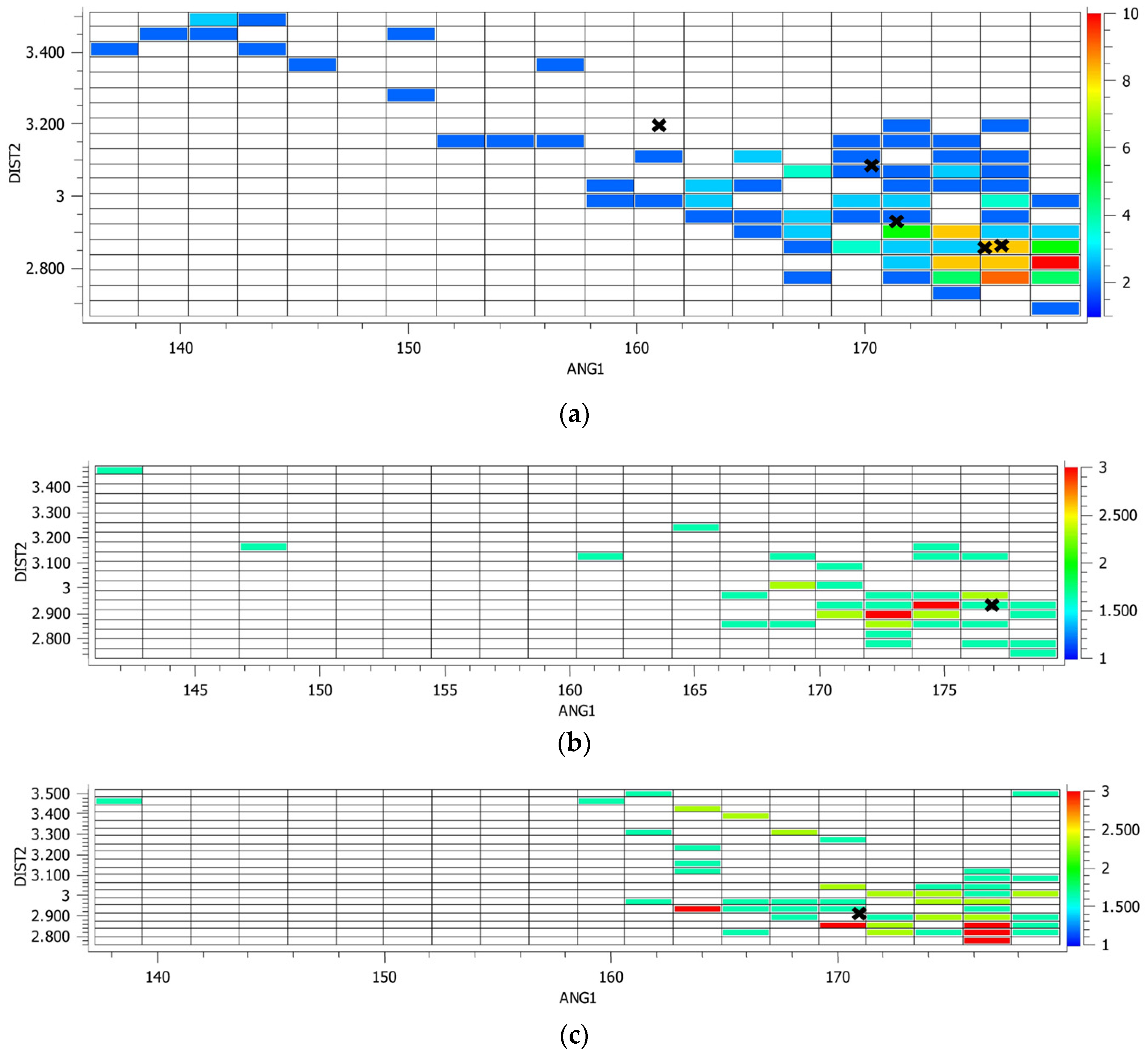
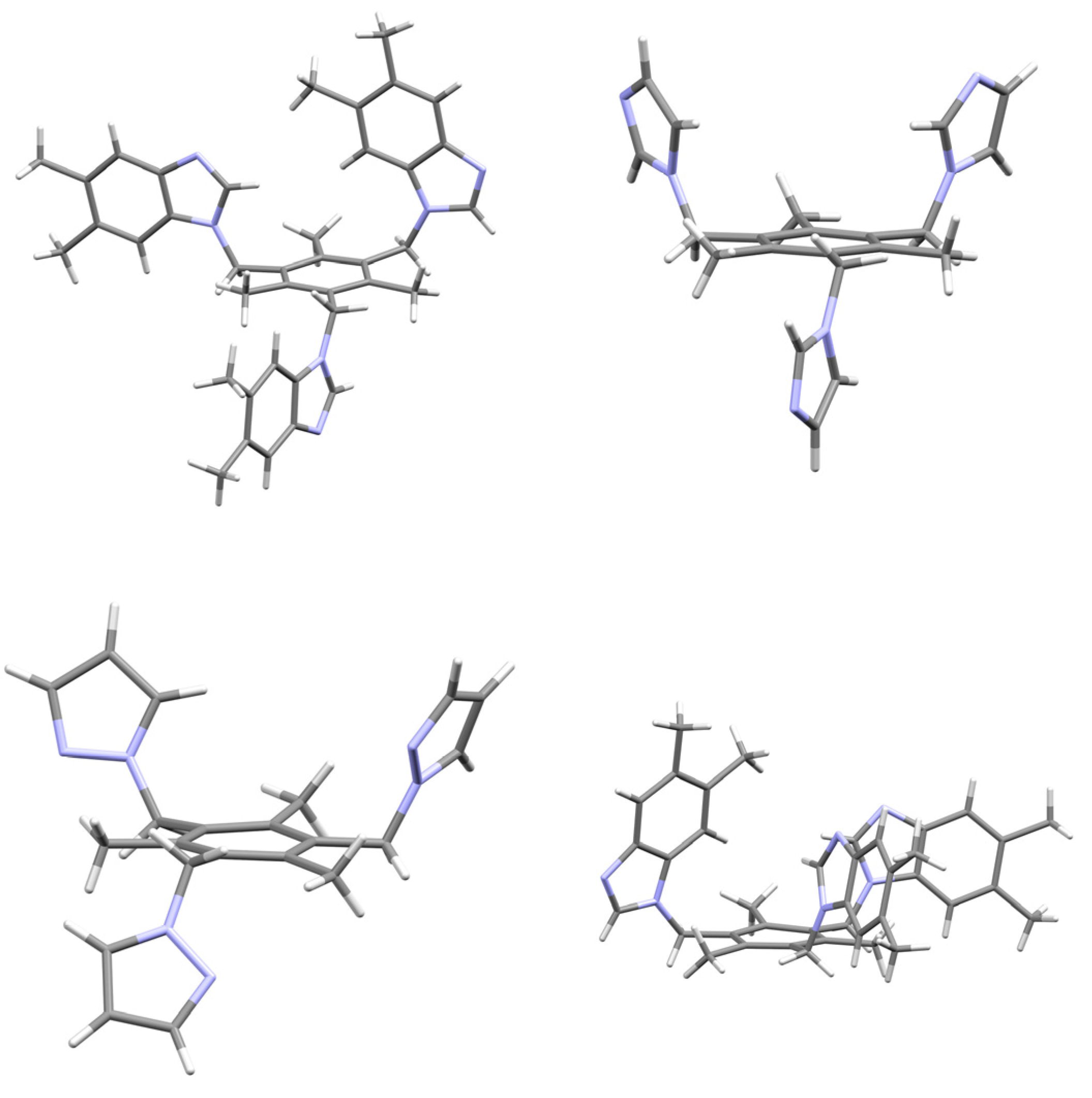
2.1. Description of Solid State Architectures
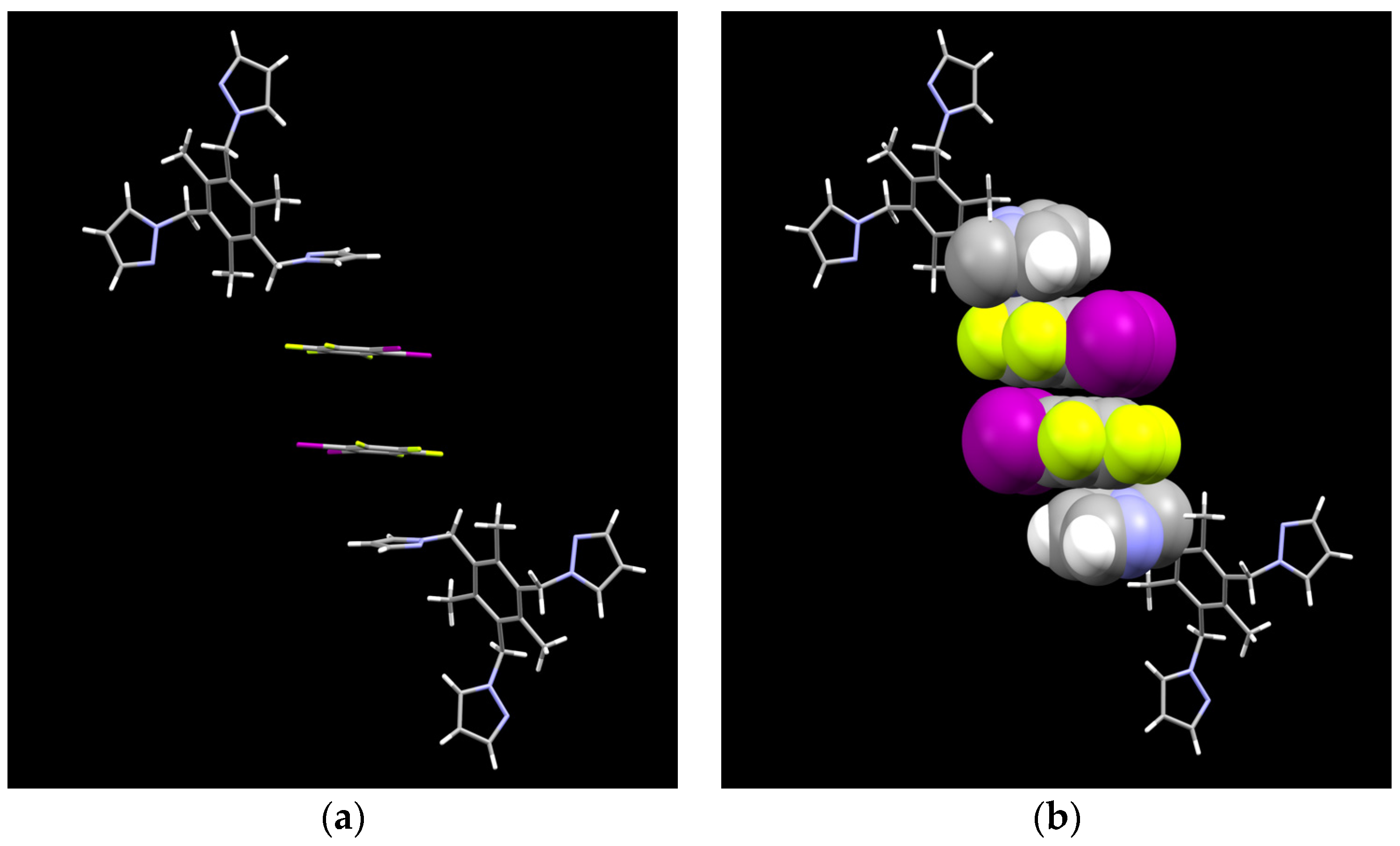
2.2. Aromatic Stacking
3. Discussion
4. Materials and Methods
4.1. Molecular Electrostatic Potential Calculations
4.2. CSD Search
4.3. Synthesis of Tritopic Acceptors
4.3.1. Synthesis of 1,3,5-Tris(bromomethyl)-2,4,6-trimethylbenzene (α)
4.3.2. Synthesis of 1,3,5-Tris(imidazole-1-yl-methyl)-2,4,6-trimethylbenzene (A)
4.3.3. Synthesis of 1,3,5-Tris(pyrazole-1-yl-methyl)-2,4,6-trimethylbenzene (B)
4.3.4. Synthesis of 1,3,5-Tris(3,5-dimethylpyrazole-1-yl-methyl)-2,4,6-trimethylbenzene (C)
4.3.5. Synthesis of 1,3,5-Tris(benzimidazole-1-yl-methyl)-2,4,6-trimethylbenzene (D)
4.3.6. Synthesis of 1,3,5-Tris(5,6-dimethylbenziimidazole-1-yl-methyl)-2,4,6-trimethylbenzene (E)
4.3.7. Synthesis of 1,3,5-Tris(bromomethyl)benzene (β)
4.3.8. Synthesis of 1,3,5-Tris(imidazole-1-yl-methyl)benzene (A′)
4.3.9. Synthesis of 1,3,5-Tris(pyrazole -1-yl-methyl)benzene (B′)
4.3.10. Synthesis of 1,3,5-Tris(3,5-dimethylpyrazole -1-yl-methyl)benzene (C′)
4.3.11. Synthesis of 1,3,5-Tris(benzimidazole -1-yl-methyl)benzene (D′)
4.3.12. Synthesis of 1,3,5-Tris(5,6-dimethylbenziimidazole -1-yl-methyl)benzene (E′)
4.4. Grinding Experiments
4.5. Synthesis of Co-Crystals
4.6. X-ray Crystallography
5. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Auffinger, P.; Hays, F.A.; Westhof, E.; Ho, P.S. Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. USA 2004, 101, 16789–16794. [Google Scholar] [CrossRef] [PubMed]
