Sorbitol as a Chain Extender of Polyurethane Prepolymers to Prepare Self-Healable and Robust Polyhydroxyurethane Elastomers



Abstract
:
1. Introduction
2. Experiment
2.1. Materials
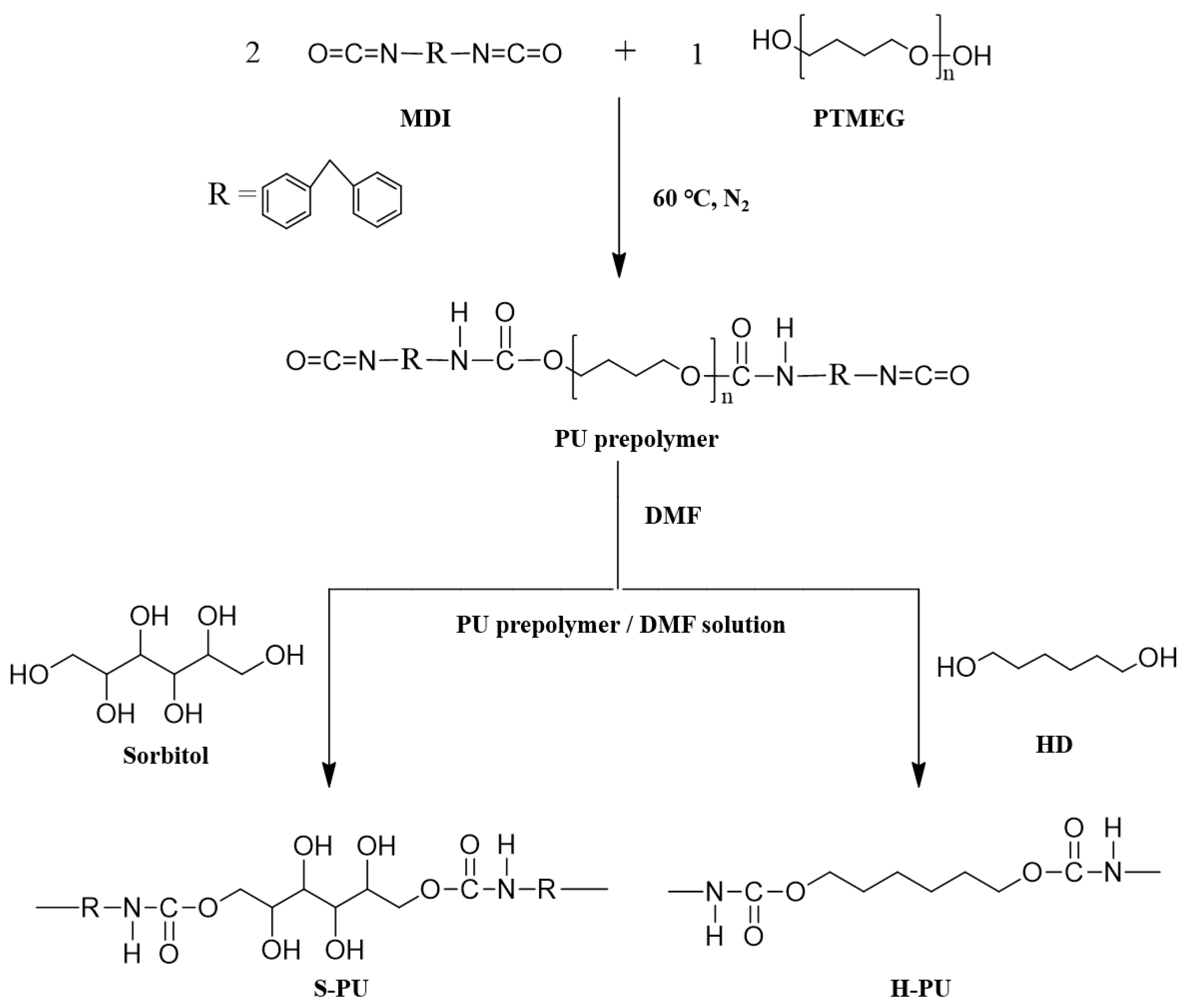
2.2. Synthesis of PU Prepolymers
2.3. Preparation of S-PU and H-PU
2.4. Characterization
3. Results and Discussion
3.1. Synthesis of S-PU and H-PU
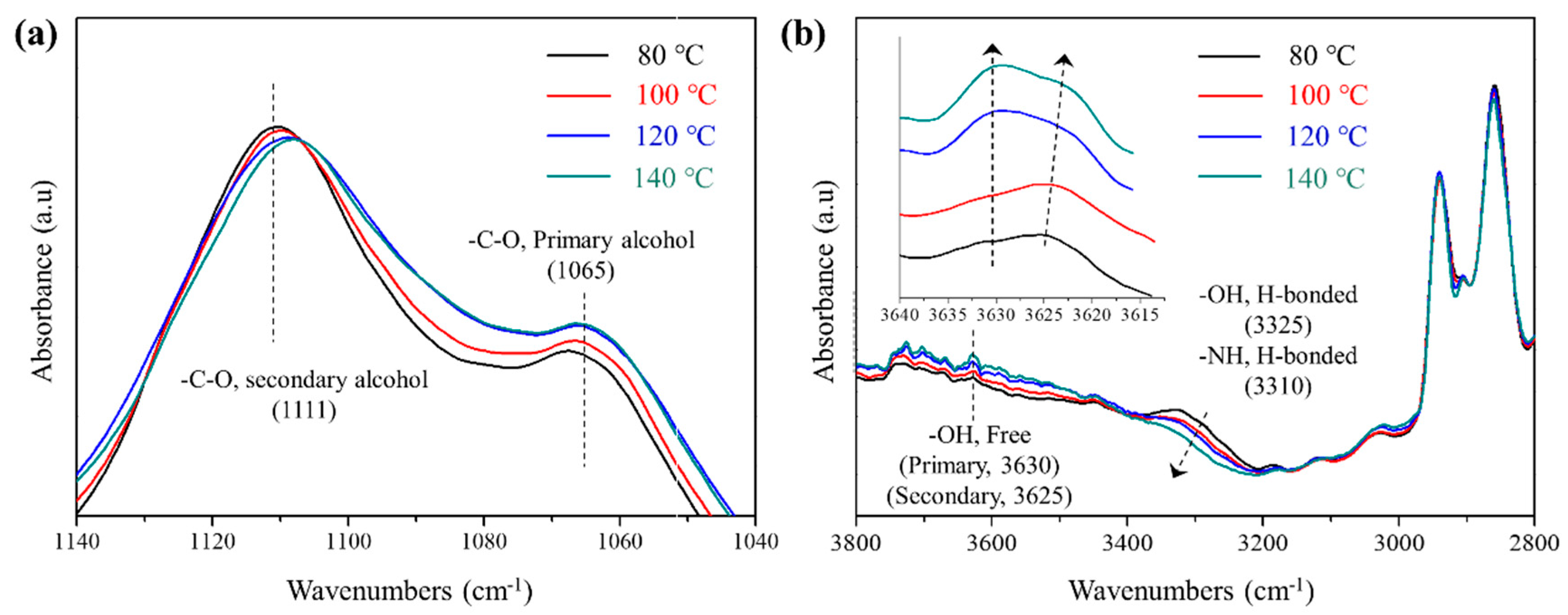
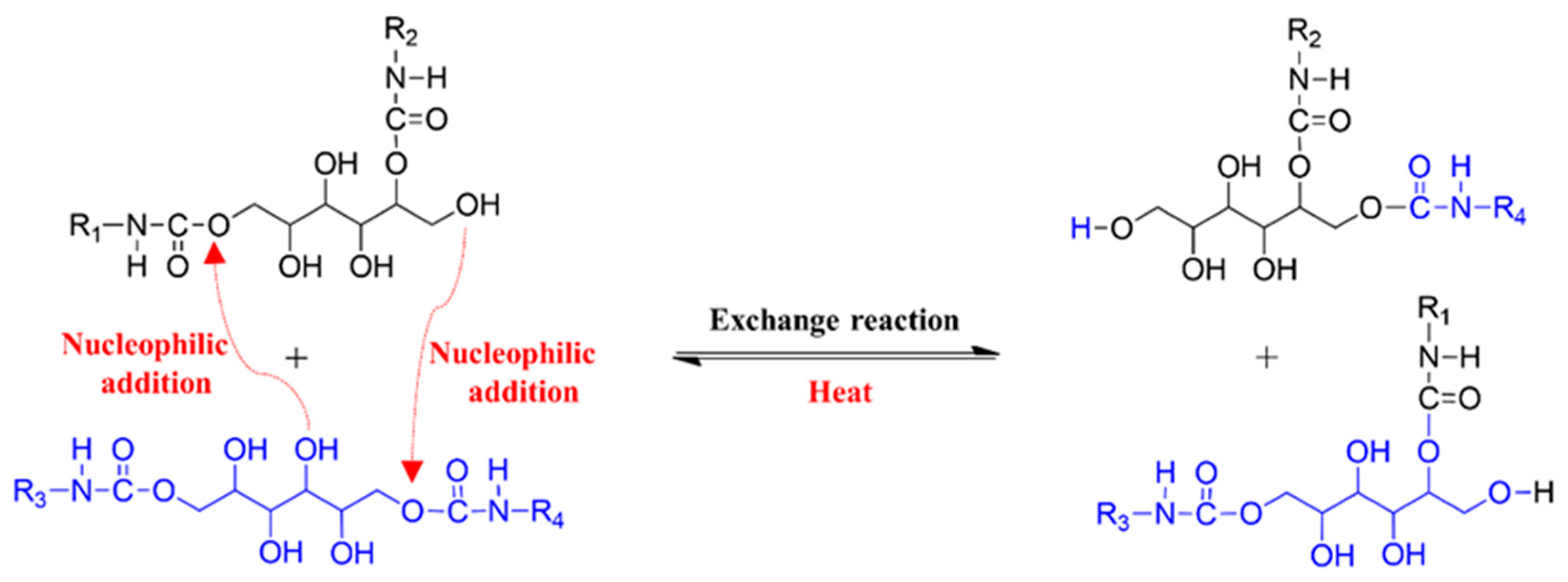
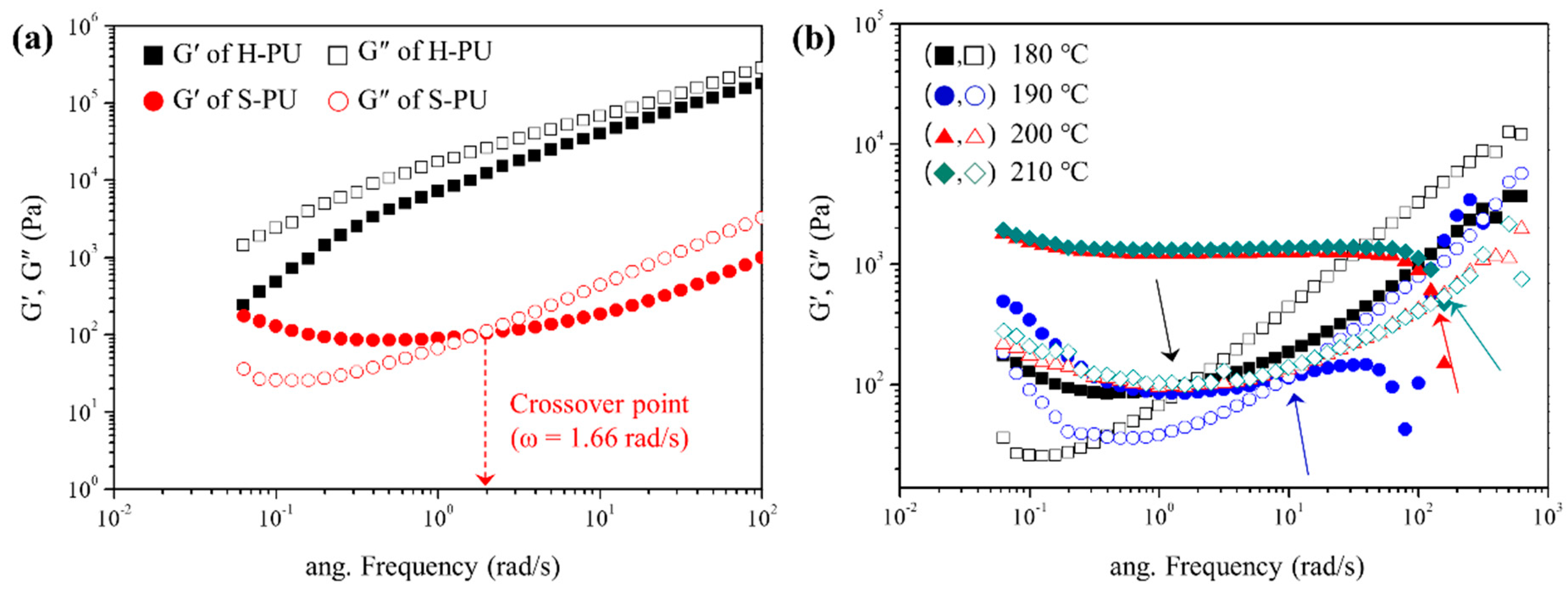
3.2. Exchange Reaction and Thermomechanical Properties of S-PU
3.3. Self-Healing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Eceiza, A.; Martin, M.D.; de la Caba, K.; Kortaberria, G.; Gabilondo, N.; Corcuera, M.A.; Mondragon, I. Thermoplastic Polyurethane Elastomers Based on Polycarbonate Diols with Different Soft Segment Molecular Weight and Chemical Structure: Mechanical and Thermal Properties. Polym. Eng. Sci. 2008, 48, 297–306. [Google Scholar] [CrossRef]
- Akindoyo, J.O.; Beg, M.D.H.; Ghazali, S.; Islam, M.R.; Jeyaratnam, N.; Yuvaraj, A.R. Polyurethane types, synthesis and applications—A review. RSC Adv. 2016, 6, 114453–114482. [Google Scholar] [CrossRef]
- Han-Do, K.; Tae-Jung, L.; Jae-Ho, H.; Dong-Jin, L. Preparation and properties of thermoplastic polyurethane elastomers with two different soft segments. J. Appl. Polym. Sci. 1999, 37, 345–352. [Google Scholar]
- James Korley, L.S.T.; Pate, B.D.; Thomas, E.L.; Hammond, P.T. Effect of the degree of soft and hard segment ordering on the morphology and mechanical behavior of semicrystalline segmented polyurethanes. Polymer 2006, 47, 3073–3082. [Google Scholar] [CrossRef]
- Liu, C.C.; Zhang, A.Y.; Ye, L.; Feng, Z.G. Self-healing biodegradable poly(urea-urethane) elastomers based on hydrogen bonding interactions. Chin. J. Polym. Sci. 2013, 31, 251–262. [Google Scholar] [CrossRef]
- Fang, Y.; Du, X.; Du, Z.; Wang, H.; Cheng, X. Light- and heat-triggered polyurethane based on dihydroxyl anthracene derivatives for self-healing applications. J. Mater. Chem. A 2017, 5, 8010–8017. [Google Scholar] [CrossRef]
- Aguirresarobe, R.H.; Martin, L.; Aramburu, N.; Irusta, L.; Fernandez-Berridi, M.J. Coumarin based light responsive healable waterborne polyurethanes. Prog. Org. Coat. 2016, 99, 314–321. [Google Scholar] [CrossRef]
