Metal-Catalyzed Cross-Coupling Reactions on Azaindole Synthesis and Functionalization



Abstract
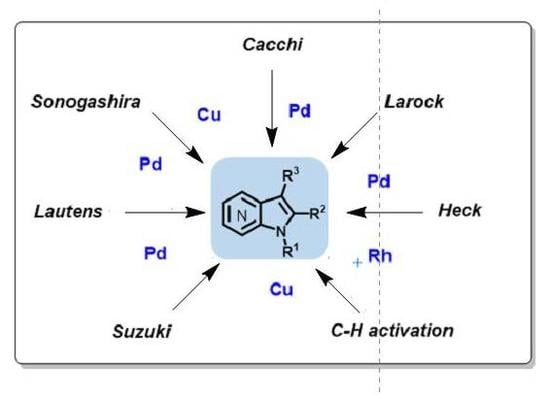
:
{kind=link}
{kind=link}
{kind=link}
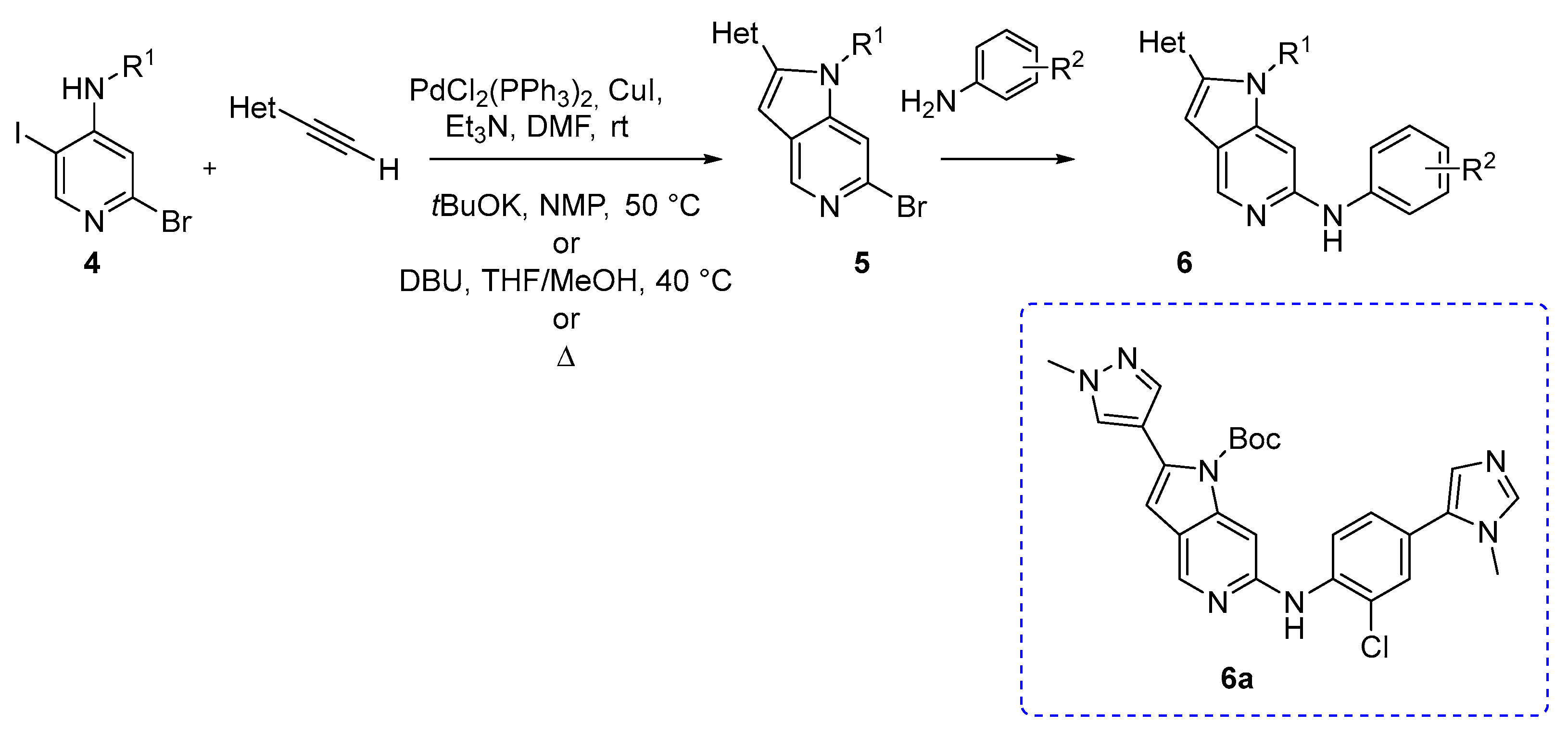{kind=link}
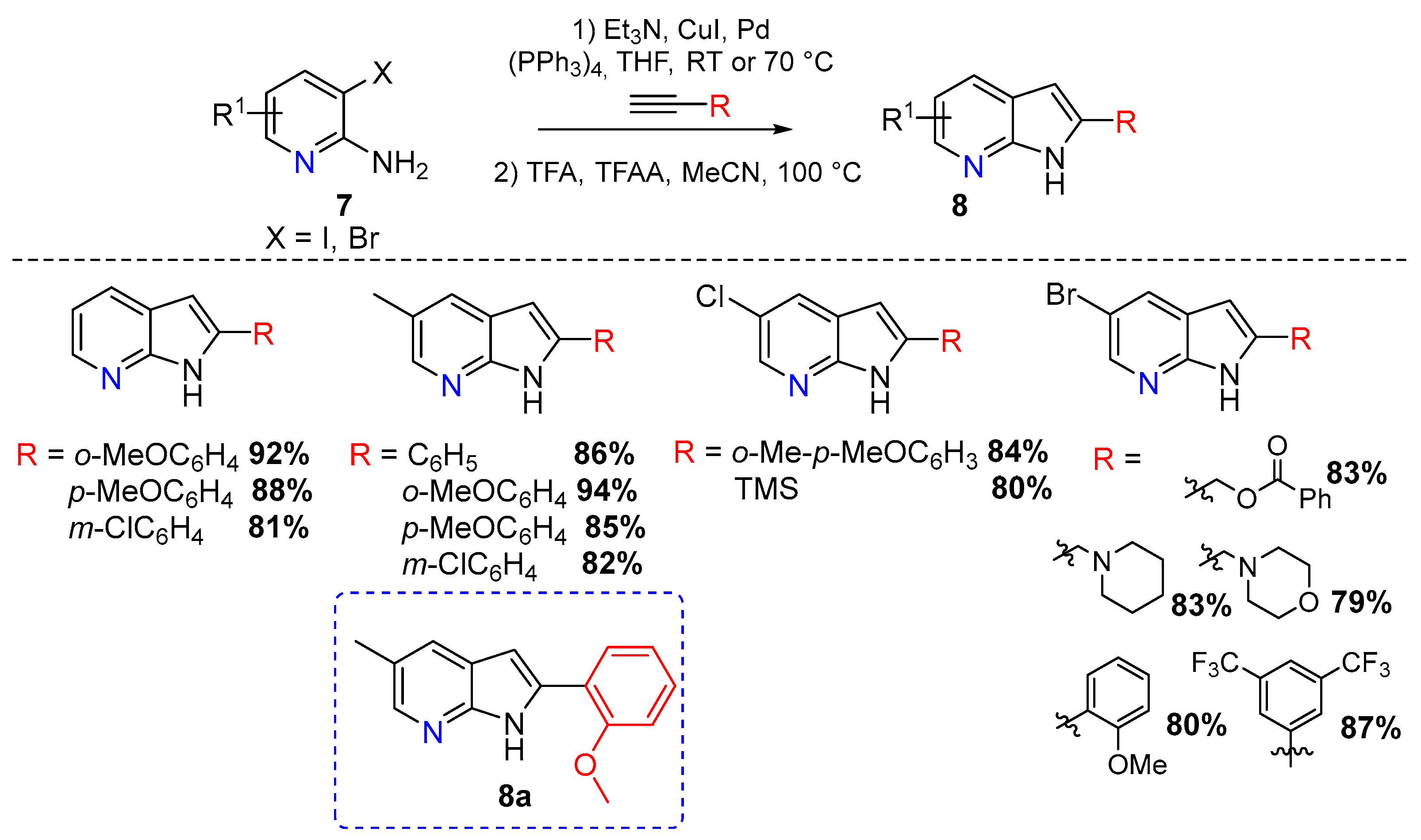{kind=link}
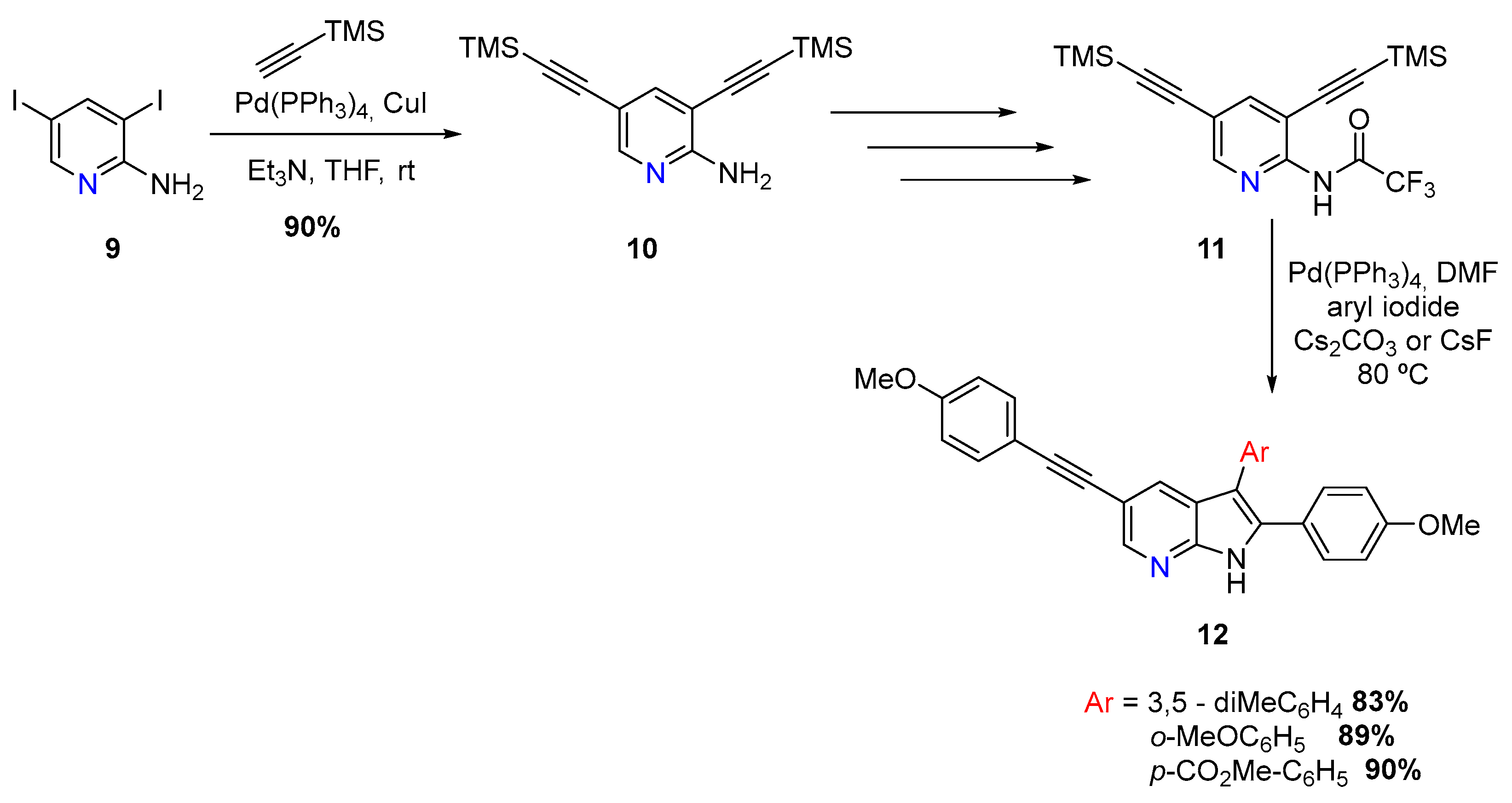{kind=link}
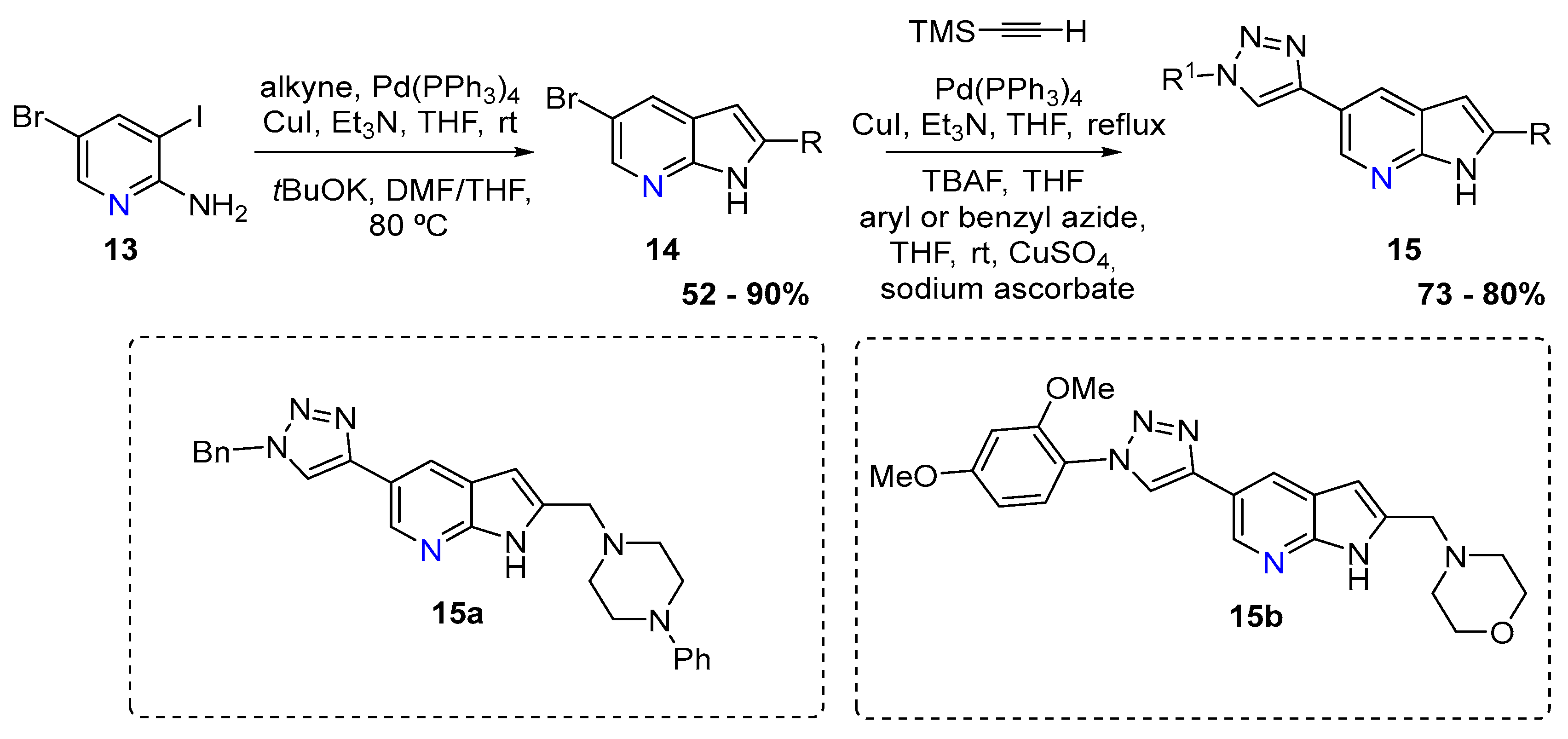{kind=link}
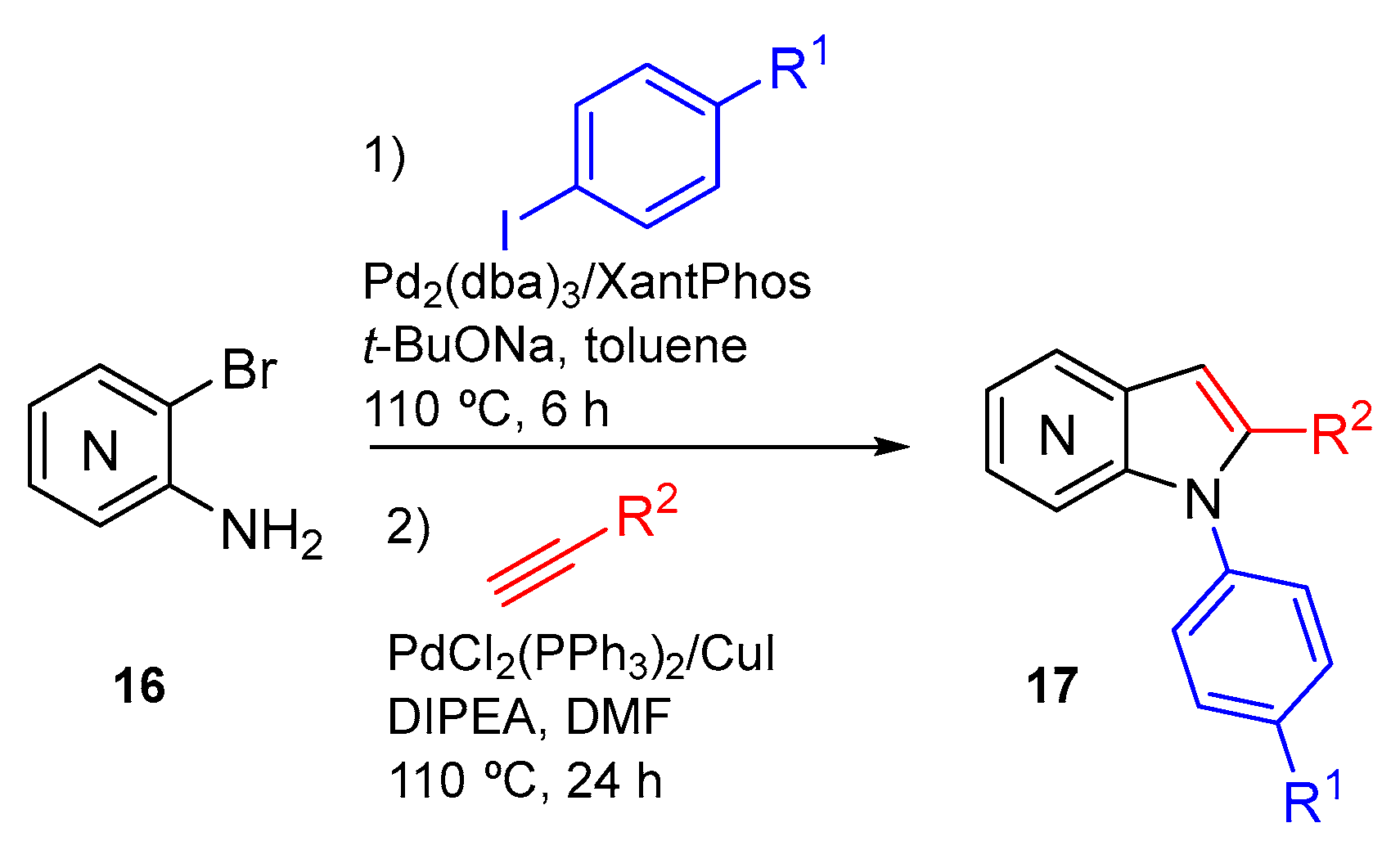{kind=link}
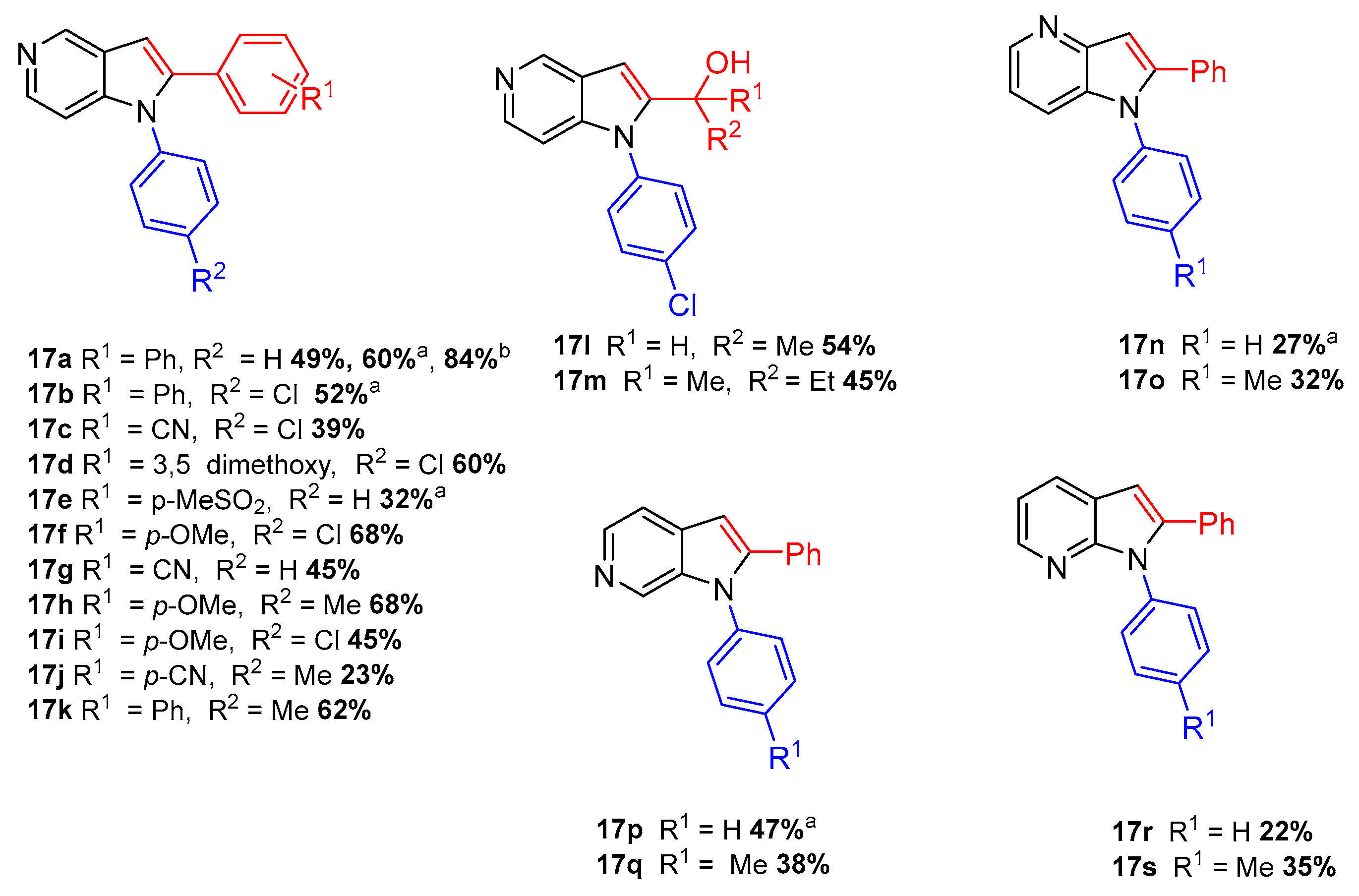{kind=link}
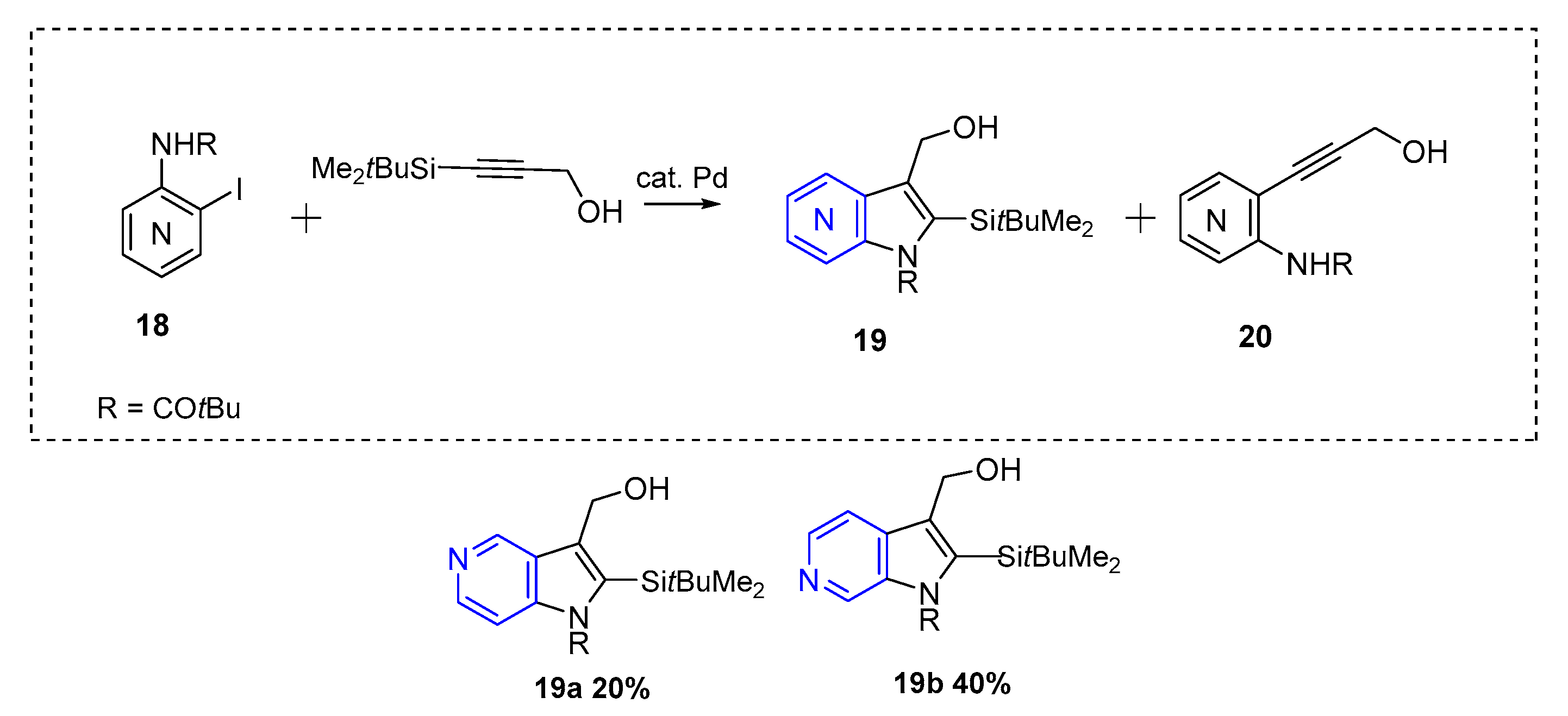{kind=link}
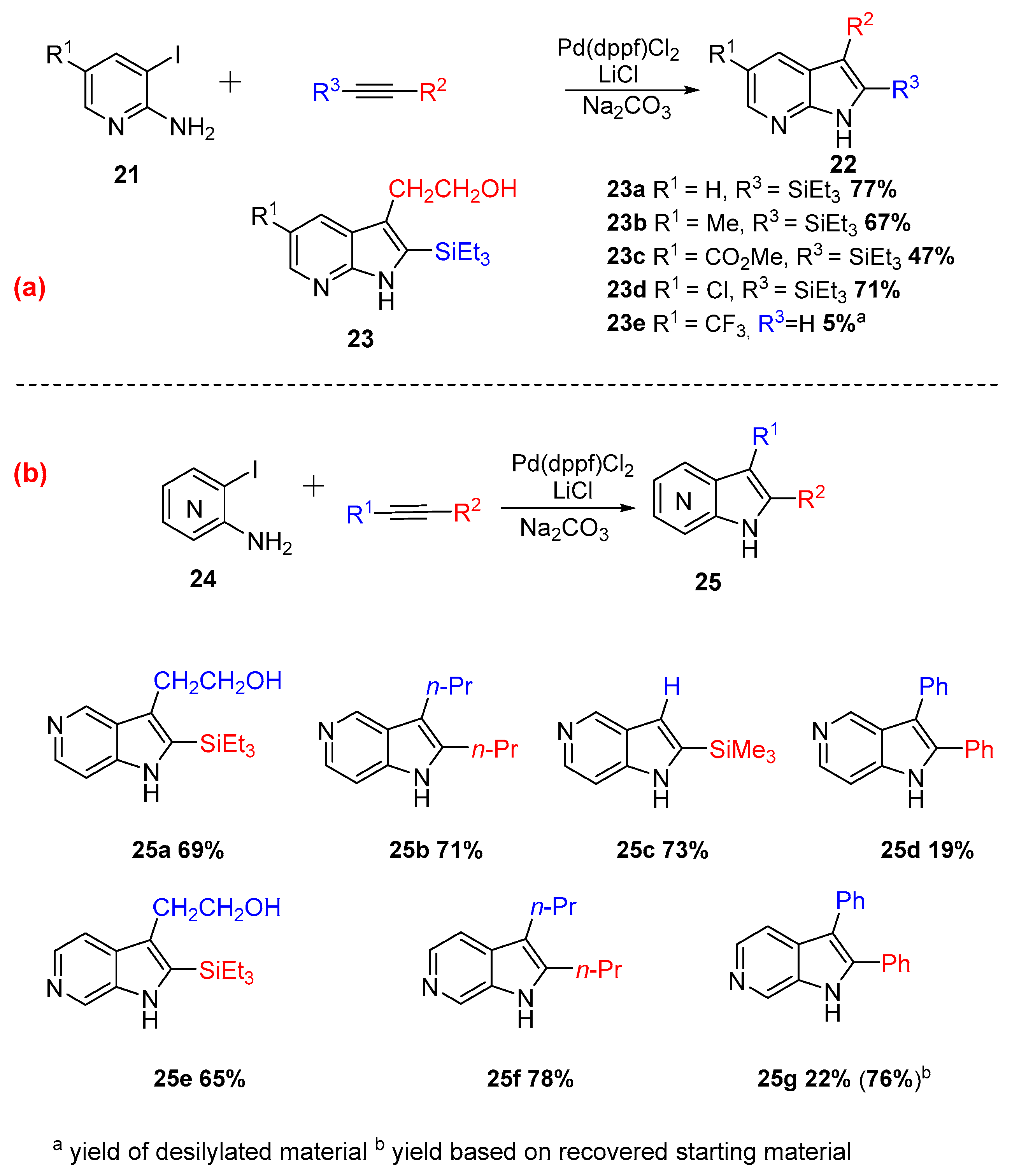{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Metal-Catalyzed Cross-Coupling Reactions
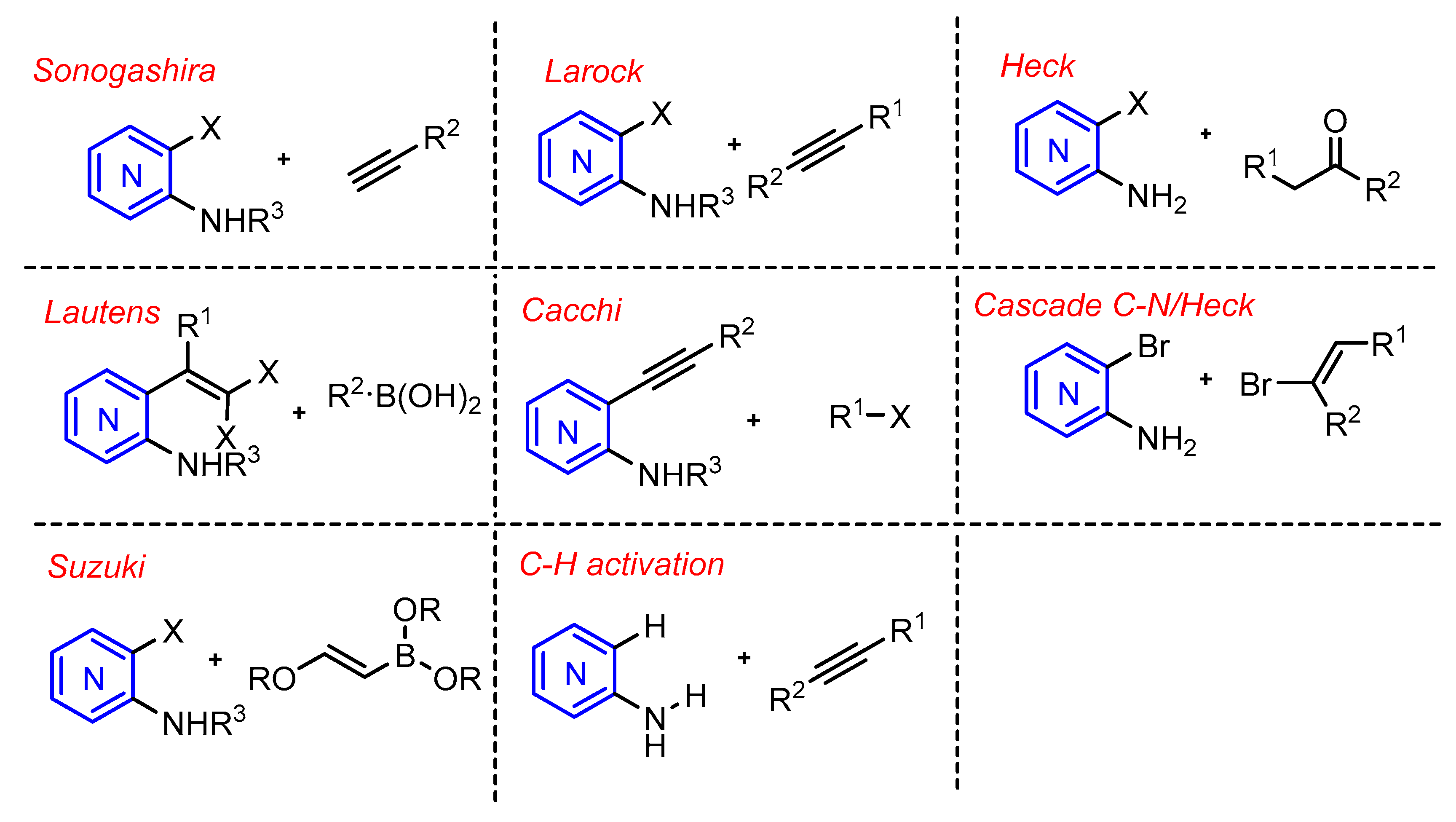
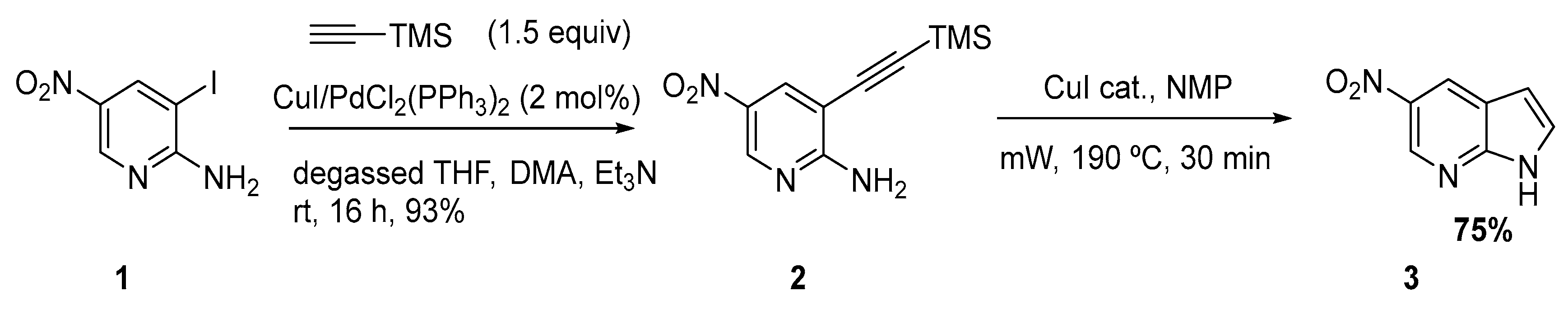
2.1. Sonogashira Reaction
2.2. Larock Reaction
2.3. Heck Reaction
2.4. Suzuki Coupling and Lautens Reaction
2.5. Cacchi Reaction
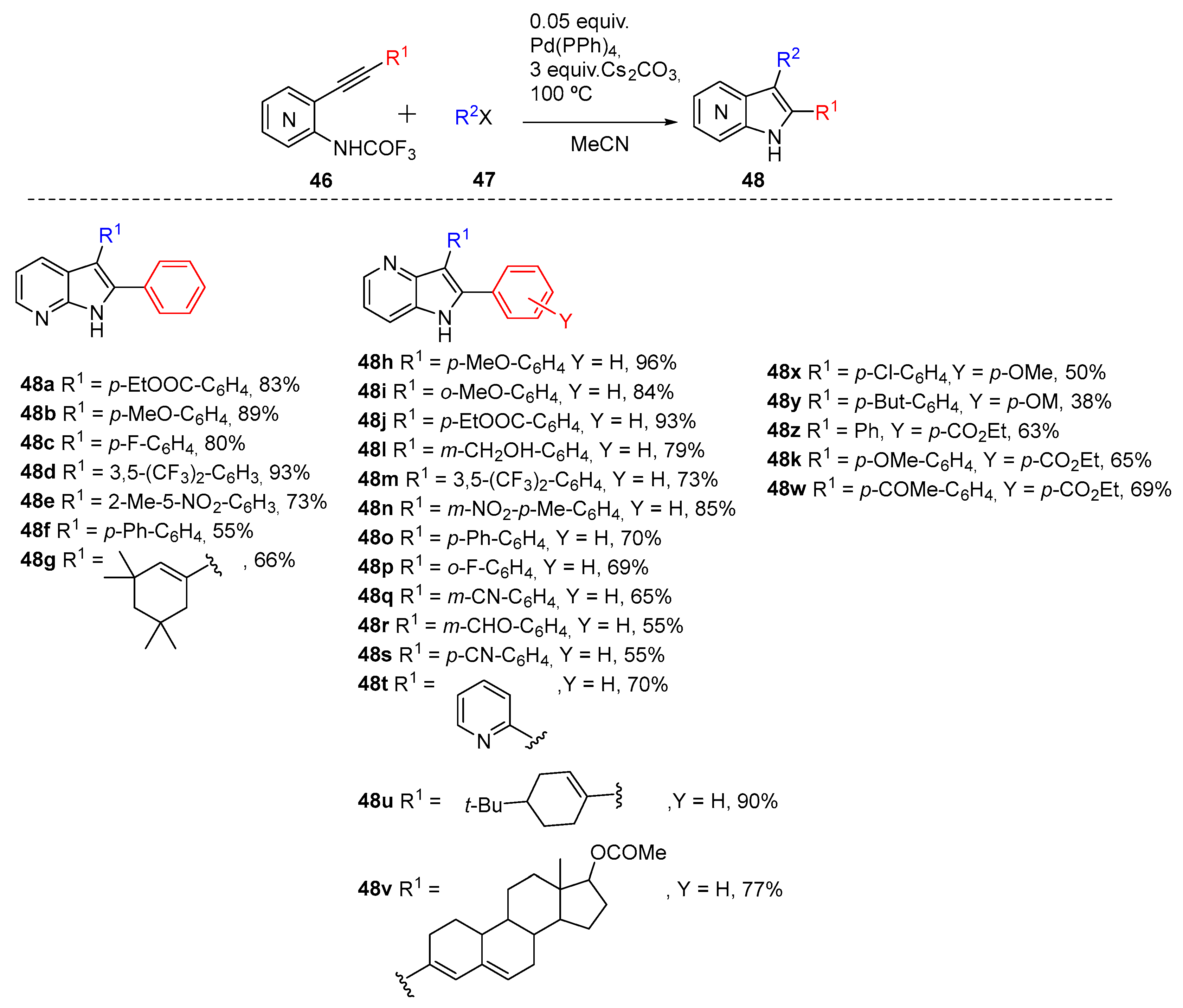
2.6. C-H Activation Reaction
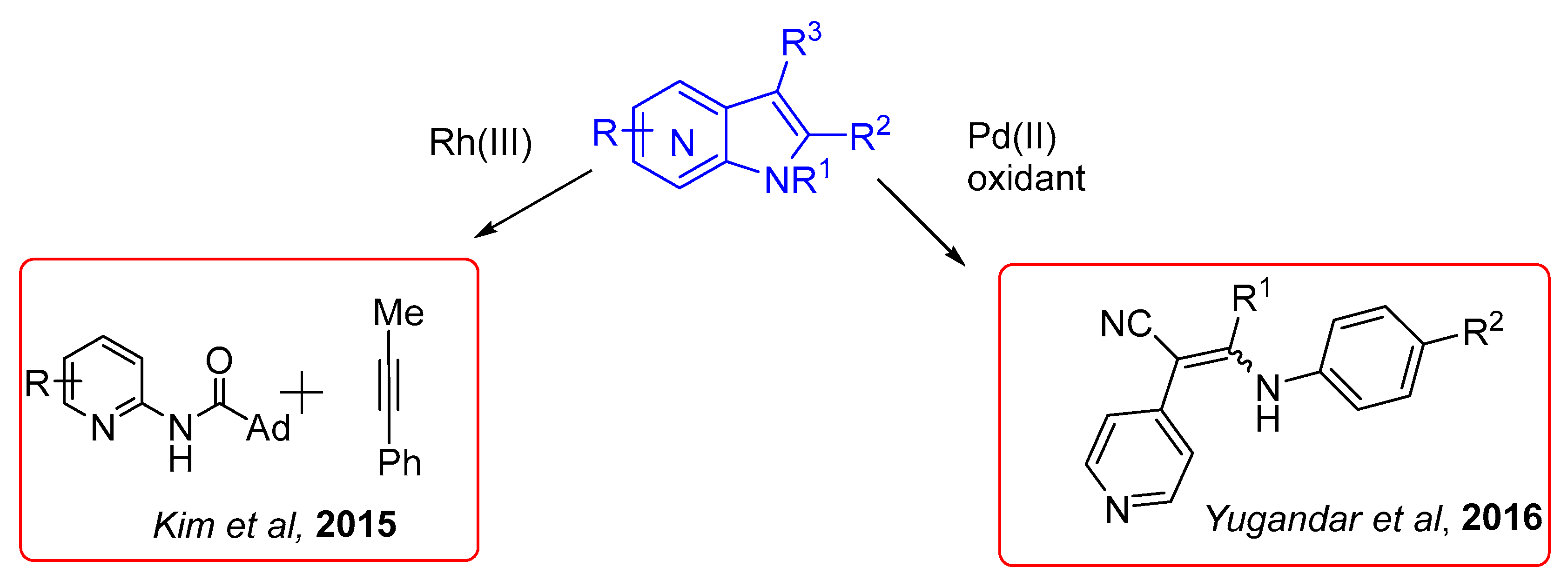
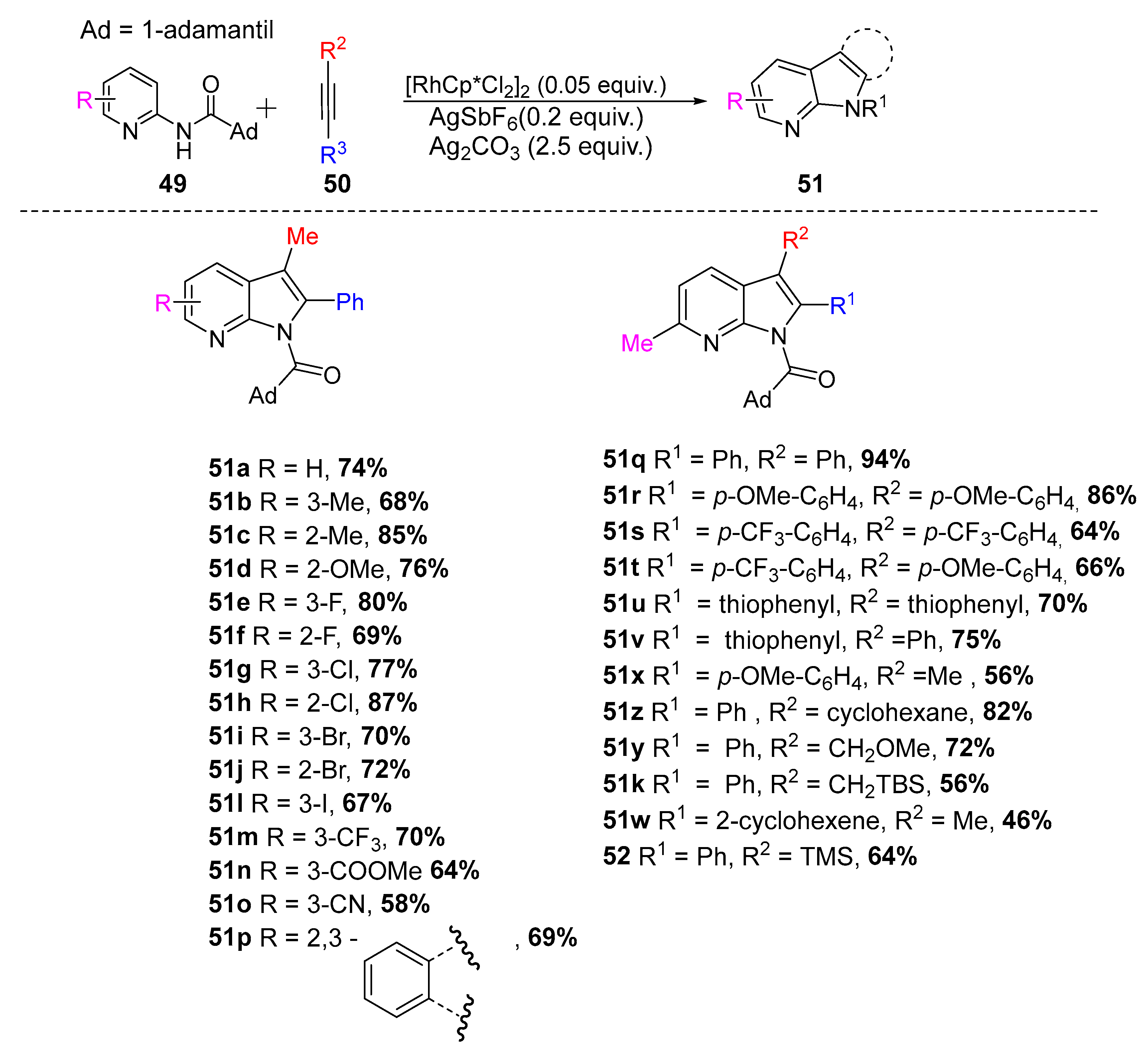
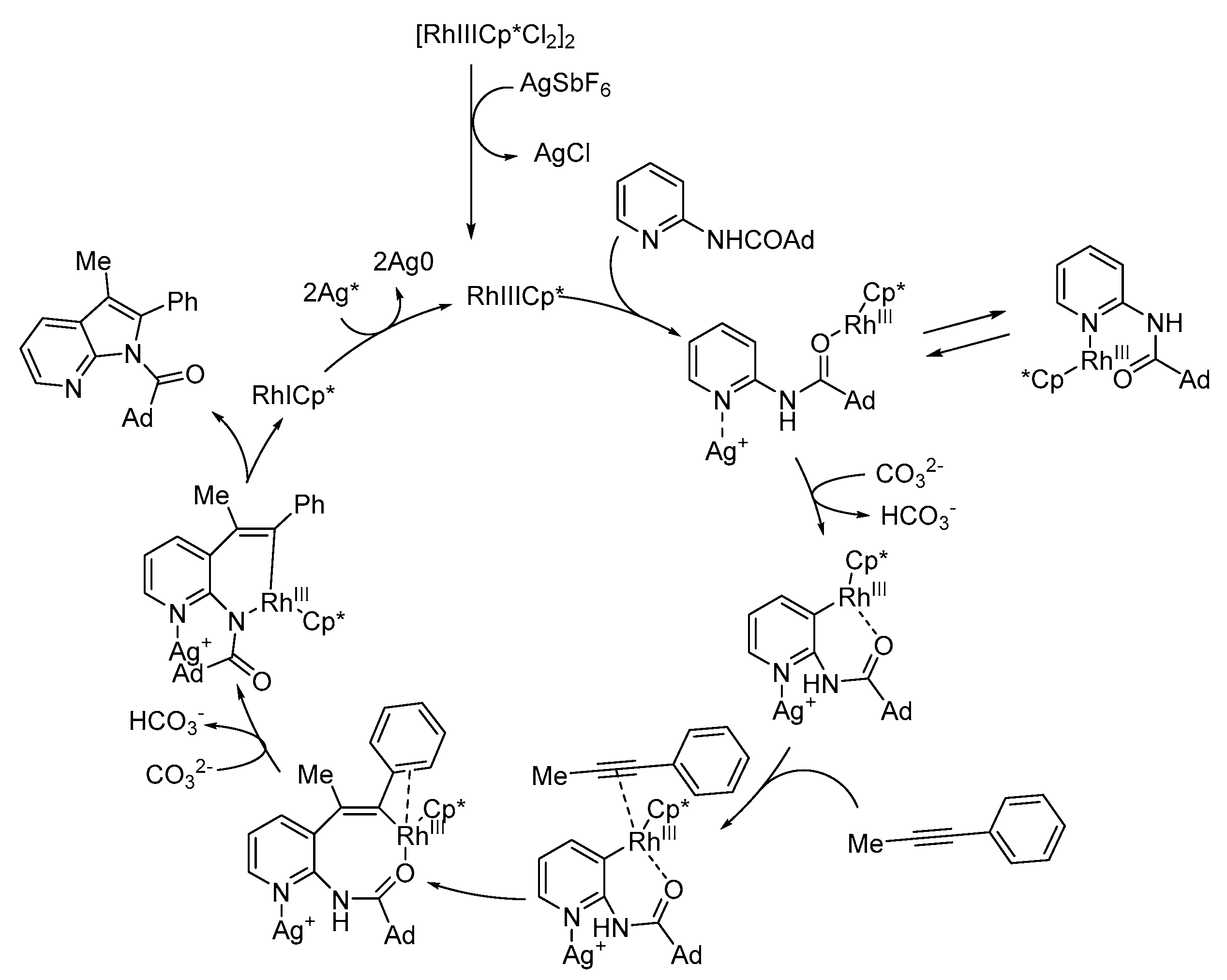
2.6.1. C–H via Rhodium Catalysis
2.6.2. C-H Activation Reaction via Palladium Catalysis
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mérour, J.Y.; Buron, F.; Plé, K.; Bonnet, P.; Routier, S. The azaindole framework in the design of kinase inhibitors. Molecules 2014, 19, 19935–19979. [Google Scholar] [CrossRef] [PubMed]
- Song, J.J.; Reeves, J.T.; Gallou, F.; Tan, Z.; Yee, N.K.; Senanayake, C.H. Organometallic methods for the synthesis and functionalization of azaindoles. Chem. Soc. Rev. 2007, 36, 1120–1132. [Google Scholar] [CrossRef] [PubMed]