- Brinck, T.; Murray, J.S.; Politzer, P. Surface electrostatic potentials of halogenated methanes as indicators of directional intermolecular interactions. Int. J. Quantum Chem. 1992, 44, 57–64. [Google Scholar] [CrossRef]
- Kolář, M.H.; Hobza, P. Computer modeling of halogen bonds and other σ-hole interactions. Chem. Rev. 2016, 116, 5155–5187. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Paulsen, K.; Politzer, P. Molecular surface electrostatic potentials in the analysis of non-hydrogen-bonding noncovalent interactions. Proc. Indian Acad. Sci. Chem. Sci. 1994, 106, 267–275. [Google Scholar]
- Politzer, P.; Lane, P.; Concha, M.C.; Ma, Y.; Murray, J.S. An overview of halogen bonding. J. Mol. Model. 2007, 13, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Metrangolo, P.; Resnati, G. Halogen bonding: A paradigm in supramolecular chemistry. Chem. Eur. J. 2001, 7, 2511–2519. [Google Scholar] [CrossRef]
- Caronna, T.; Liantonio, R.; Logothetis, T.A.; Metrangolo, P.; Pilati, T.; Resnati, G. Halogen bonding and pi center dot center dot center dot pi stacking control reactivity in the solid state. J. Am. Chem. Soc. 2004, 126, 4500–4501. [Google Scholar] [CrossRef] [PubMed]
- Crihfield, A.; Hartwell, J.; Phelps, D.; Walsh, R.B.; Harris, J.L.; Payne, J.F.; Pennington, W.T.; Hanks, T.W. Crystal engineering through halogen bonding. 2. Complexes of diacetylene-linked heterocycles with organic iodides. Cryst. Growth Des. 2003, 3, 313–320. [Google Scholar] [CrossRef]
