From Extraction of Local Structures of Protein Energy Landscapes to Improved Decoy Selection in Template-Free Protein Structure Prediction


Abstract
:1. Introduction
Related Work
2. Results
2.1. Evaluation Setup
2.1.1. Evaluation Metrics
2.1.2. Experimental Setup
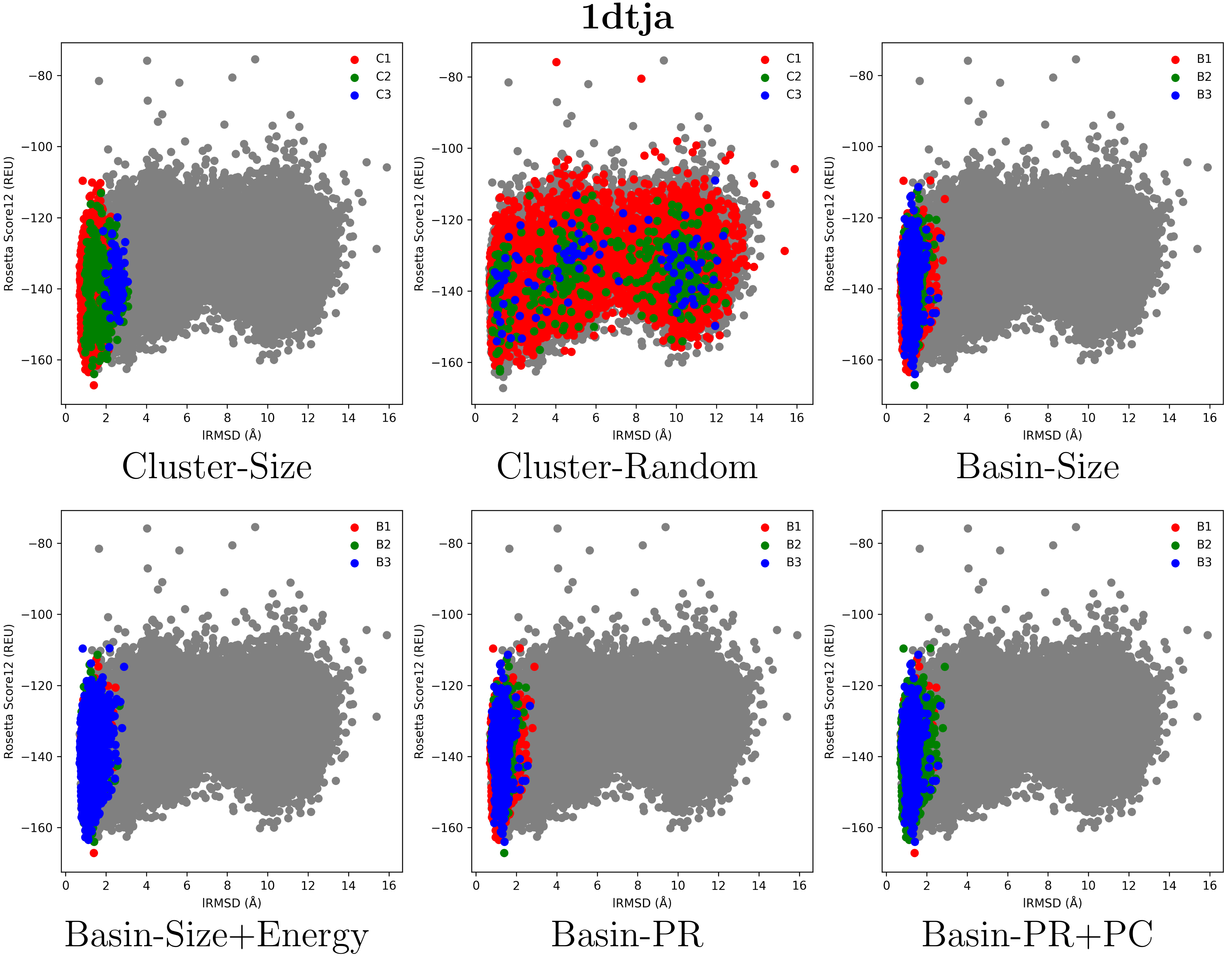
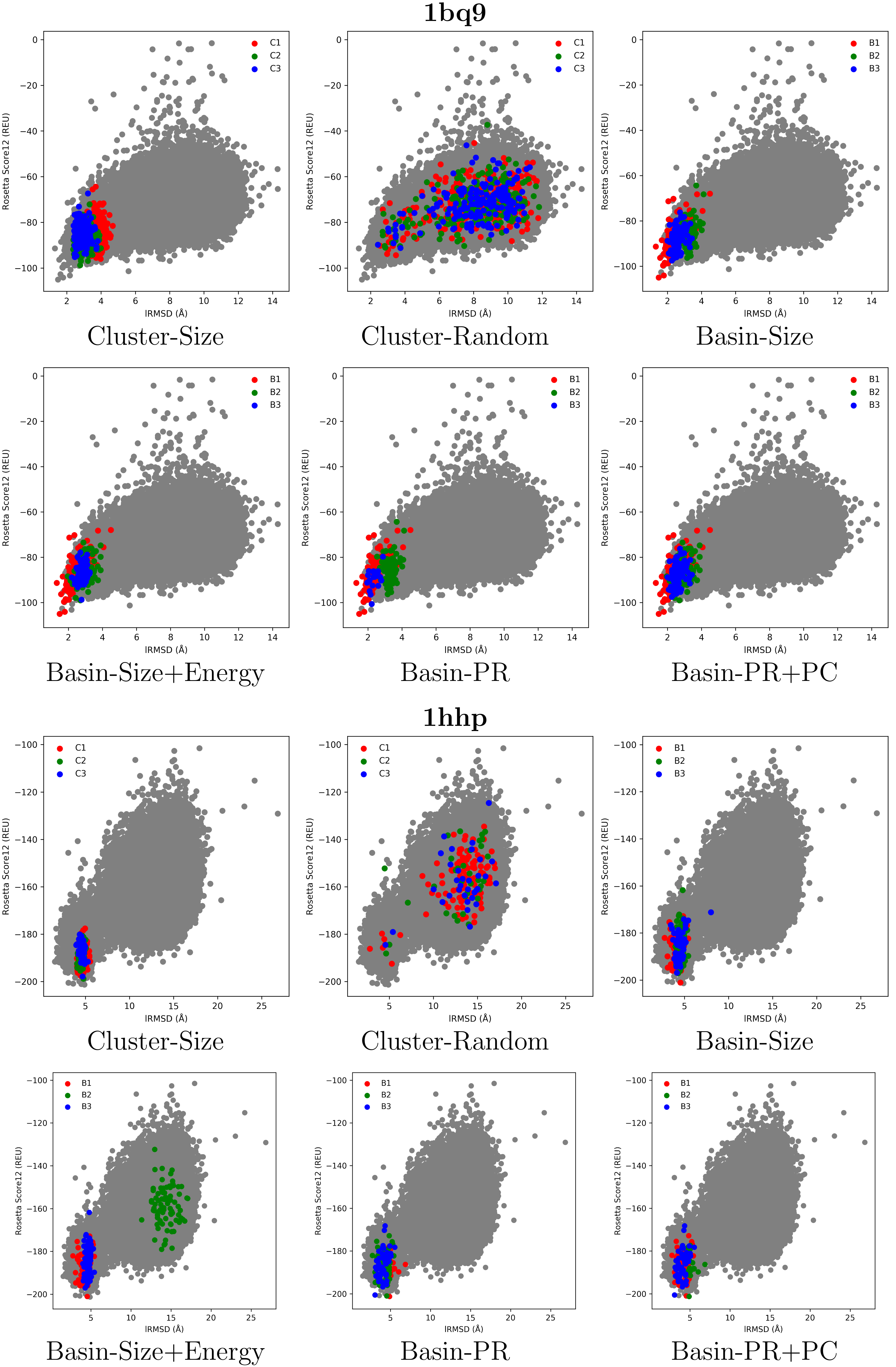
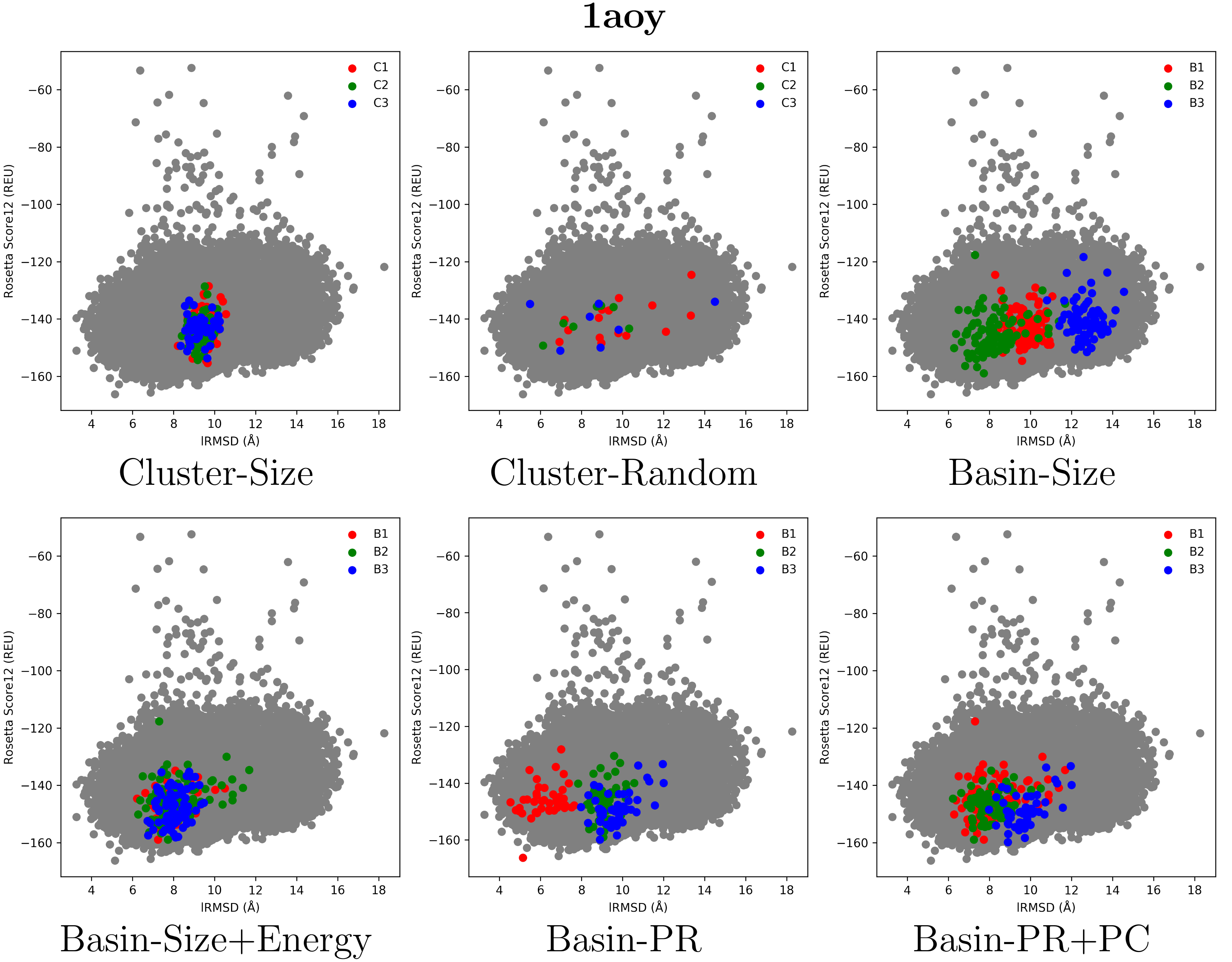
2.2. Visual Comparison of Decoy Selection Strategies
2.3. Quantitative Comparison of Decoy Selection Strategies
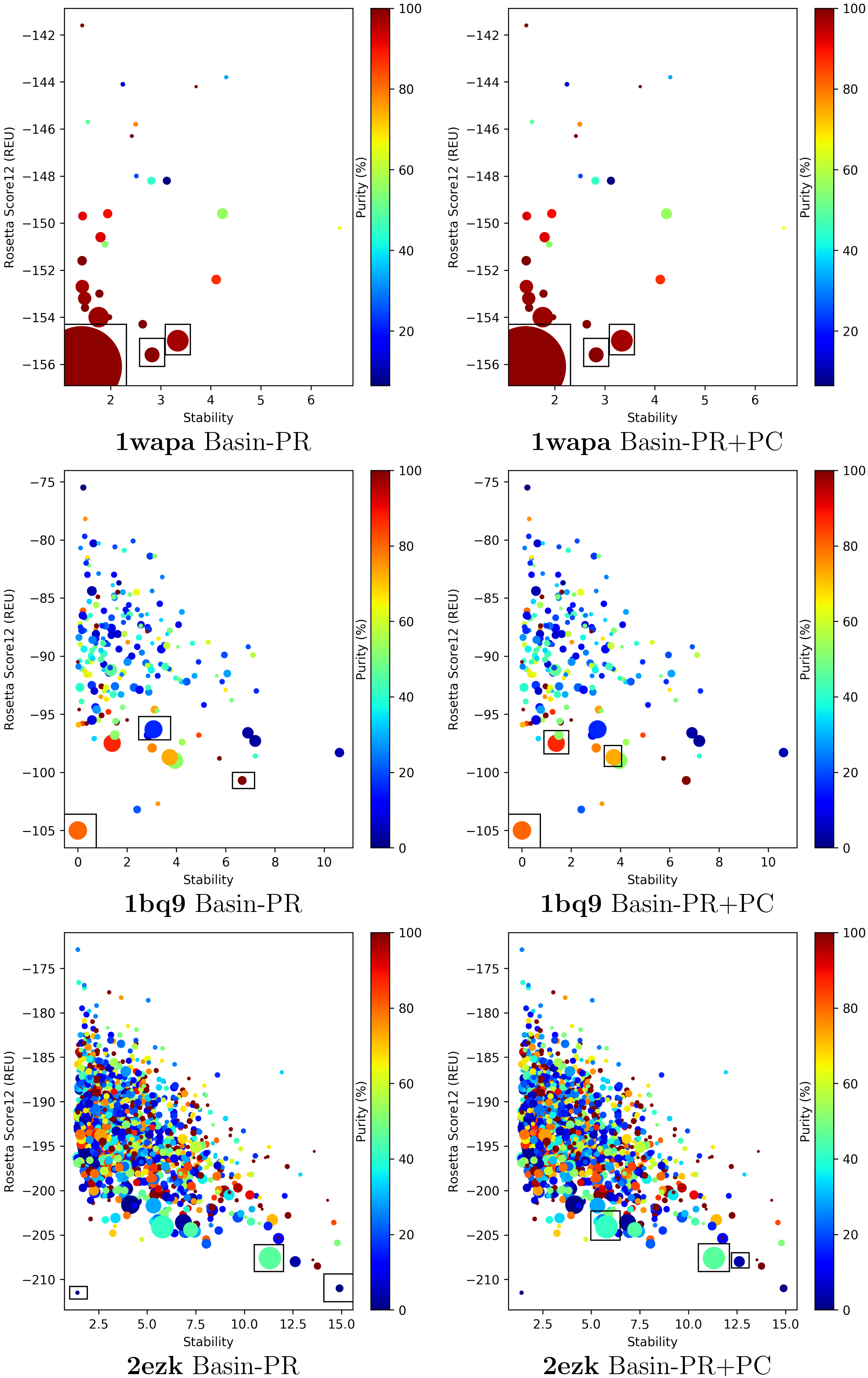
2.4. Visual Analysis of Pareto-Based Selection Strategies
3. Discussion
4. Materials and Methods
4.1. Energy-Less Decoy Selection
4.2. Energy (Landscape)-Based Decoy Selection
4.2.1. Energy Landscapes
4.2.2. Elucidating Basins via Graph Embeddings of Landscapes
4.2.3. Characteristics of Basins
4.2.4. Basin-Based Selection Strategies
4.3. Multi-Objective, Pareto-Based Basin Selection Strategies
- For all optimization objectives i,
- For at least one optimization objective i,
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| PDB | Protein Data Bank |
| CASP | Critical Assessment of protein Structure Prediction |
| lRMSD | least Root-Mean-Squared-Deviation |
| ML | Machine Learning |
| PC | Pareto Count |
| PR | Pareto Rank |
| SVM | Support Vector Machines |
References
- Boehr, D.D.; Wright, P.E. How do proteins interact? Science 2008, 320, 1429–1430. [Google Scholar] [CrossRef] [PubMed]
- Blaby-Haas, C.E.; de Crécy-Lagard, V. Mining high-throughput experimental data to link gene and function. Trends Biotechnol. 2013, 29, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 2003, 10, 980. [Google Scholar] [CrossRef] [PubMed]
- Shehu, A. A Review of Evolutionary Algorithms for Computing Functional Conformations of Protein Molecules. In Computer-Aided Drug Discovery; Zhang, W., Ed.; Methods in Pharmacology and Toxicology; Springer: New York, NY, USA, 2015. [Google Scholar]
- Leaver-Fay, A.; Tyka, M.; Lewis, S.M.; Lange, O.F.; Thompson, J.; Jacak, R.; Kaufman, K.; Renfrew, P.D.; Smith, C.A.; Sheffler, W.; et al. ROSETTA3: An object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011, 487, 545–574. [Google Scholar] [PubMed]
- Xu, D.; Zhang, Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins Struct. Funct. Bioinf. 2012, 80, 1715–1735. [Google Scholar] [CrossRef] [PubMed]
- Shehu, A. Probabilistic Search and Optimization for Protein Energy Landscapes. In Handbook of Computational Molecular Biology; Aluru, S., Singh, A., Eds.; Chapman & Hall/CRC Computer & Information Science Series; CRC Press: Boca Raton, FL, USA, 2013. [Google Scholar]
- Moult, J.; Fidelis, K.; Kryshtafovych, A.; Schwede, T.; Tramontano, A. Critical assessment of methods of protein structure prediction (CASP)—Round x. Proteins Struct. Funct. Bioinf. 2014, 82, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Moult, J.; Fidelis, K.; Kryshtafovych, A.; Schwede, T.; Tramontano, A. Critical Assessment of Methods of Protein Structure Prediction (CASP)—Round XII. Proteins 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Ginalski, K.; Elofsson, A.; Fischer, D.; Rychlewski, L. 3D-Jury: A simple approach to improve protein structure predictions. Bioinformatics 2003, 19, 1015–1018. [Google Scholar] [CrossRef] [PubMed]
