
Selective Double Addition Reaction of an E‒H Bond (E = Si, B) to a C≡N Triple Bond of Organonitriles


Abstract
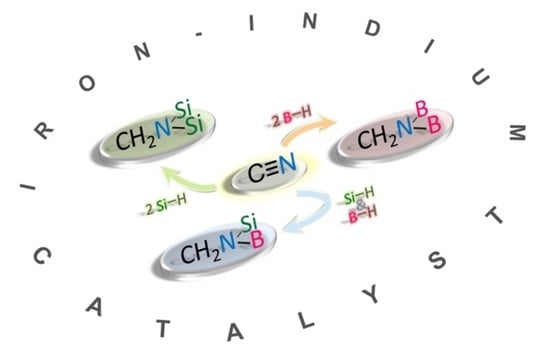
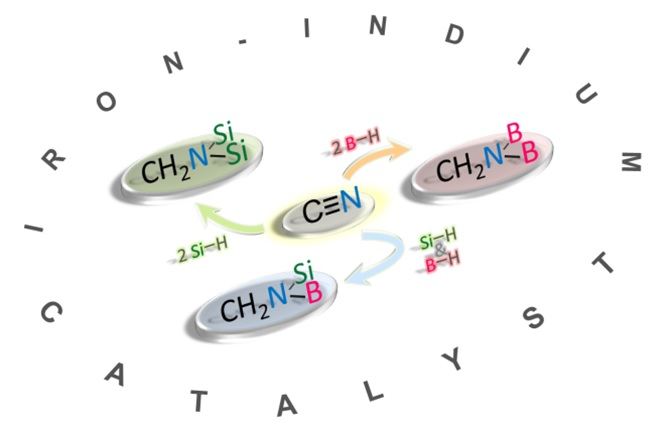
:
1. Introduction
