
Synthesis and Ceramic Conversion of a New Organodecaborane Preceramic Polymer with High-Ceramic-Yield

Abstract
:1. Introduction
2. Results
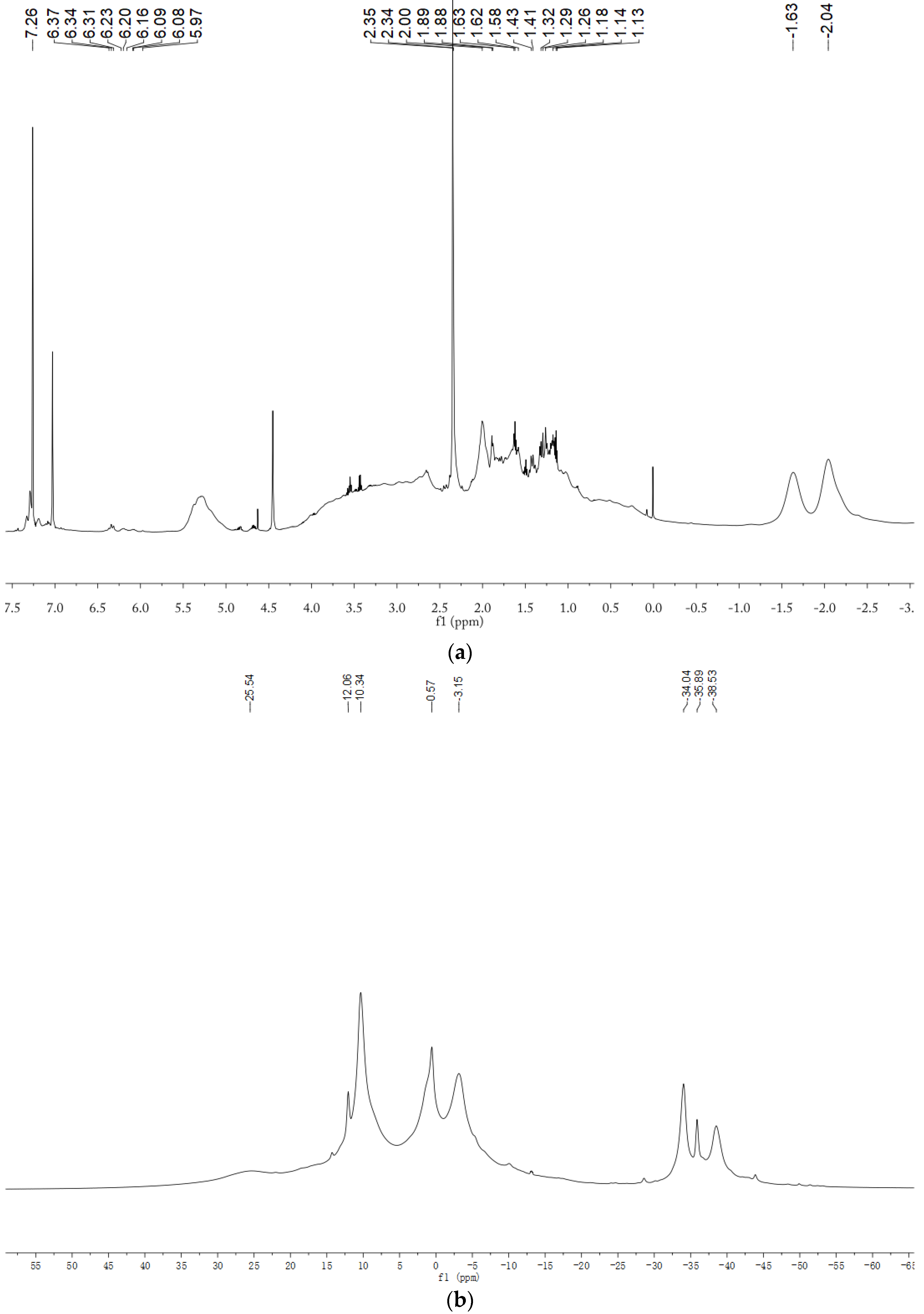
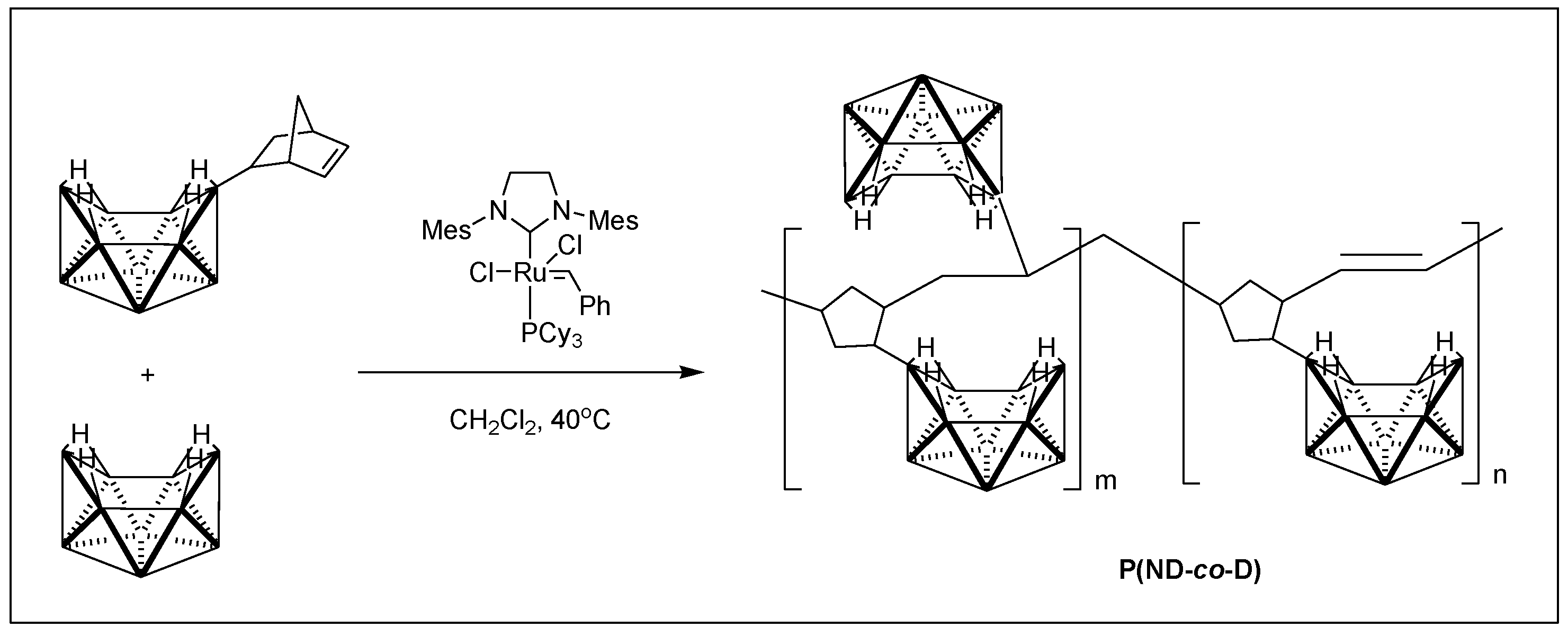
2.1. Characterization of P(ND-co-D) Precursor
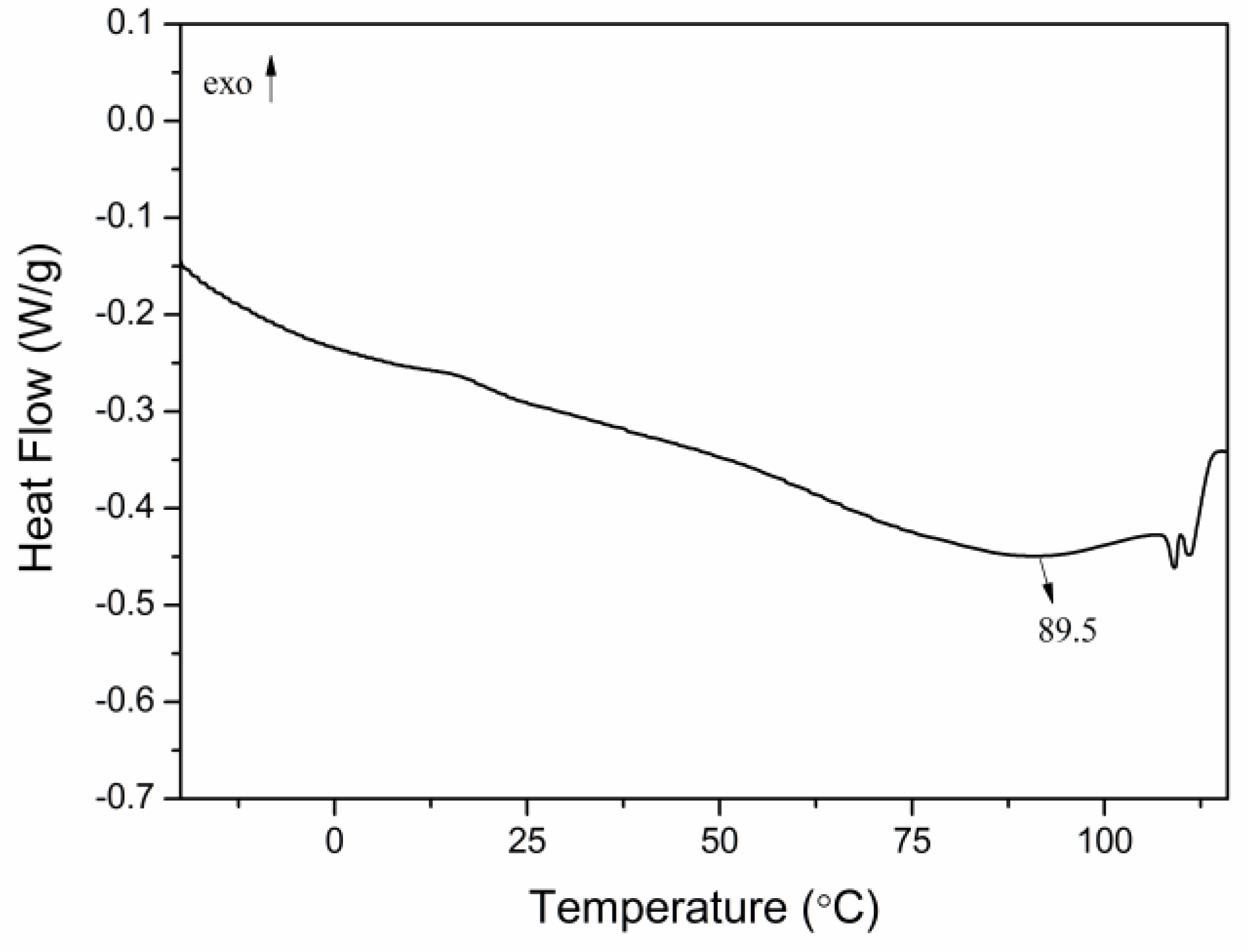
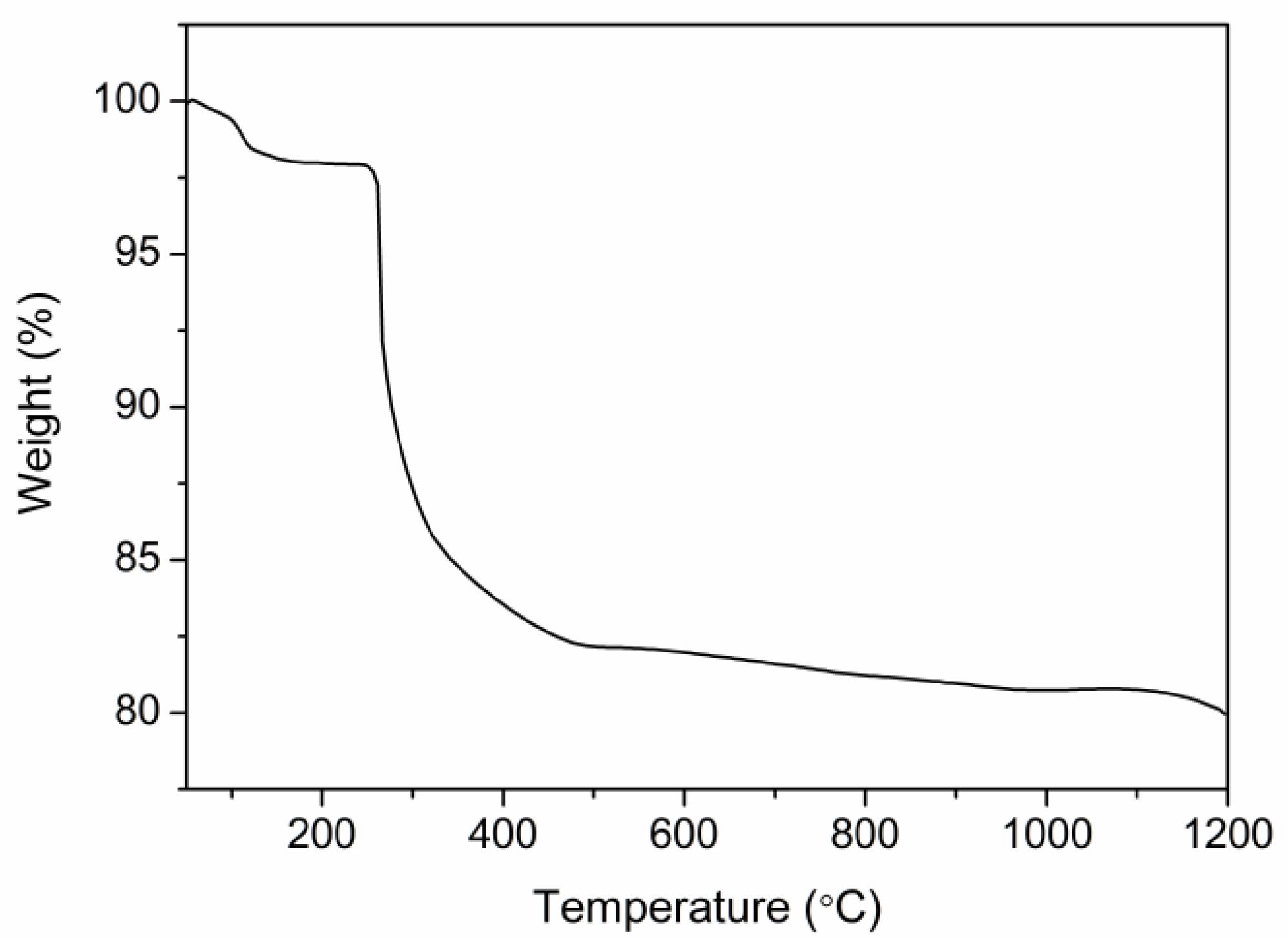
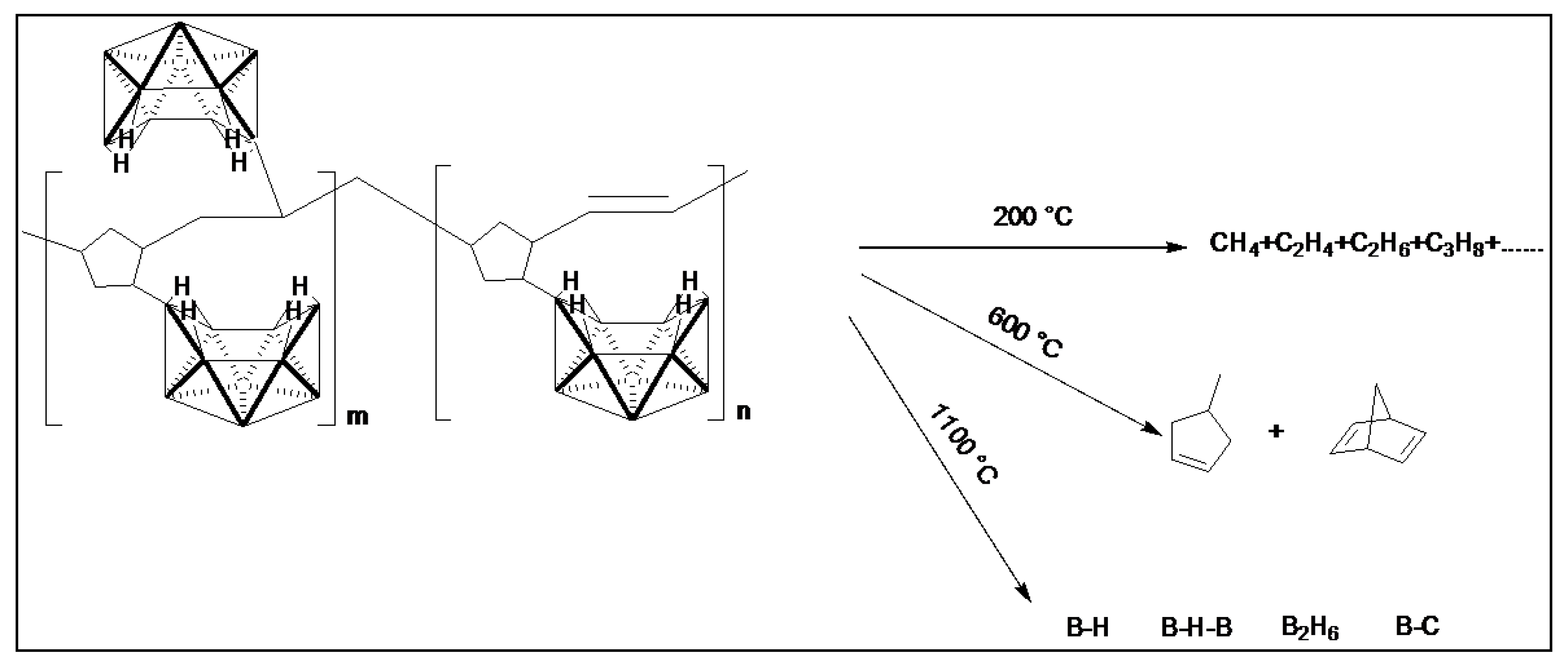
2.2. Thermal Analysis of P(ND-co-D)
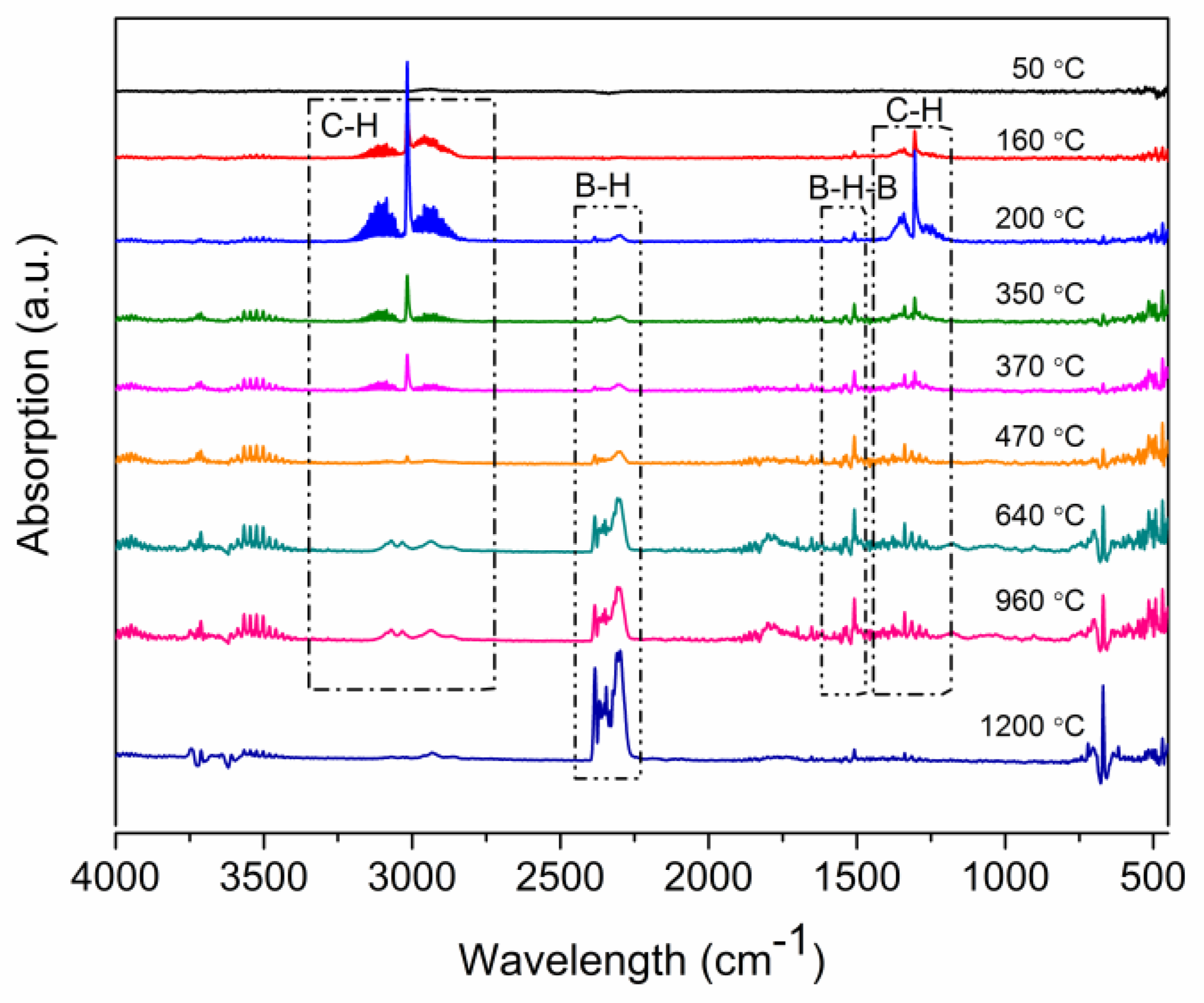
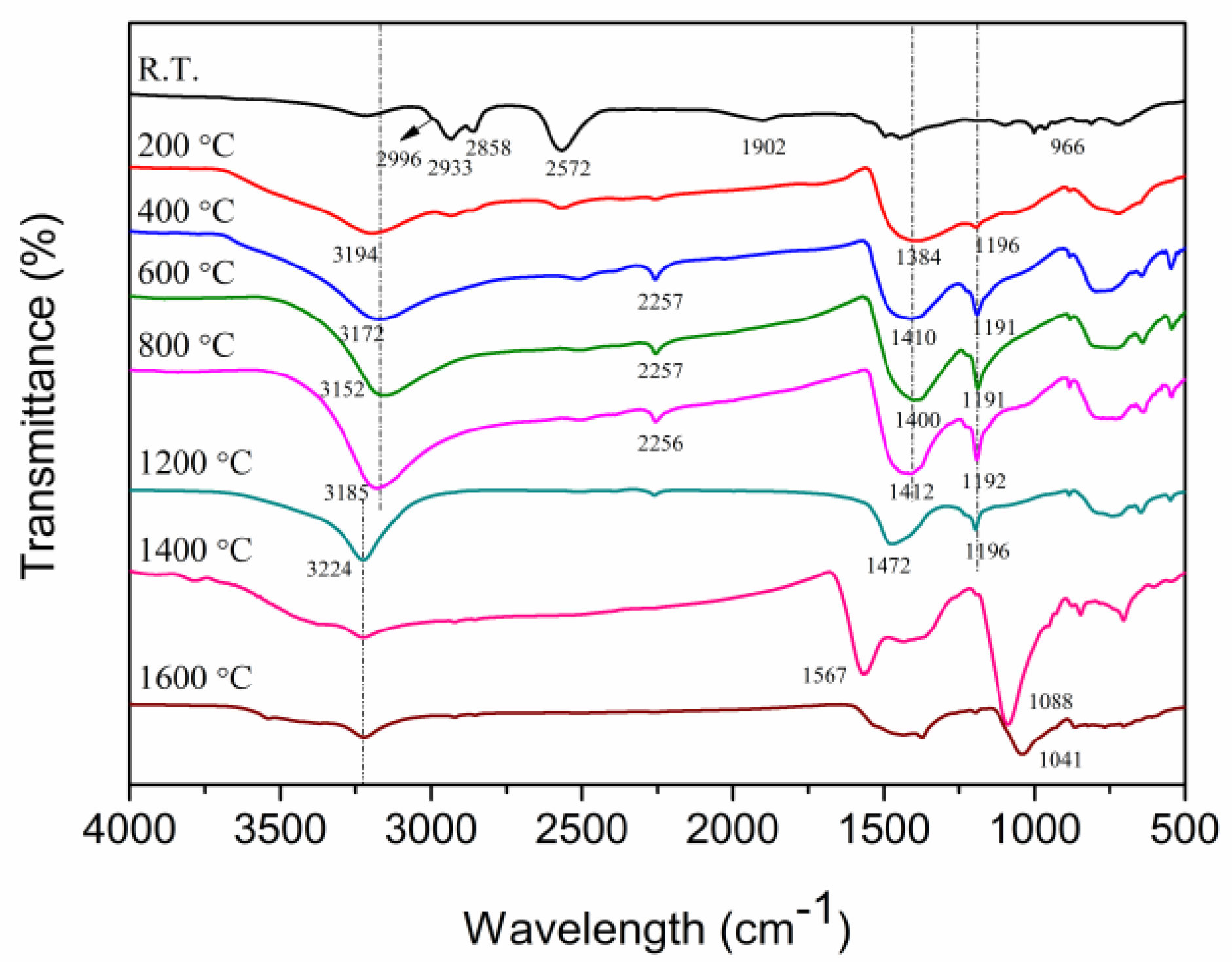
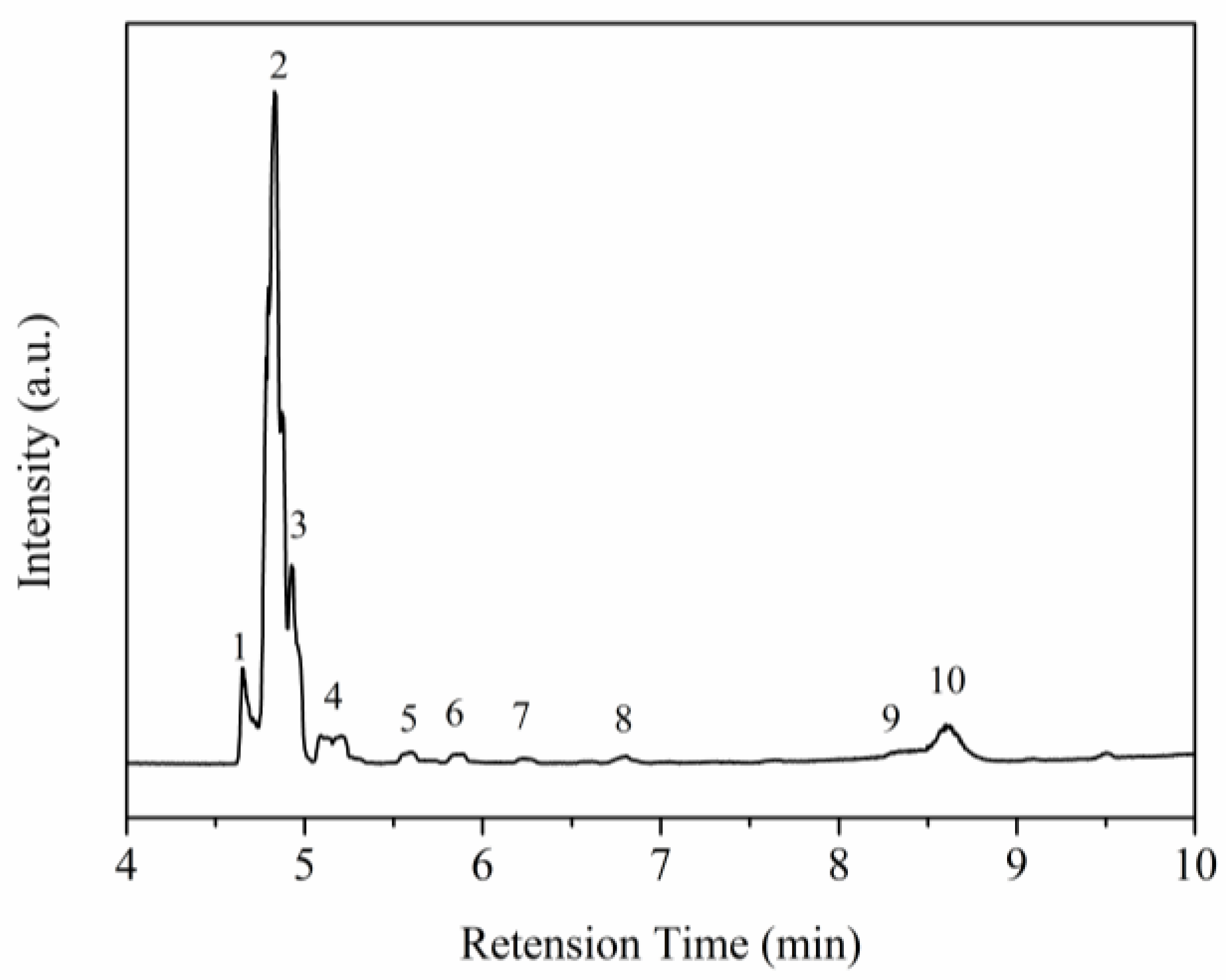
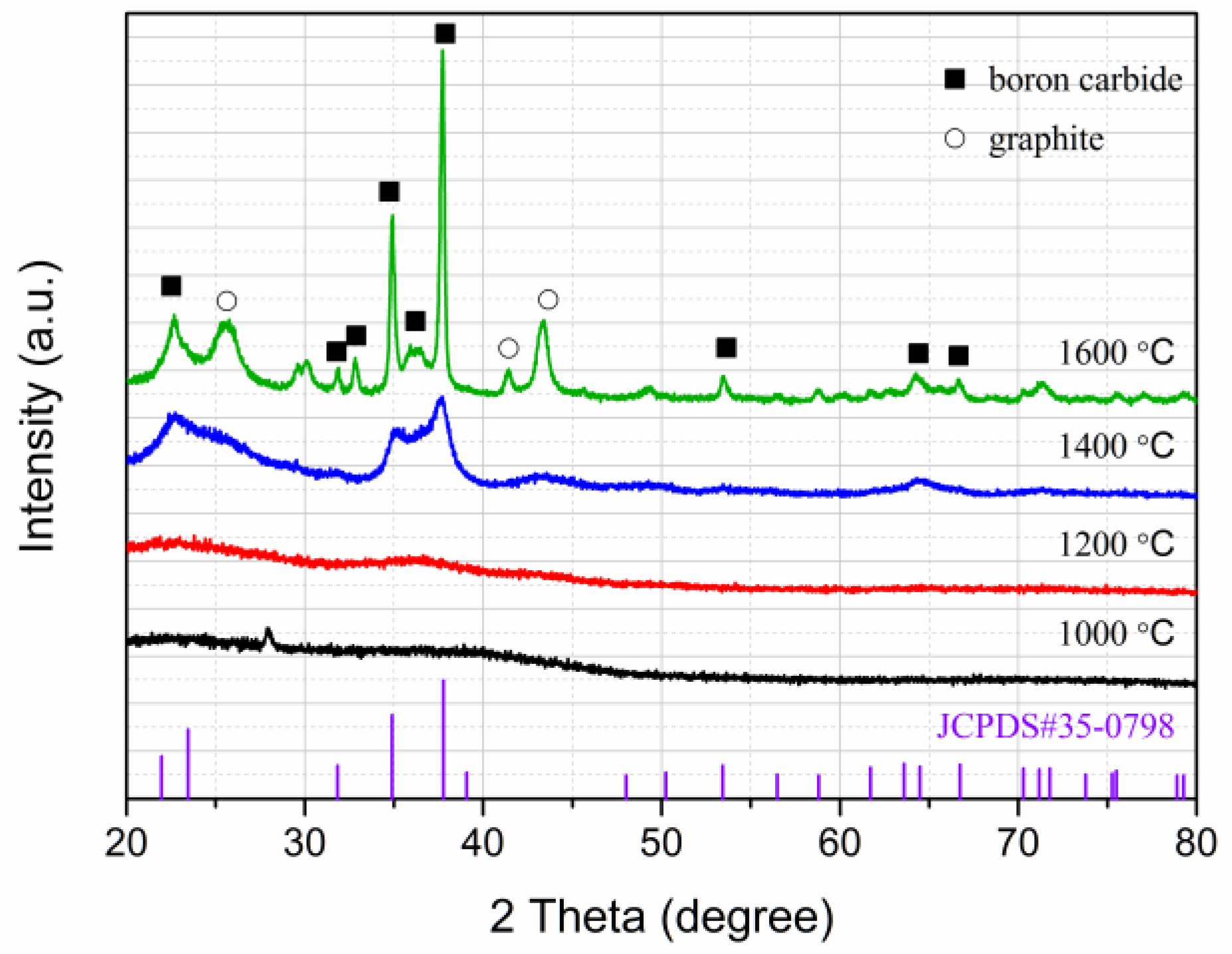
2.3. Ceramic Conversion Reactions of the P(ND-co-D)
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Synthesis of P(ND-co-D)
4.2.1. Synthesis of 6-Norbornenyldecaborane
4.2.2. Synthesis of P(ND-co-D)
4.3. Preparation of Ceramic
4.4. Characterization
Author Contributions
Funding
Conflicts of Interest
References
- Thévenot, F. Boron carbide—A comprehensive review. J. Eur. Ceram. Soc. 1990, 6, 205–225. [Google Scholar] [CrossRef]