- Wallner, B.; Elofsson, A. Identification of correct regions in protein models using structural, alignment, and consensus information. Protein Sci. 2006, 15, 900–913. [Google Scholar] [CrossRef] [PubMed]
- Molloy, K.; Saleh, S.; Shehu, A. Probabilistic Search and Energy Guidance for Biased Decoy Sampling in Ab-initio Protein Structure Prediction. IEEE/ACM Trans. Bioinform. Comput. Biol. 2013, 10, 1162–1175. [Google Scholar] [CrossRef] [PubMed]
- Shehu, A.; Plaku, E. A Survey of omputational Treatments of Biomolecules by Robotics-inspired Methods Modeling Equilibrium Structure and Dynamics. J. Artif. Intell. Res. 2016, 597, 509–572. [Google Scholar]
- Maximova, T.; Moffatt, R.; Ma, B.; Nussinov, R.; Shehu, A. Principles and Overview of Sampling Methods for Modeling Macromolecular Structure and Dynamics. PLoS Comput. Biol. 2016, 12, e1004619. [Google Scholar]
- Shehu, A.; Clementi, C.; Kavraki, L.E. Sampling Conformation Space to Model Equilibrium Fluctuations in Proteins. Algorithmica 2007, 48, 303–327. [Google Scholar] [CrossRef]
- Okazaki, K.; Koga, N.; Takada, S.; Onuchic, J.N.; Wolynes, P.G. Multiple-basin energy landscapes for large-amplitude conformational motions of proteins: Structure-based molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2006, 103, 11844–11849. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Xu, J. A position-specific distance-dependent statistical potential for protein structure and functional study. Structure 2012, 20, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhang, J.; Xu, Y.; Shang, Y.; Xu, D. Protein structural model selection based on protein-dependent scoring function. Stat. Interface 2012, 5, 109–115. [Google Scholar] [CrossRef]
- Mirzaei, S.; Sidi, T.; Keasar, C.; Crivelli, S. Purely Structural Protein Scoring Functions Using Support Vector Machine and Ensemble Learning. IEEE/ACM Trans. Comput. Biol. 2016, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Wolynes, P.G. A second molecular biology revolution? The energy landscapes of biomolecular function. Phys. Chem. Chem. Phys. 2014, 16, 6321–6322. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Kumar, S.; Tsai, C.; Nussinov, R. Folding funnels and binding mechanisms. Protein Eng. 1999, 12, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Bryngelson, J.D.; Onuchic, J.N.; Socci, N.D.; Wolynes, P.G. Funnels, pathways, and the energy landscape of protein folding: A synthesis. Proteins Struct. Funct. Genet. 1995, 21, 167–195. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.; Kumar, S.; Ma, B.; Nussinov, R. Folding funnels, binding funnels, and protein function. Protein Sci. 1999, 8, 1181–1190. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.; Ma, B.; Nussinov, R. Folding and binding cascades: Shifts in energy landscapes. Proc. Natl. Acad. Sci. USA 1999, 96, 9970–9972. [Google Scholar] [CrossRef] [PubMed]