- Ji, S.; Cao, W.; Yu, Y.; Xu, H. Visible-Light-Induced Self-Healing Diselenide-Containing Polyurethane Elastomer. Adv. Mater. 2015, 27, 7740–7745. [Google Scholar] [CrossRef] [PubMed]
- Seyed Shahabadi, S.I.; Kong, J.; Lu, X. Aqueous-Only, Green Route to Self-Healable, UV-Resistant, and Electrically Conductive Polyurethane/Graphene/Lignin Nanocomposite Coatings. ACS Sustain. Chem. Eng. 2017, 5, 3148–3157. [Google Scholar] [CrossRef]
- Fang, L.; Chen, J.; Zou, Y.; Xu, Z.; Lu, C. Thermally-induced self-healing behaviors and properties of four epoxy coatings with different network architectures. Polymers 2017, 9, 333. [Google Scholar] [CrossRef]
- Yang, L.; Lu, X.; Wang, Z.; Xia, H. Diels-Alder dynamic crosslinked polyurethane/polydopamine composites with NIR triggered self-healing function. Polym. Chem. 2018, 9, 2166–2172. [Google Scholar] [CrossRef]
- Lin, C.; Sheng, D.; Liu, X.; Xu, S.; Ji, F.; Dong, L.; Zhou, Y.; Yang, Y. NIR induced self-healing electrical conductivity polyurethane/graphene nanocomposites based on Diels−Alder reaction. Polymer 2018, 140, 150–157. [Google Scholar] [CrossRef]
- Zheng, K.; Tian, Y.; Fan, M.; Zhang, J.; Cheng, J. Recyclable, shape-memory, and self-healing soy oil-based polyurethane crosslinked by a thermoreversible Diels–Alder reaction. J. Appl. Polym. Sci. 2018, 135, 1–10. [Google Scholar] [CrossRef]
- Ke, X.; Liang, H.; Xiong, L.; Huang, S.; Zhu, M. Synthesis, curing process and thermal reversible mechanism of UV curable polyurethane based on Diels-Alder structure. Prog. Org. Coat. 2016, 100, 63–69. [Google Scholar] [CrossRef]
- Yang, Y.; Lu, X.; Wang, W. A tough polyurethane elastomer with self-healing ability. Mater. Des. 2017, 127, 30–36. [Google Scholar] [CrossRef]
- Rekondo, A.; Martin, R.; Ruiz De Luzuriaga, A.; Cabañero, G.; Grande, H.J.; Odriozola, I. Catalyst-free room-temperature self-healing elastomers based on aromatic disulfide metathesis. Mater. Horiz. 2014, 1, 237–240. [Google Scholar] [CrossRef]
- Xu, Y.; Chen, D. A Novel Self-healing polyurethane based on disulfide bonds. Macromol. Chem. Phys. 2016, 217, 1191–1196. [Google Scholar] [CrossRef]