- Domnich, V.; Reynaud, S.; Haber, R.A.; Chhowalla, M. Boron carbide: Structure, properties, and stability under stress. J. Am. Ceram. Soc. 2011, 94, 3605–3628. [Google Scholar] [CrossRef]
- Andrievski, R.A. Micro- and nanosized boron carbide: synthesis, structure and properties. Russ. Chem. Rev. 2012, 81, 549–559. [Google Scholar] [CrossRef]
- Schmalzried, R.; Schwetz, K.A. Silicon Carbide and Boron Carbide Based Hard Materials. Ceramic Science and Technology; Riedel, R., Chen, C., Eds.; Wiley VCH: Hoboken, NJ, USA, 2010; pp. 131–225. [Google Scholar]
- Pender, M.J.; Sneddon, L.G. An efficient template synthesis of aligned boron carbide nanofibers using a single-source molecular precursor. Chem. Mater. 2000, 12, 280–283. [Google Scholar] [CrossRef]
- Sneddon, L.G.; Pender, M.J.; Forsthoefel, K.M.; Kusarl, U.; Wei, X. Design, syntheses and applications of chemical precursors to advanced ceramic materials in nanostructured forms. J. Eur. Ceram. Soc. 2005, 25, 91–97. [Google Scholar] [CrossRef]