- Syssa-Magale, J.L.; Boubekeur, K.; Palvadeau, P.; Meerschaut, A.; Schollhorn, B. The tailoring of crystal structures via the self-assembly of organic coordination compounds by n center dot center dot center dot i non-covalent halogen bonds: Co-crystals of sterically hindered n-heterocycles and 1,4-diiodo-tetrafluorobenzene. CrystEngComm 2005, 7, 302–308. [Google Scholar] [CrossRef]
- Triguero, S.; Llusar, R.; Polo, V.; Fourmigué, M. Halogen bonding interactions ofsym-triiodotrifluorobenzene with halide anions: A combined structural and theoretical study. Cryst. Growth Des. 2008, 8, 2241–2247. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (iupac recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Kolář, M.; Hostaš, J.; Hobza, P. The strength and directionality of a halogen bond are co-determined by the magnitude and size of the σ-hole. Phys. Chem. Chem. Phys. 2014, 16, 9987–9996. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. PCCP 2010, 12, 7748–7757. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, S.; Wakisaka, A.; Ono, T.; Sonoda, T. Magnitude and origin of the attraction and directionality of the halogen bonds of the complexes of c6f5x and c6h5x (x = i, br, cl and f) with pyridine. Chem. Eur. J. 2012, 18, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Metrangolo, P.; Neukirch, H.; Pilati, T.; Resnati, G. Halogen bonding-based recognition processes: A world parallel to hydrogen bonding. Acc. Chem. Res. 2005, 38, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S. Halogen bonding: An interim discussion. ChemPhysChem 2013, 14, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Quiñonero, D.; Frontera, A.; Deyà, P.M. Substituent effects in halogen bonding complexes between aromatic donors and acceptors: A comprehensive ab initio study. PCCP 2011, 13, 20371–20379. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Sánchez-Sanz, G.; Elguero, J. Linear free energy relationships in halogen bonds. CrystEngComm 2013, 15, 3178–3186. [Google Scholar] [CrossRef]