- Chen, J.H.; Hu, D.D.; Li, Y.D.; Meng, F.; Zhu, J.; Zeng, J.B. Castor oil derived poly(urethane urea) networks with reprocessibility and enhanced mechanical properties. Polymer 2018, 143, 79–86. [Google Scholar] [CrossRef]
- Jian, X.; Hu, Y.; Zhou, W.; Xiao, L. Self-healing polyurethane based on disulfide bond and hydrogen bond. Polym. Adv. Technol. 2018, 29, 463–469. [Google Scholar] [CrossRef]
- Kim, S.M.; Jeon, H.; Shin, S.H.; Park, S.A.; Jegal, J.; Hwang, S.Y.; Oh, D.X.; Park, J. Superior Toughness and Fast Self-Healing at Room Temperature Engineered by Transparent Elastomers. Adv. Mater. 2018, 30, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wan, T.; Chen, D. Mechanical enhancement of self-healing waterborne polyurethane by graphene oxide. Prog. Org. Coat. 2018, 121, 73–79. [Google Scholar] [CrossRef]
- Begines, B.; Zamora, F.; De Paz, M.V.; Hakkou, K.; Galbis, J.A. Polyurethanes derived from carbohydrates and cystine-based monomers. J. Appl. Polym. Sci. 2015, 132, 1–8. [Google Scholar] [CrossRef]
- Ling, L.; Li, J.; Zhang, G.; Sun, R.; Wong, C.P. Self-Healing and Shape Memory Linear Polyurethane Based on Disulfide Linkages with Excellent Mechanical Property. Macromol. Res. 2018, 26, 365–373. [Google Scholar] [CrossRef]
- Yuan, C.; Rong, M.Z.; Zhang, M.Q. Self-healing polyurethane elastomer with thermally reversible alkoxyamines as crosslinkages. Polymer 2014, 55, 1782–1791. [Google Scholar] [CrossRef]
- Erice, A.; Ruiz de Luzuriaga, A.; Matxain, J.M.; Ruipérez, F.; Asua, J.M.; Grande, H.J.; Rekondo, A. Reprocessable and recyclable crosslinked poly(urea-urethane)s based on dynamic amine/urea exchange. Polymer 2018, 145, 127–136. [Google Scholar] [CrossRef]
- Matsukizono, H.; Endo, T. Reworkable Polyhydroxyurethane Films with Reversible Acetal Networks Obtained from Multifunctional Six-Membered Cyclic Carbonates. J. Am. Chem. Soc. 2018, 140, 884–887. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Zhao, W.; Fu, X.; Liu, Z.; Kong, W.; Zhou, C.; Lei, J. Multifunctional polyurethane-vitrimers completely based on transcarbamoylation of carbamates: Thermally-induced dual-shape memory effect and self-welding. RSC Adv. 2017, 7, 26858–26866. [Google Scholar] [CrossRef]