- Kusarl, U.; Carroll, P.J.; Sneddon, L.G. Ionic-liquid-promoted decaborane olefin-hydroboration: A new efficient route to 6-R-B10H13 derivatives. Inorg. Chem. 2008, 47, 9203–9215. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Carroll, P.J.; Sneddon, L.G. Iridium and ruthenium catalyzed syntheses hydroborations, and metathesis reactions of alkenyl-decaboranes. Inorg. Chem. 2013, 52, 9119–9130. [Google Scholar] [CrossRef] [PubMed]
- Costakis, W.J., Jr.; Rueschhoff, L.M.; Diaz-Cano, A.I.; Youngblood, J.P.; Trice, R.W. Additive manufacturing of boron carbide via continuous filament direct ink writing of aqueous ceramic suspensions. J. Eur. Ceram. Soc. 2016, 36, 3249–3256. [Google Scholar] [CrossRef]
- Chen, R.; Shi, Q.; Su, L.; Yang, M.; Huang, Z.; Shi, Y.; Zhang, Q.; Liao, Z.; Lu, T. Preparation of a B4C hollow microsphere through gel-casting for an Inertial Confinement Fusion (ICF) target. Ceram. Int. 2017, 43, 571–577. [Google Scholar] [CrossRef]
- Wang, J.; Gou, Y.; Zhang, Q.; Jian, K.; Chen, Z.; Wang, H. Linear organodecaborane block copolymer as a single-source precursor for porous boron carbide ceramics. J. Eur. Ceram. Soc. 2017, 37, 1937–1943. [Google Scholar] [CrossRef]
- Yajima, S.; Okamura, K.; Hayash, J.; Omori, M. Synthesis of continuous SiC fibers with high tensile strength. J. Am. Ceram. Soc. 1976, 5, 324–327. [Google Scholar] [CrossRef]
- Yu, X.; Cao, K.; Huang, Y.; Yang, J.; Li, J.; Chang, G. Platinum catalyzed sequential hydroboration of decaborane: A facile approach to poly(alkenyldecaborane) with decaborane in the mainchain. Chem. Commum. 2014, 50, 4585–4587. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Carroll, P.J.; Sneddon, L.G. Ruthenium-catalyzed ring-opening polymerization syntheses of poly(organodecaboranes): New single-source boron-carbide precursors. Chem. Mater. 2006, 18, 1113–1123. [Google Scholar] [CrossRef]
- Zhang, X.; Li, J.; Cao, K.; Yi, Y.; Yang, J.; Li, B. Synthesis and characterization of B–C polymer hollow microspheres from a new organodecaborane preceramic polymer. RSC Adv. 2015, 5, 86214–86218. [Google Scholar] [CrossRef]