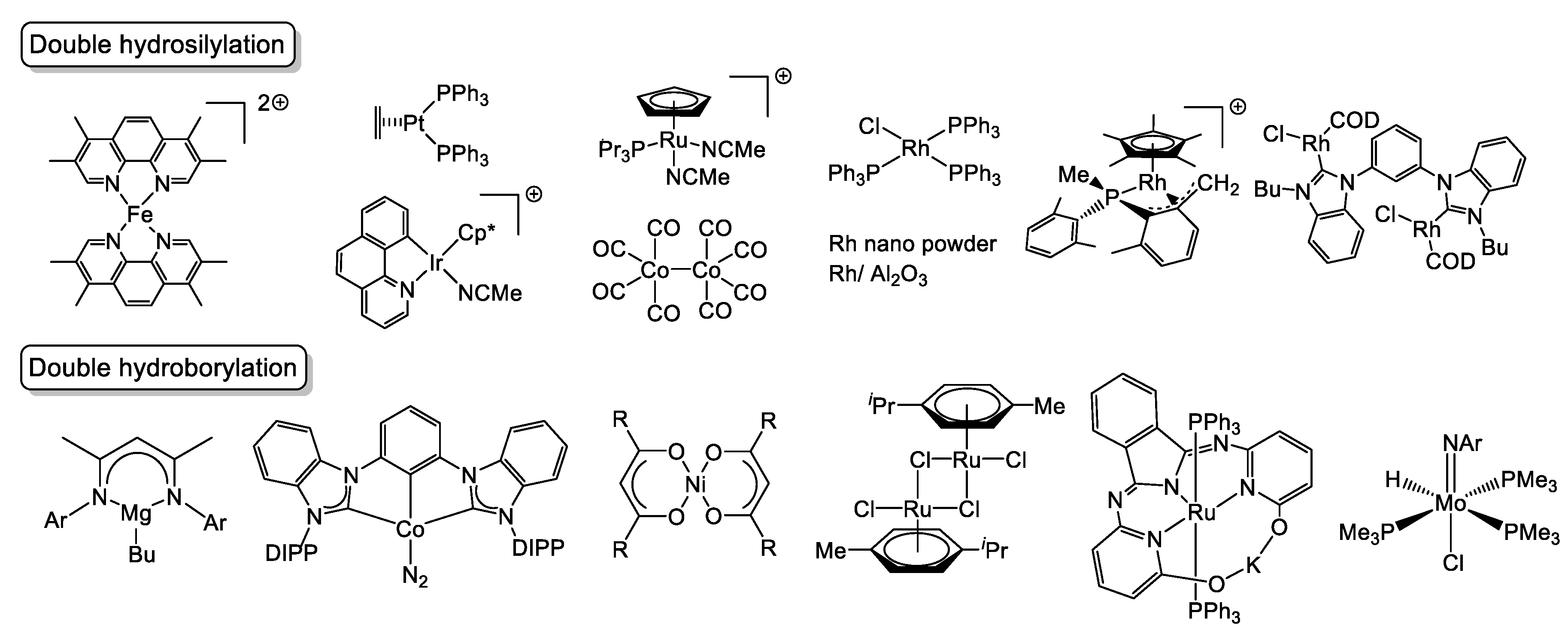
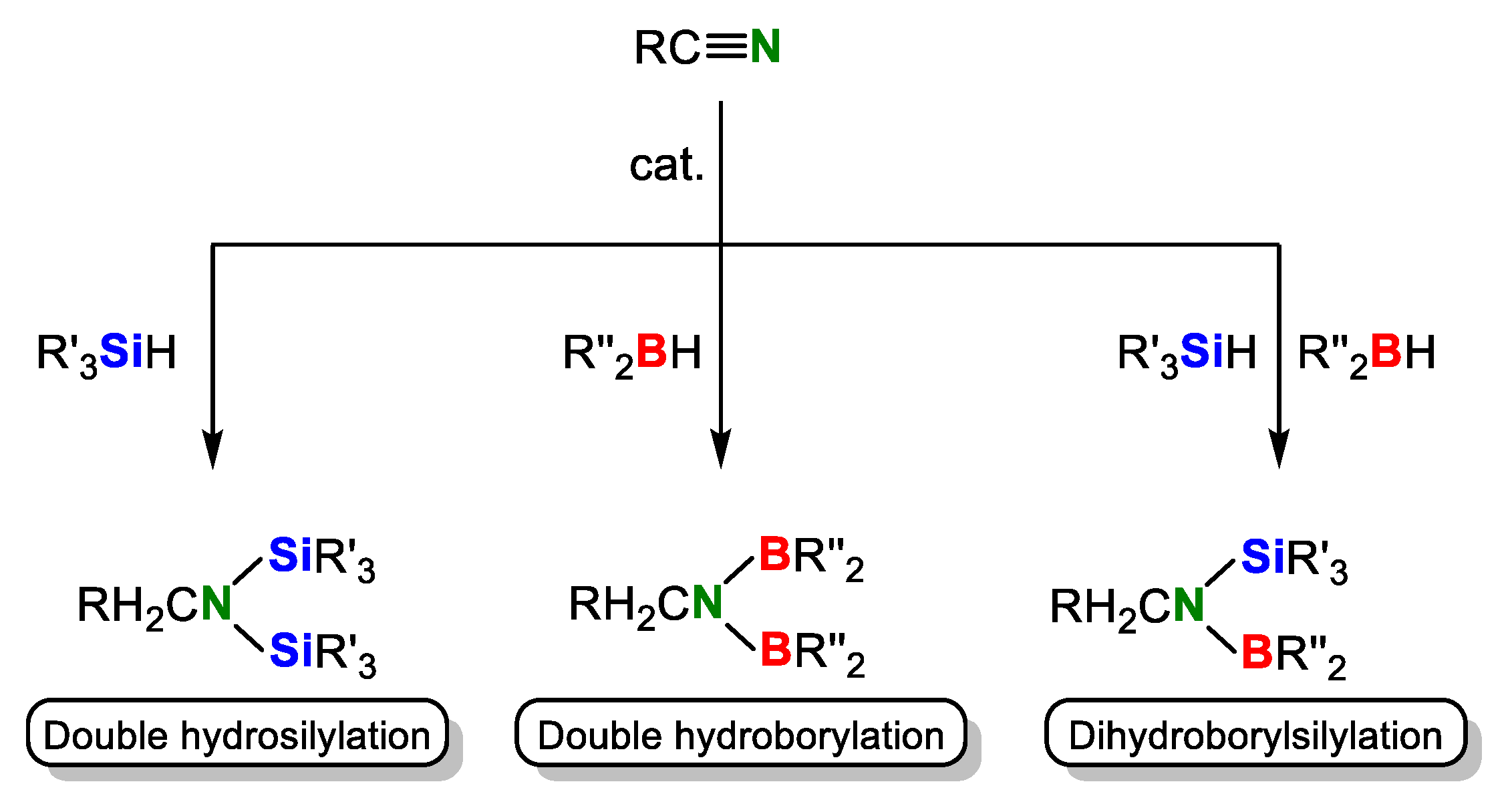
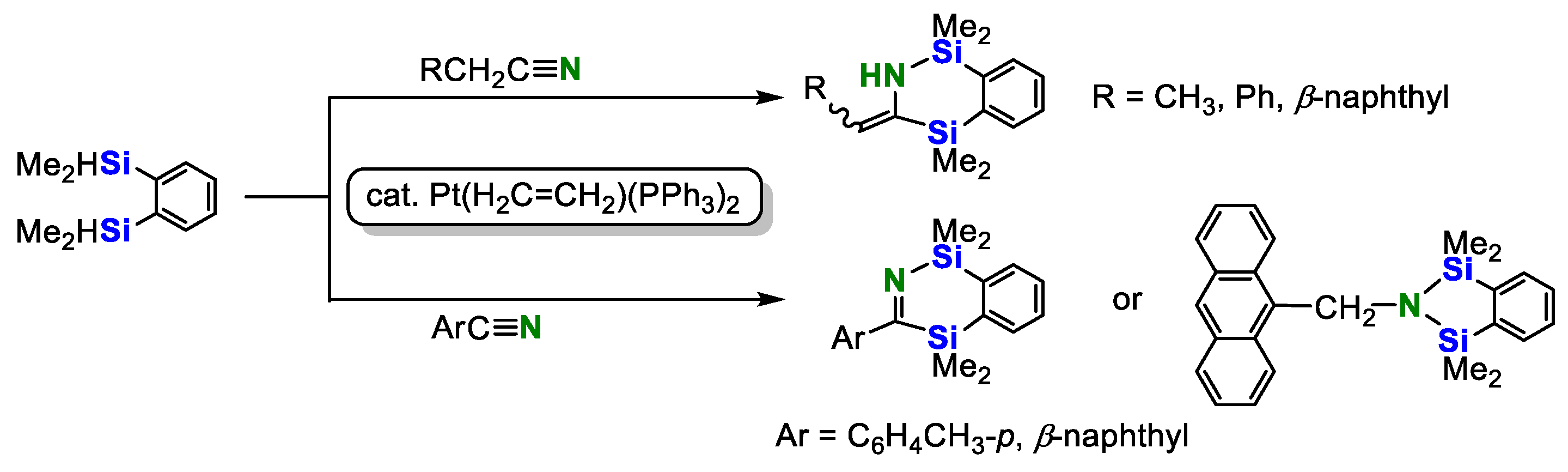