- Chen, X.; Li, L.; Jin, K.; Torkelson, J.M. Reprocessable Polyhydroxyurethane Network Exhibiting Full Property Recovery and Concurrent Associative and Dissociative Dynamic Chemistry via Transcarbamoylation and Reversible Cyclic Carbonate Aminolysis. Polym. Chem. 2017, 8, 6349–6355. [Google Scholar] [CrossRef]
- Fortman, D.J.; Brutman, J.P.; Hillmyer, M.A.; Dichtel, W.R. Structural effects on the reprocessability and stress relaxation of crosslinked polyhydroxyurethanes. J. Appl. Polym. Sci. 2017, 134, 1–11. [Google Scholar] [CrossRef]
- Zheng, N.; Fang, Z.; Zou, W.; Zhao, Q.; Xie, T. Thermoset Shape-Memory Polyurethane with Intrinsic Plasticity Enabled by Transcarbamoylation. Angew. Chem. Int. Ed. 2016, 55, 11421–11425. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, N.; Abend, M.; Geitner, R.; Vitz, J.; Zechel, S.; Schmitt, M.; Popp, J.; Schubert, U.S.; Hager, M.D. Urethanes as reversible covalent moieties in self-healing polymers. Eur. Polym. J. 2018, 104, 45–50. [Google Scholar] [CrossRef]
- Fortman, D.J.; Brutman, J.P.; Cramer, C.J.; Hillmyer, M.A.; Dichtel, W.R. Mechanically Activated, Catalyst-Free Polyhydroxyurethane Vitrimers. J. Am. Chem. Soc. 2015, 137, 14019–14022. [Google Scholar] [CrossRef] [PubMed]
- Carré, C.; Bonnet, L.; Avérous, L. Original biobased nonisocyanate polyurethanes: Solvent- and catalyst-free synthesis, thermal properties and rheological behaviour. RSC Adv. 2014, 4, 54018–54025. [Google Scholar] [CrossRef]
- Carré, C.; Zoccheddu, H.; Delalande, S.; Pichon, P.; Avérous, L. Synthesis and characterization of advanced biobased thermoplastic nonisocyanate polyurethanes, with controlled aromatic-aliphatic architectures. Eur. Polym. J. 2016, 84, 759–769. [Google Scholar] [CrossRef]
- Beniah, G.; Uno, B.E.; Lan, T.; Jeon, J.; Heath, W.H.; Scheidt, K.A.; Torkelson, J.M. Tuning nanophase separation behavior in segmented polyhydroxyurethane via judicious choice of soft segment. Polymer 2017, 110, 218–227. [Google Scholar] [CrossRef]
- Van Velthoven, J.L.J.; Gootjes, L.; Van Es, D.S.; Noordover, B.A.J.; Meuldijk, J. Poly(hydroxy urethane)s based on renewable diglycerol dicarbonate. Eur. Polym. J. 2015, 70, 125–135. [Google Scholar] [CrossRef]