- Wang, J.; Gou, Y.; Jian, K.; Huang, J.; Wang, H. Boron carbide ceramic hollow microspheres prepared from poly(6-CH2CH(CH2)4-B10H13) precursor. Mater. Des. 2016, 109, 408–414. [Google Scholar] [CrossRef]
- Makhoukhi, N.; Péré, E.; Creff, R.; Pouchan, C. Determination of the composition of a mixture of gases by infrared analysis and chemomethric methods. J. Mol. Struct. 2005, 744–747, 855–859. [Google Scholar] [CrossRef]
- Tan, Y.B.; Guo, Y.H.; Li, S.F.; Sun, W.W.; Zhu, Y.H.; Li, Q.; Yu, X.B. A liquid-based eutectic system: LiBH4∙NH3-nNH3BH3 with high dehydrogenation capacity at moderate temperature. J. Mater. Chem. 2011, 21, 14509–14515. [Google Scholar] [CrossRef]
- Romanos, J.; Beckner, M.; Stall, D. Infrared study of boron–carbon chemical bonds in boron-doped activated carbon. Carbon 2013, 54, 208–214. [Google Scholar] [CrossRef]
- Guron, M.M.; Wei, X.; Welna, D.; Krogman, N.; Kim, M.J.; Allcock, H.; Sneddon, L.G. Preceramic polymer blends as precursors for boron-carbide/silicon-carbide composite ceramics and ceramic fibers. Chem. Mater. 2009, 21, 1708–1715. [Google Scholar] [CrossRef]