- Duarte, D.J.R.; Sosa, G.L.; Peruchena, N.M.; Alkorta, I. Halogen bonding. The role of the polarizability of the electron-pair donor. Physical Chem. Chem. Phys. 2016, 18, 7300–7309. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.E.; Murray, J.S.; Fanfrlík, J.; Řezáč, J.; Solá, R.J.; Concha, M.C.; Ramos, F.M.; Politzer, P. Halogen bond tunability i: The effects of aromatic fluorine substitution on the strengths of halogen-bonding interactions involving chlorine, bromine, and iodine. J. Mol. Model. 2011, 17, 3309–3318. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.E.; Murray, J.S.; Fanfrlík, J.; Řezáč, J.; Solá, R.J.; Concha, M.C.; Ramos, F.M.; Politzer, P. Halogen bond tunability ii: The varying roles of electrostatic and dispersion contributions to attraction in halogen bonds. J. Mol. Model. 2012, 19, 4651–4659. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirst, A.R.; Escuder, B.; Miravet, J.F.; Smith, D.K. High-tech applications of self-assembling supramolecular nanostructured gel-phase materials: From regenerative medicine to electronic devices. Angew. Chem. Int. Ed. 2008, 47, 8002–8018. [Google Scholar] [CrossRef] [PubMed]
- Piepenbrock, M.-O.M.; Lloyd, G.O.; Clarke, N.; Steed, J.W. Metal- and anion-binding supramolecular gels. Chem. Rev. 2010, 110, 1960–2004. [Google Scholar] [CrossRef] [PubMed]