- Romero, A.; Alonso, E.; Sastre, Á.; Nieto-Márquez, A. Conversion of biomass into sorbitol: Cellulose hydrolysis on MCM-48 and d-Glucose hydrogenation on Ru/MCM-48. Microporous Mesoporous Mater. 2016, 224, 1–8. [Google Scholar] [CrossRef]
- Mishra, D.K.; Lee, J.M.; Chang, J.S.; Hwang, J.S. Liquid phase hydrogenation of d-glucose to d-sorbitol over the catalyst (Ru/NiO-TiO2) of ruthenium on a NiO-modified TiO2 support. Catal. Today 2012, 185, 104–108. [Google Scholar] [CrossRef]
- Bin Kassim, A.; Rice, C.L.; Kuhn, A.T. Formation of sorbitol by cathodic reduction of glucose. J. Appl. Electrochem. 1981, 11, 261–267. [Google Scholar] [CrossRef]
- Wells, J.G.; Davis, B.R.; Wachsmuth, I.K.; Riley, L.W.; Remis, R.S.; Sokolow, R.; Morris, G.K. Laboratory investigation of hemorrhagic colitis outbreaks associated with a rare Escherichia coli serotype. J. Clin. Microbiol. 1983, 18, 512–520. [Google Scholar] [PubMed]
- Van Gorp, K.; Boerman, E.; Cavenaghi, C.V.; Berben, P.H. Catalytic hydrogenation of fine chemicals: Sorbitol production. Catal. Today 1999, 52, 349–361. [Google Scholar] [CrossRef]
- Gallezot, P.; Nicolaus, N.; Flèche, G.; Fuertes, P.; Perrard, A. Glucose hydrogenation on ruthenium catalysts in a trickle-bed reactor. J. Catal. 1998, 180, 51–55. [Google Scholar] [CrossRef]
- Anand, A.; Kulkarni, R.D.; Gite, V.V. Preparation and properties of eco-friendly two pack PU coatings based on renewable source (sorbitol) and its property improvement by nano ZnO. Prog. Org. Coat. 2012, 74, 764–767. [Google Scholar] [CrossRef]
- Ugarte, L.; Gómez-Fernández, S.; Peña-Rodríuez, C.; Prociak, A.; Corcuera, M.A.; Eceiza, A. Tailoring Mechanical Properties of Rigid Polyurethane Foams by Sorbitol and Corn Derived Biopolyol Mixtures. ACS Sustain. Chem. Eng. 2015, 3, 3382–3387. [Google Scholar] [CrossRef]
- Rand, L.; Thir, B.; Reegen, S.L.; Frisch, K.C. Kinetics of alcohol–isocyanate reactions with metal catalysts. J. Appl. Polym. Sci. 1965, 9, 1787–1795. [Google Scholar] [CrossRef]
- Ajithkumar, S.; Kansara, S.S.; Patel, N.K. Kinetics of castor oil based polyol-toluene diisocyanate reactions. Eur. Polym. J. 1998, 34, 1273–1276. [Google Scholar] [CrossRef]