- Pearson, S.E.; Nandan, S. A practical, efficient synthesis of 5-amino-7-azaindole. Synthesis (Stuttg) 2005, 2503–2506. [Google Scholar] [CrossRef]
- Naud, S.; Westwood, I.M.; Faisal, A.; Sheldrake, P.; Bavetsias, V.; Atrash, B.; Cheung, K.M.J.; Liu, M.; Hayes, A.; Schmitt, J.; et al. Structure-based design of orally bioavailable 1 h -Pyrrolo[3,2-c]pyridine inhibitors of mitotic kinase monopolar spindle 1 (MPS1). J. Med. Chem. 2013, 56, 10045–10065. [Google Scholar] [CrossRef] [PubMed]
- Leboho, T.C.; Van Vuuren, S.F.; Michael, J.P.; De Koning, C.B. The acid-catalysed synthesis of 7-azaindoles from 3-alkynyl-2- aminopyridines and their antimicrobial activity. Org. Biomol. Chem. 2014, 12, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Leboho, T.C.; Giri, S.; Popova, I.; Cock, I.; Michael, J.P.; De Koning, C.B. Double Sonogashira reactions on dihalogenated aminopyridines for the assembly of an array of 7-azaindoles bearing triazole and quinoxaline substituents at C-5: Inhibitory bioactivity against Giardia duodenalis trophozoites. Bioorg. Med. Chem. 2015, 23, 4943–4951. [Google Scholar] [CrossRef] [PubMed]
- Purificação, S.I.; Pires, M.J.D.; Rippel, R.; Santos, A.S.; Marques, M.M.B. One-Pot Synthesis of 1,2-Disubstituted 4-, 5-, 6-, and 7-Azaindoles from Amino- o -halopyridines via N-Arylation/Sonogashira/Cyclization Reaction. Org. Lett. 2017, 19, 5118–5121. [Google Scholar] [CrossRef] [PubMed]
- Dority, J.A.; Bacon, E.R.; Lesher, G.Y.; Kumar, V.; Singh, B. Synthesis of 7-Azaindole and 7-Azaoxindole Derivatives through a Palladium-Catalyzed Cross-Coupling Reaction. J. Org. Chem. 1992, 57, 6995–6998. [Google Scholar] [CrossRef]