- Sippl, M.J. Knowledge-based potentials for proteins. Curr. Opin. Struct. Biol. 1995, 5, 229–235. [Google Scholar] [CrossRef]
- Bahar, I.; Jernigan, R.L. Inter-residue potentials in globular proteins and the dominance of highly specific hydrophilic interactions at close separation. J. Mol. Biol. 1997, 266, 195–214. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.A.; Finkelstein, A.V.; Sanner, M.F.; Olson, A.J. Residue-residue mean-force potentials for protein structure recognition. Protein Eng. 1997, 10, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Özkan, B.; Bahar, I. Recognition of native structure from complete enumeration of low-resolution models with constraints. Proteins Struct. Funct. Genet. 1998, 32, 211–222. [Google Scholar] [CrossRef]
- Miyazawa, S.; Jernigan, R.L. An empirical energy potential with a reference state for protein fold and sequence recognition. Proteins Struct. Funct. Bioinform. 1999, 36, 357–369. [Google Scholar] [CrossRef]
- Eyrich, V.A.; Standley, D.M.; Felts, A.K.; Friesner, R.A. Protein tertiary structure prediction using a branch and bound algorithm. Proteins Struct. Funct. Bioinform. 1999, 35, 41–57. [Google Scholar] [CrossRef]
- Simons, K.T.; Ruczinski, I.; Kooperberg, C.; Fox, B.A.; Bystroff, C.; Baker, D. Improved recognition of native-like protein structures using a combination of sequence-dependent and sequence-independent features of proteins. Proteins Struct. Funct. Bioinform. 1999, 34, 82–95. [Google Scholar] [CrossRef]
- Lazaridis, T.; Karplus, M. Discrimination of the native from misfolded protein models with an energy function including implicit solvation. J. Mol. Biol. 1999, 288, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Petrey, D.; Honig, B. Free energy determinants of tertiary structure and the evaluation of protein models. Protein Sci. 2000, 9, 2181–2191. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, S.; Zhang, Y. Identification of near-native structures by clustering protein docking conformations. Proteins Struct. Funct. Bioinform. 2007, 68, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Shortle, D.; Simons, K.T.; Baker, D. Clustering of low-energy conformations near the native structures of small proteins. Proc. Natl. Acad. Sci. USA 1998, 95, 11158–11162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Skolnick, J. SPICKER: A clustering approach to identify near-native protein folds. J. Comput. Chem. 2004, 25, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Estrada, T.; Armen, R.; Taufer, M. Automatic selection of near-native protein-ligand conformations using a hierarchical clustering and volunteer computing. In Proceedings of the First ACM International Conference on Bioinformatics and Computational Biology, Niagara Falls, NY, USA, 2–4 August 2010; ACM: New York, NY, USA, 2010; pp. 204–213. [Google Scholar]