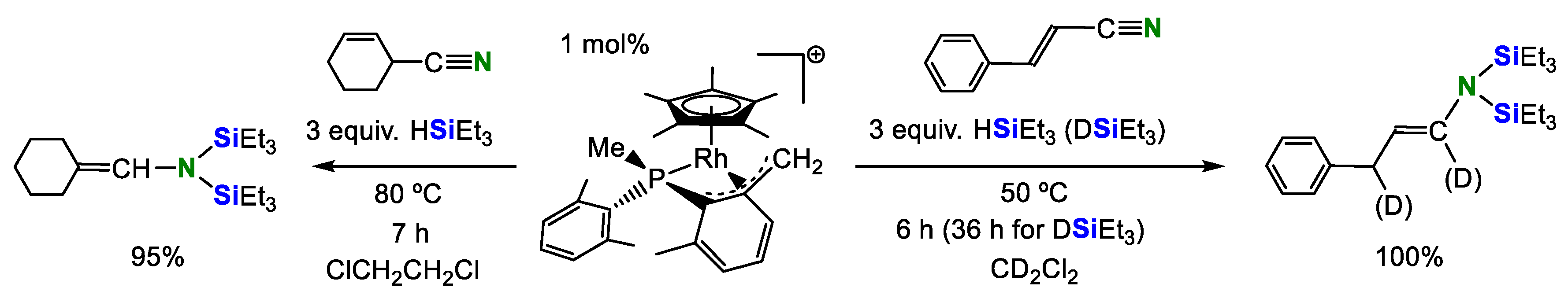
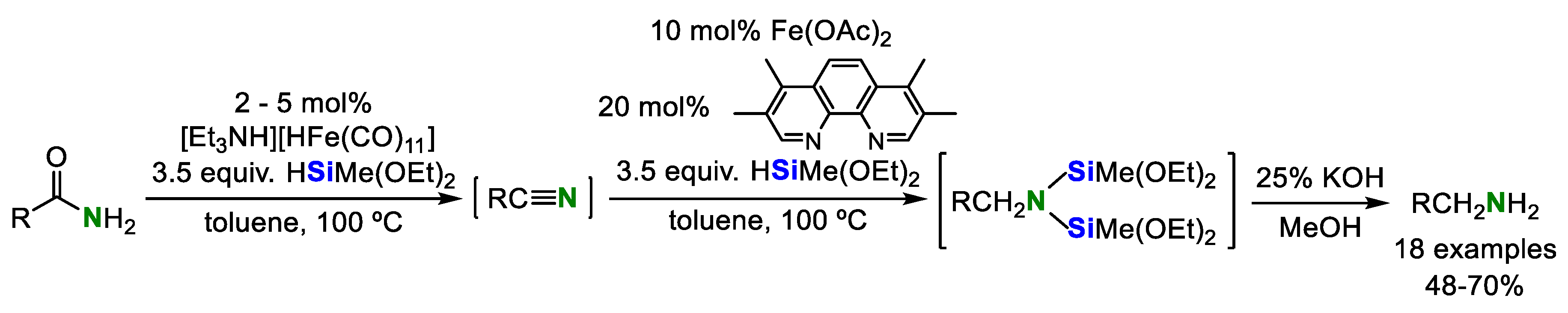