- Xu, L.; Lewis, I.R.; Davidsen, S.K.; Summers, J.B. Transition metal catalyzed synthesis of 5-azaindoles. Tetrahedron Lett. 1998, 39, 5159–5162. [Google Scholar] [CrossRef]
- Trejo, A.; Arzeno, H.; Browner, M.; Chanda, S.; Cheng, S.; Comer, D.D.; Dalrymple, S.A.; Dunten, P.; Lafargue, J.A.; Lovejoy, B.; et al. Design and Synthesis of 4-Azaindoles as Inhibitors of p38 MAP Kinase. J. Med. Chem. 2003, 46, 4702–4713. [Google Scholar] [CrossRef] [PubMed]
- Chi, S.M.; Choi, J.K.; Yum, E.K.; Chi, D.Y. Palladium-catalyzed functionalization of 5- and 7-azaindoles. Tetrahedron Lett. 2000, 41, 919–922. [Google Scholar] [CrossRef]
- Ujjainwalla, F.; Warner, D. Synthesis of 5-, 6- and 7-azaindoles via palladium-catalyzed heteroannulation of internal alkynes. Tetrahedron Lett. 1998, 39, 5355–5358. [Google Scholar] [CrossRef]
- Park, S.S.; Choi, J.K.; Yum, E.K.; Ha, D.C. A facile synthesis of 2,3-disubstituted pyrrolo[2,3-b]pyridines via palladium-catalyzed heteroannulation with internal alkynes. Tetrahedron Lett. 1998, 39, 627–630. [Google Scholar] [CrossRef]
- Wensbo, D.; Eriksson, A.; Jeschke, T.; Annby, U.; Gronowitz, S.; Cohen, L.A. Palladium-catalysed synthesis of heterocondensed pyrroles. Tetrahedron Lett. 1993, 34, 2823–2826. [Google Scholar] [CrossRef]
- Larock, R.C.; Yum, E.K.; Refvik, M.D. Synthesis of 2,3-disubstituted indoles via palladium-catalyzed annulation of internal alkynes. J. Org. Chem. 1998, 63, 7652–7662. [Google Scholar] [CrossRef]