- Li, H.; Zhou, Y. SCUD: Fast structure clustering of decoys using reference state to remove overall rotation. J. Comput. Chem. 2005, 26, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Li, S.C.; Ng, Y.K. Calibur: A tool for clustering large numbers of protein decoys. BMC Bioinform. 2010, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Berenger, F.; Zhou, Y.; Shrestha, R.; Zhang, K.Y. Entropy-accelerated exact clustering of protein decoys. Bioinformatics 2011, 27, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wishart, D.S. An improved method to detect correct protein folds using partial clustering. BMC Bioinform. 2013, 14, 11. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Sheffler, W.; Baker, D.; Noble, W.S. Ranking predicted protein structures with support vector regression. Proteins Struct. Funct. Bioinform. 2008, 71, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Lindahl, E.; Wallner, B. Improved model quality assessment using ProQ2. BMC Bioinform. 2012, 13, 224. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Skolnick, J. GOAP: A generalized orientation-dependent, all-atom statistical potential for protein structure prediction. Biophys. J. 2011, 101, 2043–2052. [Google Scholar] [CrossRef] [PubMed]
- Faraggi, E.; Kloczkowski, A. A global machine learning based scoring function for protein structure prediction. Proteins Struct. Funct. Bioinform. 2014, 82, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Cazals, F.; Dreyfus, T. The structural bioinformatics library: Modeling in biomolecular science and beyond. Bioinformatics 2017, 33, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- McLachlan, A.D. A mathematical procedure for superimposing atomic coordinates of proteins. Acta Crystallogr. A 1972, 26, 656–657. [Google Scholar] [CrossRef]
- Wright, S. The roles of mutation, inbreeding, crossbreeding, and selection in evolution. In Proceedings of the International Congress of Genetics, Zurich, Switzerland, 24–31 July 1934; pp. 356–366. [Google Scholar]
- Samoilenko, S. Fitness Landscapes of Complex Systems: Insights and Implications On Managing a Conflict Environment of Organizations. Complex. Organ. 2008, 10, 38–45. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB ID | Fold | Length | min_dist (Å) | |||
|---|---|---|---|---|---|---|
| Easy | 1ail | 70 | 53,568 | |||
| 1dtdb | 61 | 57,839 | ||||
| 1wapa | 68 | 51,841 | ||||
| 1tig | 88 | 52,099 | ||||
| 1dtja | 74 | 53,526 | ||||
| Medium | 1hz6a | 64 | 57,474 | |||
| 1c8ca | * | 64 | 53,322 | |||
| 2ci2 | 65 | 52,220 | ||||
| 1bq9 | 53 | 53,663 | ||||
| 1hhp | * | 99 | 52,159 | |||
| 1fwp | 69 | 53,133 | ||||
| 1sap | 66 | 51,209 | ||||
| Hard | 2h5nd | 123 | 51,475 | |||
| 2ezk | 93 | 50,192 | ||||
| 1aoy | 78 | 52,218 | ||||
| 1cc5 | 83 | 51,687 | ||||
| 1isua | 62 | 60,360 | ||||
| 1aly | 146 | 53,274 |
| 1ail | 1dtdb | 1wapa | 1tig | 1dtja | ||
|---|---|---|---|---|---|---|
| Cluster-Random | C | n: 4% | n: 17.8% | n: 5.2% | n: 8.8% | n: 21.4% |
| p: 6.2% | p: 18.2% | p: 10.1% | p: 15.2% | p: 22.3% | ||
| s: 4.1% | s: 22.3% | s: 5.2% | s: 8.7% | s: 21.6% | ||
| C | n: 6.6% | n: 18.6% | n: 9.8% | n: 11.3% | n: 22.2% | |