- Gilday, L.C.; Robinson, S.W.; Barendt, T.A.; Langton, M.J.; Mullaney, B.R.; Beer, P.D. Halogen bonding in supramolecular chemistry. Chem. Rev. 2015, 115, 7118–7195. [Google Scholar] [CrossRef] [PubMed]
- Lucassen, A.C.B.; Karton, A.; Leitus, G.; Shimon, L.J.W.; Martin, J.M.L.; van der Boom, M.E. Co-crystallization of sym-triiodo-trifluorobenzene with bipyridyl donors: Consistent formation of two instead of anticipated three n⋯i halogen bonds. Cryst. Growth Des. 2007, 7, 386–392. [Google Scholar] [CrossRef]
- Nicolas, I.; Barrière, F.; Jeannin, O.; Fourmigué, M. Sequential halogen bonding with ditopic donors: Σ-hole evolutions upon halogen bond formation. Cryst. Growth Des. 2016, 16, 2963–2971. [Google Scholar] [CrossRef]
- Carter, M.; Voth, A.R.; Scholfield, M.R.; Rummel, B.; Sowers, L.C.; Ho, P.S. Enthalpy–entropy compensation in biomolecular halogen bonds measured in DNA junctions. Biochemistry 2013, 52, 4891–4903. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, M.; Lucassen, A.C.B.; Shimon, L.J.W.; van der Boom, M.E. Cocrystallization of a tripyridyl donor with perfluorinated iodobenzene derivatives: Formation of different n⋯I halogen bonds determining network vs plain packing crystals. Cryst. Growth Des. 2008, 8, 786–790. [Google Scholar] [CrossRef]
- Van der Made, A.W.; Van der Made, R.H. A convenient procedure for bromomethylation of aromatic compounds. Selective mono-, bis-, or trisbromomethylation. J. Org. Chem. 1993, 58, 1262–1263. [Google Scholar] [CrossRef]
- Yuan, Y.; Jiang, Z.-L.; Yan, J.-M.; Gao, G.; Chan, A.S.C.; Xie, R.-G. A convenient and effective synthesis of tris-bridged tricationic azolophanes. Synth. Commun. 2007, 30, 4555–4561. [Google Scholar] [CrossRef]