- Larock, R.C.; Yum, E.K. Synthesis of Indoles via Palladium-Catalyzed Heteroannulation of Internal Alkynes. J. Am. Chem. Soc. 1991, 113, 6689–6690. [Google Scholar] [CrossRef]
- Koolman, H.; Heinrich, T.; Böttcher, H.; Rautenberg, W.; Reggelin, M. Syntheses of novel 2,3-diaryl-substituted 5-cyano-4-azaindoles exhibiting c-Met inhibition activity. Bioorg. Med. Chem. Lett. 2009, 19, 1879–1882. [Google Scholar] [CrossRef] [PubMed]
- Blache, Y.; Sinibaldi-Troin, M.E.; Hichour, M.; Benezech, V.; Chavignon, O.; Gramain, J.C.; Teulade, J.C.; Chapat, J.P. Heterocyclic enaminones: Photochemical synthesis of 6,7,8,9-tetrahydro- 5H-pyrido[2,3-b]indol-9-ones. Tetrahedron 1999, 55, 1959–1970. [Google Scholar] [CrossRef]
- Nazaré, M.; Schneider, C.; Lindenschmidt, A.; Will, D.W. A flexible, palladium-catalyzed indole and azaindole synthesis by direct annulation of chloroanilines and chloroaminopyridines with ketones. Angew. Chem. Int. Ed. 2004, 43, 4526–4528. [Google Scholar] [CrossRef] [PubMed]
- Kgun Yum, E.; Sung Hong, C.; Yong Seo, J.; Sung, N.-D. Synthesis of Aromatic Ring Fused Pyrrole Derivatives by Palladium-catalyzed Annulation of o-Iodoarylamines with Allyl Acetate. Heterocycles 2004, 63, 631. [Google Scholar] [CrossRef]
- Lachance, N.; April, M.; Joly, M.-A. Rapid and Efficient Microwave-Assisted Synthesis of 4-, 5-, 6- and 7-Azaindoles. Synthesis (Stuttg) 2005, 2005, 2571–2577. [Google Scholar] [CrossRef]
- Spergel, S.H.; Okoro, D.R.; Pitts, W. One-pot synthesis of azaindoles via palladium-catalyzed α-heteroarylation of ketone enolates. J. Org. Chem. 2010, 75, 5316–5319. [Google Scholar] [CrossRef] [PubMed]