| p: 6.3% | p: 18.2% | p: 10% | p: 15.2% | p: 22.2% | ||
| s: 6.7% | s: 23.3% | s: 10% | s: 11.2% | s: 22.4% | ||
| C | n: 8.5% | n: 19.1% | n: 10% | n: 13.6% | n: 22.4% | |
| p: 6.3% | p: 18.2% | p: 10% | p: 15.1% | p: 22.3% | ||
| s: 8.7% | s: 23.9% | s: 10.2% | s: 13.6% | s: 22.6% | ||
| Cluster-Size | C | n: 63.9% | n: 97.6% | n: 50.8% | n: 57.3% | n: 95.5% |
| p: 99.5% | p: 99.9% | p: 99.9% | p: 99.1% | p: 99.2% | ||
| s: 4.1% | s: 22.3% | s: 5.2% | s: 8.7% | s: 21.6% | ||
| C | n: 64.4% | n: 97.6% | n: 97.6% | n: 73% | n: 97.8% | |
| p: 61.1% | p: 95.7% | p: 99.5% | p: 98.2% | p: 98% | ||
| s: 6.7% | s: 23.3% | s: 10% | s: 11.2% | s: 22.4% | ||
| C | n: 65.6% | n: 97.6% | n: 97.6% | n: 88.4% | n: 97.8% | |
| p: 48.2% | p: 93.3% | p: 97.3% | p: 98.4% | p: 97.2% | ||
| s: 8.7% | s: 23.9% | s: 10.2% | s: 13.6% | s: 22.6% | ||
| Basin-Size | B | n: 47.2% | n: 85.3% | n: 76.8% | n: 28.8% | n: 36.9% |
| p: 100% | p: 99% | p: 98.9% | p: 100% | p: 98.9% | ||
| s: 3% | s: 19.7% | s: 7.9% | s: 4.4% | s: 8.4% | ||
| B | n: 48.4% | n: 94.9% | n: 81.8% | n: 40.1% | n: 56.7% | |
| p: 52.8% | p: 98.9% | p: 98.8% | p: 99.6% | p: 99.1% | ||
| s: 5.8% | s: 21.9% | s: 8.4% | s: 6.1% | s: 12.8% | ||
| B | n: 48.4% | n: 94.9% | n: 86.3% | n: 50.2% | n: 70.7% | |
| p: 44.8% | p: 94.8% | p: 98.7% | p: 99.7% | p: 99.2% | ||
| s: 6.9% | s: 22.9% | s: 8.9% | s: 7.6% | s: 16% | ||
| Basin-Size+Energy | B | n: 1.2% | n: 85.3% | n: 76.8% | n: 2.7% | n: 19.9% |
| p: 2.8% | p: 99% | p: 98.9% | p: 88.4% | p: 99.6% | ||
| s: 3% | s: 19.7% | s: 7.9% | s: 0.5% | s: 4.5% | ||
| B | n: 48.4% | n: 94.9% | n: 79.1% | n: 31.5% | n: 33.8% | |
| p: 52.8% | p: 98.9% | p: 98.9% | p: 98.9% | p: 99.6% | ||
| s: 5.8% | s: 21.9% | s: 8.2% | s: 4.8% | s: 7.6% | ||
| B | n: 61.9% | n: 95.9% | n: 84.1% | n: 42.8% | n: 70.7% | |
| p: 58.6% | p: 98.9% | p: 98.8% | p: 98.8% | p: 99.2% | ||
| s: 6.7% | s: 22.1% | s: 8.7% | s: 6.5% | s: 16% | ||
| Basin-PR | B | n: 47.2% | n: 85.3% | n: 76.8% | n: 28.8% | n: 36.9% |
| p: 100% | p: 99% | p: 98.9% | p: 100% | p: 98.9% | ||
| s: 3% | s: 19.7% | s: 7.9% | s: 4.4% | s: 8.4% | ||
| B | n: 48.4% | n: 94.9% | n: 79.1% | n: 31.5% | n: 56.7% | |
| p: 52.8% | p: 98.9% | p: 98.9% | p: 98.9% | p: 99.1% | ||
| s: 5.8% | s: 21.9% | s: 8.2% | s: 4.8% | s: 12.8% | ||
| B | n: 61.9% | n: 94.9% | n: 84.1% | n: 42.8% | n: 70.7% | |
| p: 58.6% | p: 98.9% | p: 98.8% | p: 98.8% | p: 99.2% | ||
| s: 6.7% | s: 21.9% | s: 8.7% | s: 6.6% | s: 16% | ||
| Basin-PR+PC | B | n: 47.2% | n: 85.3% | n: 76.8% | n: 28.8% | n: 19.9% |
| p: 100% | p: 99% | p: 98.9% | p: 100% | p: 99.6% | ||
| s: 3% | s: 19.7% | s: 7.9% | s: 4.4% | s: 4.5% | ||
| B | n: 48.4% | n: 94.9% | n: 81.8% | n: 31.5% | n: 56.7% | |
| p: 52.8% | p: 98.9% | p: 98.8% | p: 98.9% | p: 99.1% | ||
| s: 5.8% | s: 21.9% | s: 8.4% | s: 4.8% | s: 12.8% | ||
| B | n: 61.9% | n: 95.4% | n: 84.1% | n: 42.8% | n: 70.7% | |
| p: 58.6% | p: 98.8% | p: 98.8% | p: 98.8% | p: 99.2% | ||
| s: 6.7% | s: 22% | s: 8.7% | s: 6.6% | s: 16% |