- Hartshorn, C.M.; Steel, P.J. Poly(pyrazol-1-ylmethyl)benzenes: New multidentate ligands. Aust. J. Chem. 1995, 48, 1587–1599. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Smith, M.; Desper, J. Finding a single-molecule receptor for citramalic acid through supramolecular chelation. Can. J. Chem. 2015, 93, 822–825. [Google Scholar] [CrossRef]
- Wang, B.; Fang, J.; Li, B.; You, H.; Ma, D.; Hong, Z.; Li, W.; Su, Z. Soluble dendrimers europium(iii) β-diketonate complex for organic memory devices. Thin Solid Films 2008, 516, 3123–3127. [Google Scholar] [CrossRef]
- D’Anna, F.; Nimal Gunaratne, H.Q.; Lazzara, G.; Noto, R.; Rizzo, C.; Seddon, K.R. Solution and thermal behaviour of novel dicationic imidazolium ionic liquids. Org. Biomol. Chem. 2013, 11, 5836–5846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Xiao, R.; Gao, G.; Su, X.-Y.; Yu, H.; You, J.; Xie, R.-G. A direct synthetic approach to tripodal imidazole compounds. J. Chem. Res. 2002, 2002, 267–269. [Google Scholar] [CrossRef]
Sample Availability: Not available. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
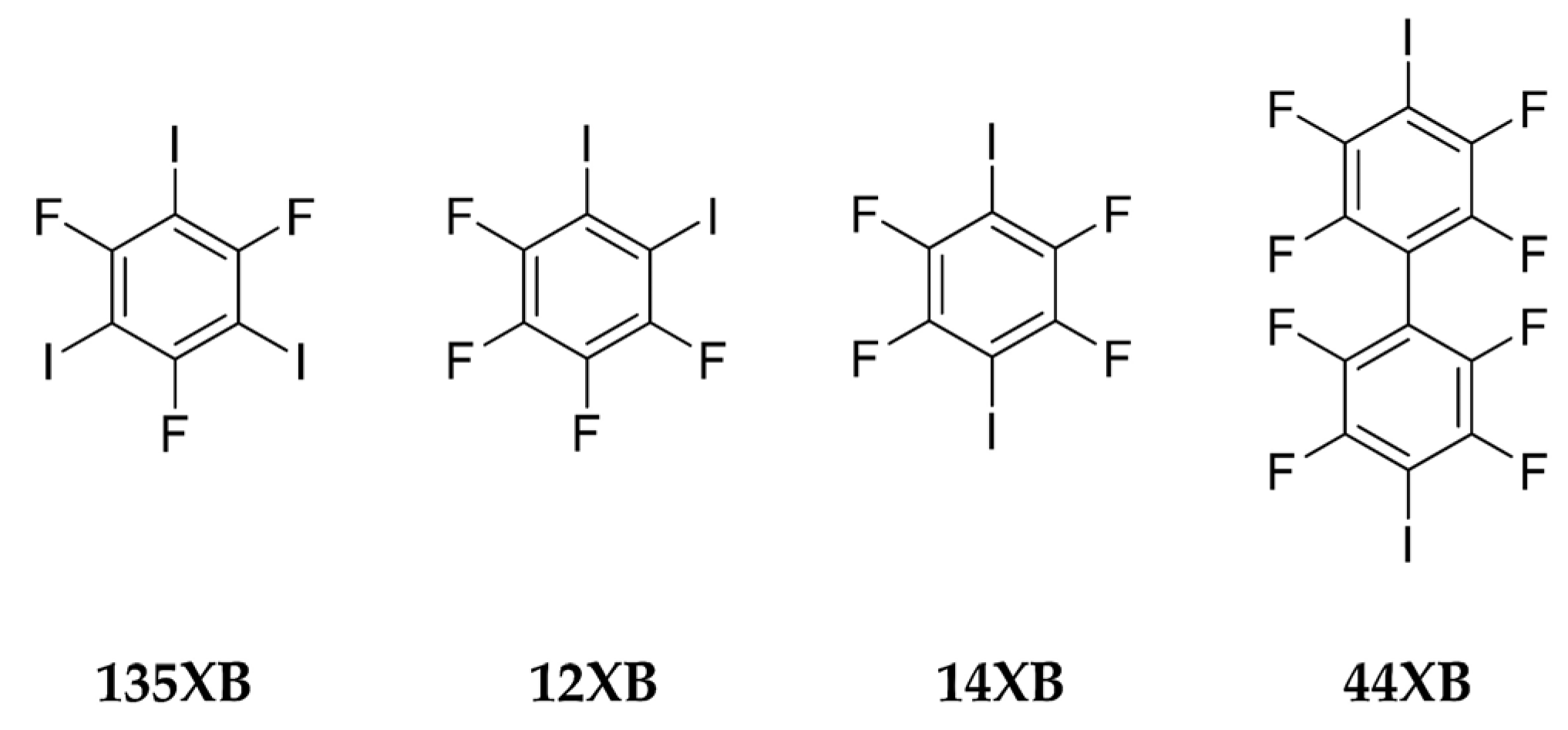{kind=link}
| Halogen Bond Donor | 135XB | 12XB | 14XB | |||
|---|---|---|---|---|---|---|
| MEP | 158 kJ/mol | 163 kJ/mol | 169 kJ/moL | |||
| No. Hits in the CSD | 30 hits | 19 hits | 100 hits | |||
| Coordination | 2 out of 3 | 3 out of 3 | 1 out of 2 | 2 out of 2 | 1 out of 2 | 2 out of 2 |
| Result | 16 | 14 | 2 | 17 | 3 | 97 |
| % outcome | 53% | 47% | 10.5% | 89.5% | 3% | 97% |