- Viciu, M.S.; Germaneau, R.F.; Nolan, S.P. Well-Defined, Air-Stable (NHC)Pd(Allyl)Cl (NHC = N-Heterocyclic Carbene) Catalysts for the Arylation of Ketones. Org. Lett. 2002, 4, 4053–4056. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.Q.; Yuen, J.; Lautens, M. A general modular method of azaindole and thienopyrrole synthesis via Pd-catalyzed tandem couplings of gem-dichloroolefins. J. Org. Chem. 2007, 72, 5152–5160. [Google Scholar] [CrossRef] [PubMed]
- Whelligan, D.K.; Thomson, D.W.; Taylor, D.; Hoelder, S. Two-step synthesis of aza- and diazaindoles from chloroamino-N-heterocycles using ethoxyvinylborolane. J. Org. Chem. 2010, 75, 11–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacchi, S.; Fabrizi, G.; Parisi, L.M. The aminopalladation-reductive elimination process as a tool for the solution-phase synthesis of 2,3-disubstituted azaindole libraries. J. Comb. Chem. 2005, 7, 510–512. [Google Scholar] [CrossRef] [PubMed]
- Antonio, A.; Sandro, C.; Fabio, M. A versatile approach to 2,3-disubstituted indoles through the palladium-catalysed cyclization of o-alkynyltrifluoroacetanilides with vinyl triflates and aryl halides. Tetrahedron Lett. 1992, 33, 3915–3918. [Google Scholar] [CrossRef]
- Shul’Pin, G.B. Selectivity enhancement in functionalization of C-H bonds: A review. Org. Biomol. Chem. 2010, 8, 4217–4228. [Google Scholar] [CrossRef] [PubMed]
- Goldman, A.S.; Goldberg, K.I. Organometallic C-H Bond Activation: An Introduction. In Activation and Functionalization of C-H Bonds; American Chemical Society: Washington, DC, USA, 2004; pp. 1–43. ISBN 0097-6156. [Google Scholar]