- Dyer, E.; Taylor, H.A.; Mason, S.J.; Samson, J. The rates of reaction of isocyanates with alcohols. I. Phenyl isocyanate with 1-and 2-butanol. J. Am. Chem. Soc. 1949, 71, 4106–4109. [Google Scholar] [CrossRef]
- Tang, J.; Zhang, S.; Lin, Y.; Zhou, J.; Pang, L.; Nie, X.; Zhou, B.; Tang, W. Engineering Cyclodextrin Clicked Chiral Stationary Phase for High-Efficiency Enantiomer Separation. Sci. Rep. 2015, 5, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Sang, Z.; Zhao, J.; Zhang, Z.; Zhang, J.; Yang, W. Crystallizable and Tough Aliphatic Thermoplastic Polyureas Synthesized through a Nonisocyanate Route. Ind. Eng. Chem. Res. 2016, 55, 1902–1911. [Google Scholar] [CrossRef]
- Pavia, D.L.; Lampman, G.M.; Kriz, G.S.; Vyvyan, J.A. Introduction to Spectroscopy, 4th ed.; Cengage Learning: Boston, MA, USA, 2009; pp. 13–70. ISBN 9780495114789. [Google Scholar]
- Das, D.; Varghese, L.R.; Das, N. Enhanced TDS removal using cyclodextrinated, sulfonated and aminated forms of bead-membrane duo nanobiocomposite via sophorolipid mediated complexation. Desalination 2015, 360, 35–44. [Google Scholar] [CrossRef]
- Kenkyu, T.N. The assignment of IR absorption bands due to free hydroxyl groups in cellulose. Cellulose 1997, 4, 281. [Google Scholar] [CrossRef]
- Coleman, M.M.; Skrovanek, D.J.; Hu, J.; Painter, P.C. Hydrogen Bonding in Polymer Blends. 1. FTIR Studies of Urethane-Ether Blends. Macromolecules 1988, 21, 59–65. [Google Scholar] [CrossRef]
- Pukánszky, B.; Bagdi, K.; Molnár, K.; Pukannszky, B. Thermal analysis of the structure of segmented polyurethane elastomers: Relation to mechanical properties. J. Therm. Anal. Calorim. 2009, 98, 825–832. [Google Scholar]
- Chattopadhyay, D.K.; Sreedhar, B.; Raju, K.V.S.N. The phase mixing studies on moisture cured polyurethane-ureas during cure. Polymer 2006, 47, 3814–3825. [Google Scholar] [CrossRef]
- Christenson, E.M.; Anderson, J.M.; Hiltner, A.; Baer, E. Relationship between nanoscale deformation processes and elastic behavior of polyurethane elastomers. Polymer 2005, 46, 11744–11754. [Google Scholar] [CrossRef]
- Narine, S.S.; Kong, X.; Bouzidi, L.; Sporns, P. Physical properties of polyurethanes produced from polyols from seed oils: I. Elastomers. J. Am. Oil Chem. Soc. 2007, 84, 55–63. [Google Scholar] [CrossRef]