| Acceptors | %Success | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | A′ | B | B′ | C | C′ | D | D′ | E | E′ | |||
| Donors | 135XB | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | 100 |
| 12XB | - | √ | √ | √ | √ | √ | - | √ | √ | √ | 80 | |
| 14XB | √ | √ | √ | √ | - | - | - | - | √ | √ | 60 | |
| 44XB | - | - | √ | - | √ | - | √ | √ | √ | - | 50 | |
| %Success | 50 | 75 | 100 | 75 | 75 | 50 | 50 | 75 | 100 | 75 | ||
| Code | 14XB:E | 135XB:E | 12XB:B | 135XB:A |
|---|---|---|---|---|
| Formula moiety | (C39H42N6), (C6F4I2) | (C39H42N6), (C6F3I3), (C4H8O2) | (C21H24N6), (C6F4I2) | (C21H24N6), (C6F3I3) |
| Empirical formula | C45H42N6F4I2 | C49H50F3I3N6O2 | C27H24N6F4I2 | C27H24F3I3N6 |
| Molecular weight | 996.65 | 1192.65 | 762.32 | 870.22 |
| Crystal system | Monoclinic | Triclinic | Triclinic | Orthorhombic |
| Space group | P21/c | P ī | P ī | Pbca |
| a, Å | 16.337(6) | 9.764(4) | 9.255(3) | 7.931(2) |
| b, Å | 16.340(5) | 11.594(4) | 11.995(4) | 20.310(5) |
| c, Å | 15.642(5) | 22.901(9) | 13.507(5) | 37.342(10) |
| α, ° | 90 | 101.90(2) | 78.82(2) | 90 |
| β, ° | 102.862(13) | 97.56(3) | 84.15(2) | 90 |
| γ, ° | 90 | 99.98(2) | 68.959(19) | 90 |
| Volume, Å3 | 4071(2) | 2460.4(16) | 1372.1(8) | 296(2) |
| Z | 4 | 2 | 2 | 8 |
| Density, g/cm3 | 1.626 | 1.610 | 1.845 | 1.922 |
| T,K | 130 | 180 | 200 | 296(2) |
| X-ray wavelength, Å | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| µ, mm−1 | 1.604 | 1.961 | 2.348 | 3.164 |
| R1 (observed) | 0.0442 | 0.0338 | 0.0497 | 0.0638 |
| wR2 (all) | 0.1198 | 0.1100 | 0.1773 | 0.1740 |
| Compound | D–I⋯A | d(I⋯A) | d(D⋯A) | <(D–I⋯A) | Symmetry Operation |