| 1hz6a | 1c8ca | 2ci2 | 1bq9 | 1hhp | 1fwp | 1sap | ||
|---|---|---|---|---|---|---|---|---|
| Cluster-Random | C | n: 4.5% | n: 3.5% | n: 0.4% | n: 0.8% | n: 0.2% | n: 1.9% | n: 9.5% |
| p: 11.4% | p: 11.4% | p: 22.5% | p: 1.9% | p: 2.8% | p: 6% | p: 2.3% | ||
| s: 4.4% | s: 3.4% | s: 0.4% | s: 0.6% | s: 0.2% | s: 1.8% | s: 9.3% | ||
| C | n: 7.7% | n: 5.3% | n: 0.6% | n: 1.4% | n: 0.3% | n: 3.2% | n: 14.6% | |
| p: 11.3% | p: 11.2% | p: 22.9% | p: 2.1% | p: 2.7% | p: 6.1% | p: 2.4% | ||
| s: 7.7% | s: 5.2% | s: 0.6% | s: 1% | s: 0.3% | s: 3.1% | s: 13.9% | ||
| C | n: 10.9% | n: 6.3% | n: 0.8% | n: 1.9% | n: 0.3% | n: 4% | n: 18.3% | |
| p: 11.4% | p: 11.2% | p: 22.2% | p: 2.1% | p: 2.3% | p: 5.8% | p: 7.4% | ||
| s: 10.8% | s: 6.2% | s: 0.8% | s: 1.4% | s: 0.3% | s: 4% | s: 17.4% | ||
| Cluster-Size | C | n: 0% | n: 10% | n: 1.3% | n: 0.6% | n: 1.5% | n: 29.1% | n: 0% |
| p: 0% | p: 32.1% | p: 82% | p: 1.5% | p: 19.8% | p: 92.8% | p: 0% | ||
| s: 4.4% | s: 3.4% | s: 0.4% | s: 0.64% | s: 0.19% | s: 1.8% | s: 9.3% | ||
| C | n: 0% | n: 11.8% | n: 2.4% | n: 9.1% | n: 2.6% | n: 36.3% | n: 44.1% | |
| p: 0% | p: 24.7% | p: 89.4% | p: 13.6% | p: 25.4% | p: 69.2% | p: 7.3% | ||
| s: 7.7% | s: 5.2% | s: 0.6% | s: 1.04% | s: 0.26% | s: 3.1% | s: 13.9% | ||
| C | n: 26.4% | n: 20.5% | n: 3.2% | n: 21% | n: 3.7% | n: 44.1% | n: 55.9 | |
| p: 27.7% | p: 36.3% | p: 92% | p: 24% | p: 28.7% | p: 63.7% | p: 7.4 | ||
| s: 10.8% | s: 6.2% | s: 0.8% | s: 1.4% | s: 0.32% | s: 4% | s: 17.4% | ||
| Basin-Size | B | n: 55.5% | n: 6.1% | n: 0.3% | n: 9.3% | n: 3.5% | n: 5.6% | n: 0% |
| p: 85.5% | p: 32.9% | p: 47.2% | p: 80.4% | p: 53.6% | p: 97.7% | p: 0% | ||
| s: 7.3% | s: 2% | s: 0.13% | s: 0.18% | s: 0.16% | s: 0.33% | s: 4.4% | ||
| B | n: 55.5% | n: 20.2% | n: 0.3% | n: 11.1% | n: 3.5% | n: 9.1% | n: 32.4% | |
| p: 50% | p: 60.8% | p: 23.6% | p: 49.2% | p: 27% | p: 97.2% | p: 9.3% | ||
| s: 12.6% | s: 3.6% | s: 0.3% | s: 0.4% | s: 0.32% | s: 0.54% | s: 8.1% | ||
| B | n: 55.5% | n: 22.3% | n: 0.3% | n: 19.8% | n: 5.6% | n: 10.7% | n: 51.4% | |
| p: 39.3% | p: 48.5% | p: 15.9% | p: 60.8% | p: 30.8% | p: 84.2% | p: 11.5% | ||
| s: 16% | s: 5% | s: 0.4% | s: 0.51% | s: 0.45% | s: 0.74% | s: 10.3% | ||
| Basin-Size+Energy | B | n: 55.5% | n: 3.3% | n: 0.42% | n: 9.3% | n: 3.5% | n: 3.5% | n: 32.4% |
| p: 85.5% | p: 47.8% | p: 100% | p: 80.4% | p: 53.6% | p: 96.4% | p: 20.2% | ||
| s: 7.3% | s: 0.8% | s: 0.1% | s: 0.18% | s: 0.16% | s: 0.21% | s: 3.7% | ||
| B | n: 55.5% | n: 17.4% | n: 0.71% | n: 14.1% | n: 5.6% | n: 3.7% | n: 51.4% | |
| p: 66.6% | p: 80.6% | p: 68.9% | p: 68.2% | p: 47.7% | p: 58.4% | p: 20% | ||
| s: 9.4% | s: 2.4% | s: 0.23% | s: 0.32% | s: 0.29% | s: 0.37% | s: 5.9% | ||
| B | n: 55.7% | n: 20.1% | n: 1.13% | n: 20.5% | n: 8.5% | n: 9.3% | n: 51.4% | |
| p: 55.7% | p: 80.4% | p: 76.9% | p: 69.6% | p: 51.4% | p: 77% | p: 18.2% | ||
| s: 11.3% | s: 2.7% | s: 0.33% | s: 0.46% | s: 0.41% | s: 0.7% | s: 6.5% | ||
| Basin-PR | B | n: 55.5% | n: 3.3% | n: 0.1% | n: 9.3% | n: 0.1% | n: 3.5% | n: 32.4% |