- Dantignana, V.; Milan, M.; Cussó, O.; Company, A.; Bietti, M.; Costas, M. Chemoselective Aliphatic C-H Bond Oxidation Enabled by Polarity Reversal. ACS Cent. Sci. 2017, 3, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Kitamura, T.; Fujiwara, Y. Catalytic functionalization of arenes and alkanes via C-H bond activation. Acc. Chem. Res. 2001, 34, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Dyker, G. Palladium-Catalyzed C-H Activation of tert-Butyl Groups: A Simple Synthesis of 1,2-Dihydrocyclobutabenzene Derivatives. Angew. Chem. Int. Ed. Engl. 1994, 33, 103–105. [Google Scholar] [CrossRef]
- Ritleng, V.; Sirlin, C.; Pfeffer, M. Ru-, Rh-, and Pd-catalyzed C-C bond formation involving C-H activation and addition on unsaturated substrates: Reactions and mechanistic aspects. Chem. Rev. 2002, 102, 1731–1769. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Hong, S. Rh(III)-catalyzed 7-azaindole synthesis via C-H activation/annulative coupling of aminopyridines with alkynes. Chem. Commun. 2015, 51, 11202–11205. [Google Scholar] [CrossRef] [PubMed]
- Stokes, B.J.; Driver, T.G. Transition metal-catalyzed formation of N-heterocycles via aryl-or vinyl C-H bond amination. Eur. J. Org. Chem. 2011, 2011, 4071–4088. [Google Scholar] [CrossRef]
- Kim, H.; Chang, S. Transition-Metal-Mediated Direct C-H Amination of Hydrocarbons with Amine Reactants: The Most Desirable but Challenging C-N Bond-Formation Approach. ACS Catal. 2016, 6, 2341–2351. [Google Scholar] [CrossRef]
- Zhang, M. Construction of heterocycle scaffolds via transition metal-catalyzed sp2 C-H functionalization. Adv. Synth. Catal. 2009, 351, 2243–2270. [Google Scholar] [CrossRef]