- Cho, D.; Lee, S.; Yang, G.; Fukushima, H.; Drzal, L.T. Dynamic mechanical and thermal properties of phenylethynyl-terminated polyimide composites reinforced with expanded graphite nanoplatelets. Macromol. Mater. Eng. 2005, 290, 179–187. [Google Scholar] [CrossRef]
- Liu, T.; Hao, C.; Wang, L.; Li, Y.; Liu, W.; Xin, J.; Zhang, J. Eugenol-Derived Biobased Epoxy: Shape Memory, Repairing, and Recyclability. Macromolecules 2017, 50, 8588–8597. [Google Scholar] [CrossRef]
- Roberts, M.C.; Hanson, M.C.; Massey, A.P.; Karren, E.A.; Kiser, P.F. Dynamically restructuring hydrogel networks formed with reversible covalent crosslinks. Adv. Mater. 2007, 19, 2503–2507. [Google Scholar] [CrossRef]
- Callies, X.; Fonteneau, C.; Pensec, S.; Chazeau, L.; Bouteiller, L.; Ducouret, G.; Creton, C. Linear Rheology of Supramolecular Polymers Center-Functionalized with Strong Stickers. Macromolecules 2015, 48, 7320–7326. [Google Scholar] [CrossRef]
- Zhao, X.; Guo, S.; Li, H.; Liu, J.; Su, C.; Song, H. One-pot synthesis of self-healable and recyclable ionogels based on polyamidoamine (PAMAM) dendrimers via Schiff base reaction. RSC Adv. 2017, 7, 38765–38772. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.; Meng, L.; Wang, Y.; Wu, X.; Liu, S.; Li, Y.; Kang, W. Superior thermal stability gel emulsion produced by low concentration Gemini surfactant. Colloids Surf. A Physicochem. Eng. Asp. 2011, 384, 194–199. [Google Scholar] [CrossRef]
- Zhou, Y.; Goossens, J.G.P.; Sijbesma, R.P.; Heuts, J.P.A. Poly(butylene terephthalate)/Glycerol-based Vitrimers via Solid-State Polymerization. Macromolecules 2017, 50, 6742–6751. [Google Scholar] [CrossRef]
- Lewis, C.L.; Dell, E.M. A review of shape memory polymers bearing reversible binding groups. J. Polym. Sci. Part B Polym. Phys. 2016, 54, 1340–1364. [Google Scholar] [CrossRef] [Green Version]
- Döhler, D.; Michael, P.; Binder, W. Self-Healing Polymers: From Principles to Applications; Binder, W.H., Ed.; Wiley-VCH Verlag GmbH & Co., KGaA: Weinheim, Germany, 2013; pp. 7–60. ISBN 9783527334391. [Google Scholar]