| p: 85.5% | p: 47.8% | p: 100% | p: 80.4% | p: 5% | p: 96.4% | p: 20.2% | ||
| s: 7.3% | s: 0.8% | s: 0.01% | s: 0.18% | s: 0.04% | s: 0.21% | s: 3.7% | ||
| B | n: 55.5% | n: 17.4% | n: 0.1% | n: 11.1% | n: 3.6% | n: 9.1% | n: 32.4% | |
| p: 58.3% | p: 80.6% | p: 7.7% | p: 49.2% | p: 44.2% | p: 97.2% | p: 9.3% | ||
| s: 10.8% | s: 2.4% | s: 0.15% | s: 0.35% | s: 0.2% | s: 0.54% | s: 8.1% | ||
| B | n: 57.7% | n: 23.5% | n: 0.3% | n: 13.3% | n: 6.9% | n: 9.3% | n: 51.4% | |
| p: 58.4% | p: 58.5% | p: 26.5% | p: 53.9% | p: 55.6% | p: 77% | p: 11.5% | ||
| s: 11.2% | s: 4.4% | s: 0.2% | s: 0.51% | s: 0.31% | s: 0.7% | s: 10.3% | ||
| Basin-PR+PC | B | n: 55.5% | n: 14% | n: 0.43% | n: 9.3% | n: 3.5% | n: 3.5% | n: 32.4% |
| p: 85.5% | p: 96.3% | p: 100% | p: 80.4% | p: 53.6% | p: 96.4% | p: 20.2% | ||
| s: 7.3% | s: 1.6% | s: 0.1% | s: 0.18% | s: 0.16% | s: 0.21% | s: 3.7% | ||
| B | n: 55.5% | n: 17.4% | n: 0.72% | n: 14.1% | n: 3.6% | n: 9.1% | n: 32.4% | |
| p: 50% | p: 80.6% | p: 68.9% | p: 68.2% | p: 44.2% | p: 97.2% | p: 9.3% | ||
| s: 12.6% | s: 2.4% | s: 0.23% | s: 0.32% | s: 0.2% | s: 0.54% | s: 8.1% | ||
| B | n: 55.5% | n: 23.5% | n: 0.93% | n: 22.7% | n: 6.9% | n: 9.3% | n: 51.4% | |
| p: 39.3% | p: 58.5% | p: 67.7% | p: 74.3% | p: 55.6% | p: 77% | p: 11.5% | ||
| s: 16% | s: 4.4% | s: 0.31% | s: 0.46% | s: 0.31% | s: 0.7% | s: 10.3% |
| 2h5nd | 2ezk | 1aoy | 1cc5 | 1isua | 1aly | ||
|---|---|---|---|---|---|---|---|
| Cluster-Random | C | n: 0% | n: 0.01% | n: 0.02% | n: 0% | n: 0.02% | n: 0% |
| p: 0% | p: 5% | p: 8.0% | p: 0% | p: 5.5% | p: 0% | ||
| s: 0.004% | s: 0.02% | s: 0.03% | s: 0.01% | s: 0.02% | s: 0.01% | ||
| C | n: 0% | n: 0.03% | n: 0.03% | n: 0% | n: 0.04% | n: 0% | |
| p: 0% | p: 7.5% | p: 8.2% | p: 0% | p: 6% | p: 0% | ||
| s: 0.008% | s: 0.05% | s: 0.04% | s: 0.02% | s: 0.03% | s: 0.02% | ||
| C | n: 0% | n: 0.05% | n: 0.04% | n: 0% | n: 0.04% | n: 0.01% | |
| p: 0% | p: 10% | p: 6.9% | p: 0% | p: 5% | p: 1.4% | ||
| s: 0.01% | s: 0.07% | s: 0.06% | s: 0.03% | s: 0.05% | s: 0.03% | ||
| Cluster-Size | C | n: 0% | n: 0% | n: 0% | n: 0% | n: 0% | n: 0% |
| p: 0% | p: 0% | p: 0% | p: 0% | p: 0% | p: 0% | ||
| s: 0.004% | s: 0.02% | s: 0.03% | s: 0.01% | s: 0.02% | s: 0.01% | ||
| C | n: 0% | n: 0% | n: 0% | n: 0% | n: 0% | n: 0.3% | |
| p: 0% | p: 0% | p: 0% | p: 0% | p: 0% | p: 40% | ||
| s: 0.008% | s: 0.05% | s: 0.04% | s: 0.02% | s: 0.03% | s: 0.02% | ||
| C | n: 0% | n: 0% | n: 0% | n: 0% | n: 0% | n: 0.4% | |
| p: 0% | p: 0% | p: 0% | p: 0% | p: 0% | p: 42.9% | ||
| s: 0.01% | s: 0.07% | s: 0.06% | s: 0.03% | s: 0.05% | s: 0.03% | ||
| Basin-Size | B | n: 0% | n: 0.96% | n: 0% | n: 0.03% | n: 0.34% | n: 0% |
| p: 0% | p: 41.2% | p: 0% | p: 1.14% | p: 14.1% | p: 0% | ||
| s: 0.27% | s: 0.3% | s: 0.2% | s: 0.17% | s: 0.13% | s: 0.06% | ||
| B | n: 0% | n: 2% | n: 0.2% | n: 0.03% | n: 0.34% | n: 0.07% | |
| p: 0% | p: 43.5% | p: 4.9% | p: 0.6% | p: 7.1% | p: 1.6% | ||
| s: 0.38% | s: 0.6% | s: 0.39% | s: 0.32% | s: 0.26% | s: 0.12% | ||