- Li, C.J. Cross-dehydrogenative coupling (CDC): Exploring C-C bond formations beyond functional group transformations. Acc. Chem. Res. 2009, 42, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhao, L.; Li, C.J. Palladium-catalyzed direct oxidative Heck-Cassar-Sonogashira type alkynylation of indoles with alkynes under oxygen. Chem. Commun. 2010, 46, 4184–4186. [Google Scholar] [CrossRef] [PubMed]
- Yeung, C.S.; Dong, V.M. Catalytic dehydrogenative cross-coupling: Forming carbon-carbon bonds by oxidizing two carbon-hydrogen bonds. Chem. Rev. 2011, 111, 1215–1292. [Google Scholar] [CrossRef] [PubMed]
- Bugaut, X.; Glorius, F. Palladium-catalyzed selective dehydrogenative cross-couplings of heteroarenes. Angew. Chem. Int. Ed. 2011, 50, 7479–7481. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Hua, R. Rhodium(III)-catalyzed c-h activation and indole synthesis with hydrazone as an auto-formed and auto-cleavable directing group. Chem. A Eur. J. 2014, 20, 2352–2356. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Han, J.; Chen, J.; Deng, H.; Shao, M.; Zhang, H.; Cao, W. Mild and Efficient One-Pot Synthesis of 2-(Perfluoroalkyl)indoles by Means of Sequential Michael-Type Addition and Pd(II)-Catalyzed Cross-Dehydrogenative Coupling (CDC) Reaction. Org. Lett. 2015, 17, 3283–3285. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.B.; Zhang, J.L.; Liu, Y.Y.; Liu, B.; Yang, X.H.; Li, J.H. Metal-free nitrative cyclization of N-aryl imines with tert-butyl nitrite: Dehydrogenative access to 3-nitroindoles. Chem. Commun. 2015, 51, 1886–1888. [Google Scholar] [CrossRef] [PubMed]