Sample Availability: Not Available. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Composition (Molar Ratio) | Mn (g/mol) | Mw (g/mol) | PDI | |||
|---|---|---|---|---|---|---|---|
| MDI | PTMEG | Sorbitol | HD | ||||
| S-PU | 2 | 1 | 1 | – | 56,600 | 126,900 | 2.24 |
| H-PU | 2 | 1 | – | 1 | 64,600 | 131,900 | 2.04 |
| CTE (1/°C) | ||||
|---|---|---|---|---|
| 1st Cycle | 2nd Cycle | |||
| S-PU | 0.484 a | 0.132 b | 0.398 a | 0.306 b |
| H-PU | 0.436 | 0.459 | ||
| S-PU | H-PU | |||||||
|---|---|---|---|---|---|---|---|---|
| 180 °C | 190 °C | 200 °C | 210 °C | 180 °C | 190 °C | 200 °C | 210 °C | |
| ω (rad/s) | 1.62 | 9.55 | 131.57 | 159.47 | – | – | – | 0.08 |
| Properties | S-PU | H-PU | ||||||
|---|---|---|---|---|---|---|---|---|
| Uncut S-PU | Self-Healed S-PU | Uncut H-PU | Self-Healed H-PU | |||||
| 1st | 2nd | 3rd | 1st | 2nd | 3rd | |||
| σ (MPa) | 22.92 | 25.24 | 23.76 | 22.67 | 22.53 | 5.182 | 4.541 | 2.554 |
| ε (%) | 660 | 620 | 580 | 590 | 430 | 290 | 260 | 130 |
| E (MPa) | 4.447 | 4.893 | 5.012 | 5.221 | 4.655 | 4.353 | 4.117 | 3.832 |
| Self-healing efficiency (%) | – | 93.79 | 88.29 | 84.24 | – | 22.68 | 19.87 | 11.17 |
| Gel-fraction a (%) | 25.4 | 52.7 | 53.2 | 53.1 | 35.8 | 36.2 | 36.3 | 35.2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.H.; Shin, S.-R.; Lee, D.-S. Sorbitol as a Chain Extender of Polyurethane Prepolymers to Prepare Self-Healable and Robust Polyhydroxyurethane Elastomers. Molecules 2018, 23, 2515. https://doi.org/10.3390/molecules23102515
Lee SH, Shin S-R, Lee D-S. Sorbitol as a Chain Extender of Polyurethane Prepolymers to Prepare Self-Healable and Robust Polyhydroxyurethane Elastomers. Molecules. 2018; 23(10):2515. https://doi.org/10.3390/molecules23102515
Chicago/Turabian StyleLee, Sang Hyub, Se-Ra Shin, and Dai-Soo Lee. 2018. "Sorbitol as a Chain Extender of Polyurethane Prepolymers to Prepare Self-Healable and Robust Polyhydroxyurethane Elastomers" Molecules 23, no. 10: 2515. https://doi.org/10.3390/molecules23102515
APA StyleLee, S. H., Shin, S. -R., & Lee, D. -S. (2018). Sorbitol as a Chain Extender of Polyurethane Prepolymers to Prepare Self-Healable and Robust Polyhydroxyurethane Elastomers. Molecules, 23(10), 2515. https://doi.org/10.3390/molecules23102515