|---|---|---|---|---|---|
| 12XB:B | C(29)–I(35)u⋯N(9)v | 2.934(7) | 5.03(1) | 177.0(2) | u = x,y,z; v = x,y,z |
| Co-crystal | |||||
| 14XB:E | C(46)–I(52)u⋯N(10)v | 2.922(4) | 5.024(6) | 171.3(2) | u = x,y,z; v = x,y,z |
| Co-crystal | |||||
| 135XB:E | C(48)–I(54)u⋯N(23)v | 2.936(5) | 5.063(7) | 171.8(1) | u = x,y,z; v = −x,1 − y,2 − z |
| Co-crystal | C(50)–I(56)u⋯N(10)v | 2.864(4) | 4.991(6) | 175.7(1) | u = x,y,z; v = −1 + x,1 + y,z |
| Solvate | |||||
| 135XB:A | C(32)–I(38)u⋯N(17)v | 2.869(9) | 4.95(1) | 175.9(3) | u = x,y,z; v = 1 + x,y,z |
| Co-crystal | C(28)–I(34)u⋯N(17)v | 3.206(9) | 5.23(1) | 162.7(3) | u = x,y,z; v = −1/2 + x, 3/2 − y,1 − z |
| C(30)–I(36)u⋯N(24)v | 3.093(2) | 5.15(2) | 170.9(4) | u = x,y,z; v = ½ − x,−1/2 + y,z |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andree, S.N.L.; Sinha, A.S.; Aakeröy, C.B. Structural Examination of Halogen-Bonded Co-Crystals of Tritopic Acceptors. Molecules 2018, 23, 163. https://doi.org/10.3390/molecules23010163
Andree SNL, Sinha AS, Aakeröy CB. Structural Examination of Halogen-Bonded Co-Crystals of Tritopic Acceptors. Molecules. 2018; 23(1):163. https://doi.org/10.3390/molecules23010163
Chicago/Turabian StyleAndree, Stefan N. L., Abhijeet S. Sinha, and Christer B. Aakeröy. 2018. "Structural Examination of Halogen-Bonded Co-Crystals of Tritopic Acceptors" Molecules 23, no. 1: 163. https://doi.org/10.3390/molecules23010163
APA StyleAndree, S. N. L., Sinha, A. S., & Aakeröy, C. B. (2018). Structural Examination of Halogen-Bonded Co-Crystals of Tritopic Acceptors. Molecules, 23(1), 163. https://doi.org/10.3390/molecules23010163