- Vijay Kumar, S.; Saraiah, B.; Parameshwarappa, G.; Ila, H.; Verma, G.K. Synthesis of N-functionalized/NH-multisubstituted indoles, thienopyrroles, pyrroloindoles, and pyrazolopyrroles via sequential one-pot base-mediated and copper-catalyzed inter- and intramolecular amination of 2-[2-bromo(het)aryl]-3-(het)aryl-3-(methylthio. J. Org. Chem. 2014, 79, 7961–7978. [Google Scholar] [CrossRef] [PubMed]
- Haffemayer, B.; Gulias, M.; Gaunt, M.J. Amine directed Pd(II)-catalyzed C-H bond functionalization under ambient conditions. Chem. Sci. 2011, 2, 312–315. [Google Scholar] [CrossRef]























© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, A.S.; Mortinho, A.C.; Marques, M.M.B. Metal-Catalyzed Cross-Coupling Reactions on Azaindole Synthesis and Functionalization. Molecules 2018, 23, 2673. https://doi.org/10.3390/molecules23102673
Santos AS, Mortinho AC, Marques MMB. Metal-Catalyzed Cross-Coupling Reactions on Azaindole Synthesis and Functionalization. Molecules. 2018; 23(10):2673. https://doi.org/10.3390/molecules23102673
Chicago/Turabian StyleSantos, A. Sofia, Ana C. Mortinho, and M. Manuel B. Marques. 2018. "Metal-Catalyzed Cross-Coupling Reactions on Azaindole Synthesis and Functionalization" Molecules 23, no. 10: 2673. https://doi.org/10.3390/molecules23102673
APA StyleSantos, A. S., Mortinho, A. C., & Marques, M. M. B. (2018). Metal-Catalyzed Cross-Coupling Reactions on Azaindole Synthesis and Functionalization. Molecules, 23(10), 2673. https://doi.org/10.3390/molecules23102673