- Qi, S.; Wang, H.; Han, G.; Yang, Z.; Zhang, X.A.; Jiang, S.; Lu, Y. Synthesis, characterization, and curing behavior of carborane-containing benzoxazine resins with excellent thermal and thermos-oxidative stability. J. Appl. Polym. Sci. 2016, 133, 43488-1–43488-11. [Google Scholar] [CrossRef]
- Tillekaratne, A.; Trenary, M. Adsorption and dehydrogenation of decaborane on the Pt(111) surface. J. Phys. Chem. C 2009, 113, 13847–13854. [Google Scholar] [CrossRef]
- Sinha, A.; Mahata, T.; Sharma, B.P. Carbothermal route for preparation of boron carbide powder from boric acid-citric acid gel precursor. J. Nucl. Mater. 2002, 301, 165–169. [Google Scholar] [CrossRef]
- Wei, X.; Carroll, P.J.; Sneddon, L.G. New routes to organodecaborane polymers via ruthenium-catalyzed ring-opening metathesis polymerization. Organometallics 2004, 23, 163–165. [Google Scholar] [CrossRef]
- Shriver, D.F.; Drezdzon, M.A. The Manipulation of Air-Sensitive Compounds, 2nd ed.; Wiley-Interscience: New York, NY, USA, 1986; ISBN 13 9780471867739. [Google Scholar]
Sample Availability: Samples of the compounds P(ND-co-D) are available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | RT (min) | Compounds | m/z | Formula |
|---|---|---|---|---|
| 1 | 4.65 | Allene | 40 | C3H4 |
| 2 | 4.83 | Propane | 44 | C3H8 |
| 3 | 4.93 | Butane | 58 | C4H10 |
| 4 | 5.09 | Butane, 2-methyl | 72 | C5H12 |
| 5 | 5.59 | n-Hexane | 86 | C6H14 |
| 6 | 5.89 | Pentane, 2,4-dimethyl | 100 | C7H16 |
| 7 | 6.22 | Cyclopentane, methyl | 84 | C6H12 |
| 8 | 6.81 | 1,4-Hexadiene | 82 | C6H10 |
| 9 | 8.34 | Cyclohexane, methyl | 98 | C7H14 |
| 10 | 8.60 | 5-Propyl-1-pentene | 112 | C8H16 |
| Temp (°C) | B% (wt.) | C% (wt.) | B:C | Nominal Composition |
|---|---|---|---|---|
| 1000 | 48.60 | 51.40 | 1.05 | B4C/C2.80 |
| 1200 | 50.82 | 49.18 | 1.15 | B4C/C2.48 |
| 1400 | 56.50 | 43.50 | 1.44 | B4C/C1.77 |