| B | n: 10% | n: 2% | n: 0.2% | n: 0.03% | n: 0.34% | n: 0.07% | |
| p: 17.4% | p: 33% | p: 3.4% | p: 0.42% | p: 4.9% | p: 1.1% | ||
| s: 0.48% | s: 0.8% | s: 0.57% | s: 0.46% | s: 0.38% | s: 0.17% | ||
| Basin-Size+Energy | B | n: 0% | n: 1.02% | n: 0.05% | n: 0% | n: 0.34% | n: 0% |
| p: 0% | p: 45.9% | p: 3.5% | p: 0% | p: 14.1% | p: 0% | ||
| s: 0.09% | s: 0.29% | s: 0.16% | s: 0.14% | s: 0.13% | s: 0.05% | ||
| B | n: 0% | n: 1.5% | n: 0.23% | n: 1.15% | n: 0.34% | n: 0% | |
| p: 0% | p: 45.7% | p: 6.9% | p: 27.3% | p: 7.6% | p: 0% | ||
| s: 0.37% | s: 0.41% | s: 0.36% | s: 0.23% | s: 0.24% | s: 0.1% | ||
| B | n: 10% | n: 2.4% | n: 0.28% | n: 1.2% | n: 0.44% | n: 0% | |
| p: 17.8% | p: 43.8% | p: 6.1% | p: 18.9% | p: 6.6% | p: 0% | ||
| s: 0.47% | s: 0.72% | s: 0.51% | s: 0.35% | s: 0.35% | s: 0.16% | ||
| Basin-PR | B | n: 0% | n: 0% | n: 0.56% | n: 0.03% | n: 0% | n: 0.27% |
| p: 0% | p: 0% | p: 78.1% | p: 1.14% | p: 0% | p: 40% | ||
| s: 0.006% | s: 0.03% | s: 0.08% | s: 0.17% | s: 0.02% | s: 0.02% | ||
| B | n: 0% | n: 1.02% | n: 0.56% | n: 0.03% | n: 0% | n: 0.27% | |
| p: 0% | p: 41.9% | p: 33% | p: 1.12% | p: 0% | p: 19.1% | ||
| s: 0.28% | s: 0.32% | s: 0.19% | s: 0.17% | s: 0.12% | s: 0.04% | ||
| B | n: 0% | n: 1.02% | n: 0.56% | n: 0.66% | n: 0.07% | n: 0.27% | |
| p: 0% | p: 41.1% | p: 21.8% | p: 15.8% | p: 4.8% | p: 8% | ||
| s: 0.31% | s: 0.32% | s: 0.28% | s: 0.23% | s: 0.21% | s: 0.09% | ||
| Basin-PR+PC | B | n: 0% | n: 1.02% | n: 0.18% | n: 0% | n: 0% | n: 0% |
| p: 0% | p: 45.9% | p: 9.8% | p: 0% | p: 0% | p: 0% | ||
| s: 0.27% | s: 0.29% | s: 0.2% | s: 0.14% | s: 0.05% | s: 0.04% | ||
| B | n: 0% | n: 2% | n: 0.23% | n: 0.63% | n: 0% | n: 0% | |
| p: 0% | p: 43.5% | p: 6.9% | p: 17.5% | p: 0% | p: 0% | ||
| s: 0.37% | s: 0.6% | s: 0.36% | s: 0.2% | s: 0.11% | s: 0.08% | ||
| B | n: 0% | n: 2.0% | n: 0.23% | n: 0.73% | n: 0.03% | n: 0% | |
| p: 0% | p: 39.7% | p: 5.5% | p: 15.8% | p: 1.2% | p: 0% | ||
| s: 0.39% | s: 0.66% | s: 0.46% | s: 0.26% | s: 0.14% | s: 0.10% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akhter, N.; Shehu, A. From Extraction of Local Structures of Protein Energy Landscapes to Improved Decoy Selection in Template-Free Protein Structure Prediction. Molecules 2018, 23, 216. https://doi.org/10.3390/molecules23010216
Akhter N, Shehu A. From Extraction of Local Structures of Protein Energy Landscapes to Improved Decoy Selection in Template-Free Protein Structure Prediction. Molecules. 2018; 23(1):216. https://doi.org/10.3390/molecules23010216
Chicago/Turabian StyleAkhter, Nasrin, and Amarda Shehu. 2018. "From Extraction of Local Structures of Protein Energy Landscapes to Improved Decoy Selection in Template-Free Protein Structure Prediction" Molecules 23, no. 1: 216. https://doi.org/10.3390/molecules23010216
APA StyleAkhter, N., & Shehu, A. (2018). From Extraction of Local Structures of Protein Energy Landscapes to Improved Decoy Selection in Template-Free Protein Structure Prediction. Molecules, 23(1), 216. https://doi.org/10.3390/molecules23010216