| 1600 | 67.78 | 27.74 | 2.52 | B4C/C0.59 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Cao, K.; Li, J.; Liu, M.; Zhang, S.; Yang, J.; Zhang, Z.; Li, B. Synthesis and Ceramic Conversion of a New Organodecaborane Preceramic Polymer with High-Ceramic-Yield. Molecules 2018, 23, 2461. https://doi.org/10.3390/molecules23102461
Li J, Cao K, Li J, Liu M, Zhang S, Yang J, Zhang Z, Li B. Synthesis and Ceramic Conversion of a New Organodecaborane Preceramic Polymer with High-Ceramic-Yield. Molecules. 2018; 23(10):2461. https://doi.org/10.3390/molecules23102461
Chicago/Turabian StyleLi, Jing, Ke Cao, Jie Li, Meifang Liu, Shuai Zhang, Junxiao Yang, Zhanwen Zhang, and Bo Li. 2018. "Synthesis and Ceramic Conversion of a New Organodecaborane Preceramic Polymer with High-Ceramic-Yield" Molecules 23, no. 10: 2461. https://doi.org/10.3390/molecules23102461
APA StyleLi, J., Cao, K., Li, J., Liu, M., Zhang, S., Yang, J., Zhang, Z., & Li, B. (2018). Synthesis and Ceramic Conversion of a New Organodecaborane Preceramic Polymer with High-Ceramic-Yield. Molecules, 23(10), 2461. https://doi.org/10.3390/molecules23102461