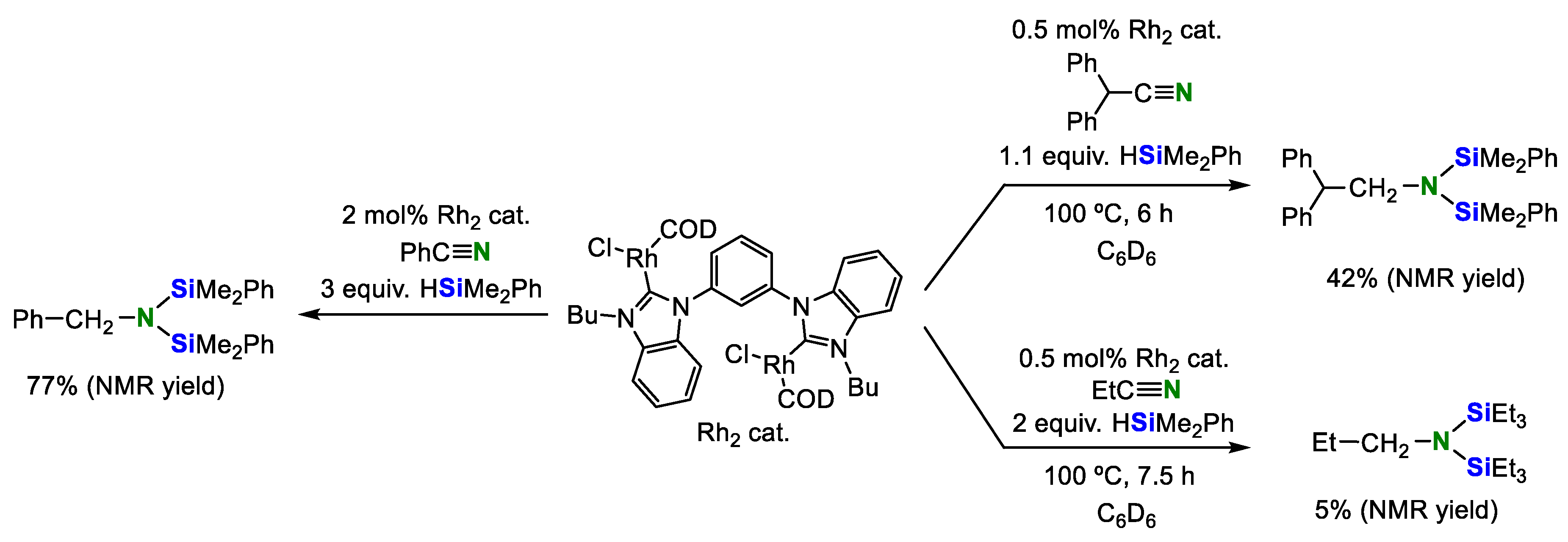
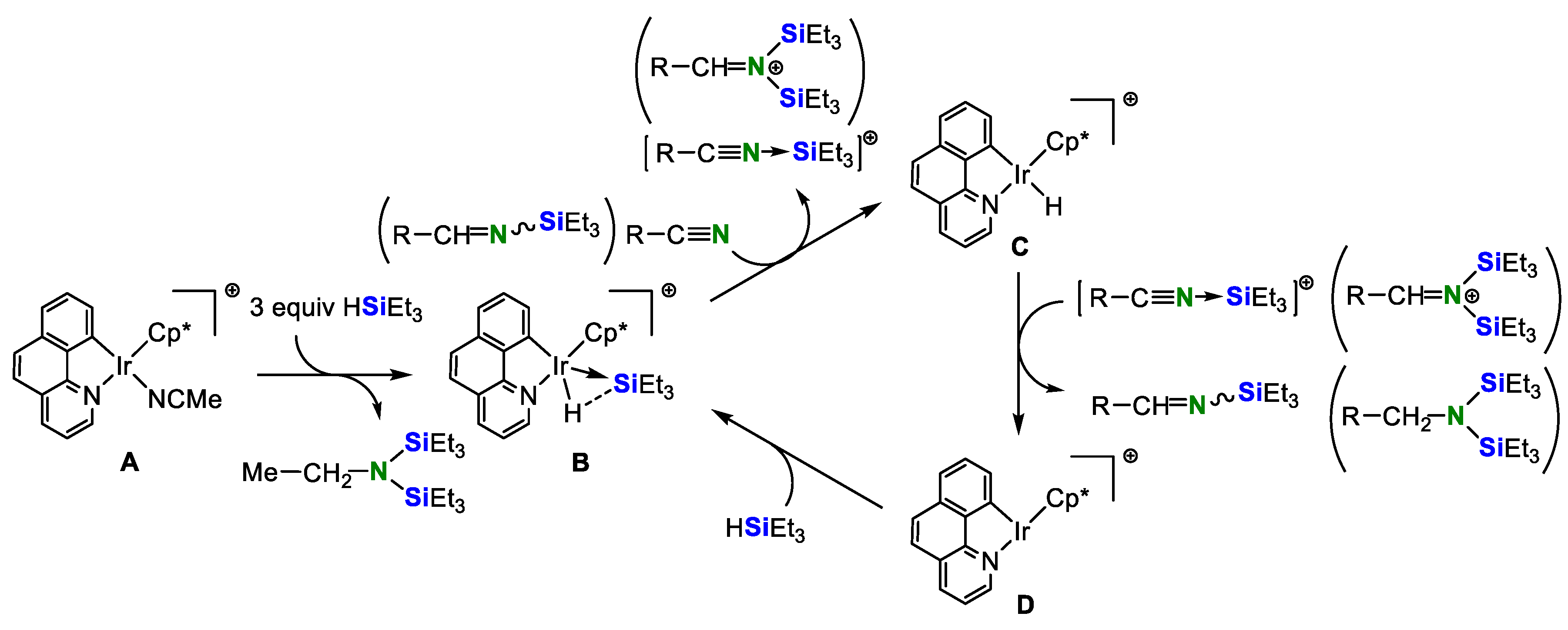
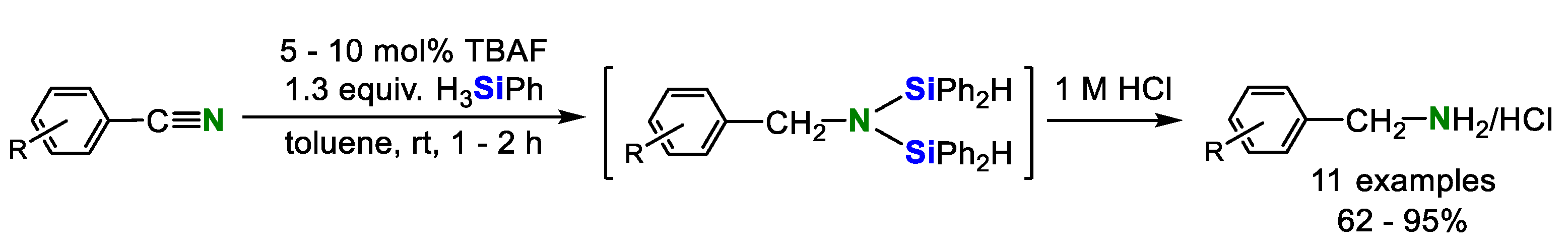
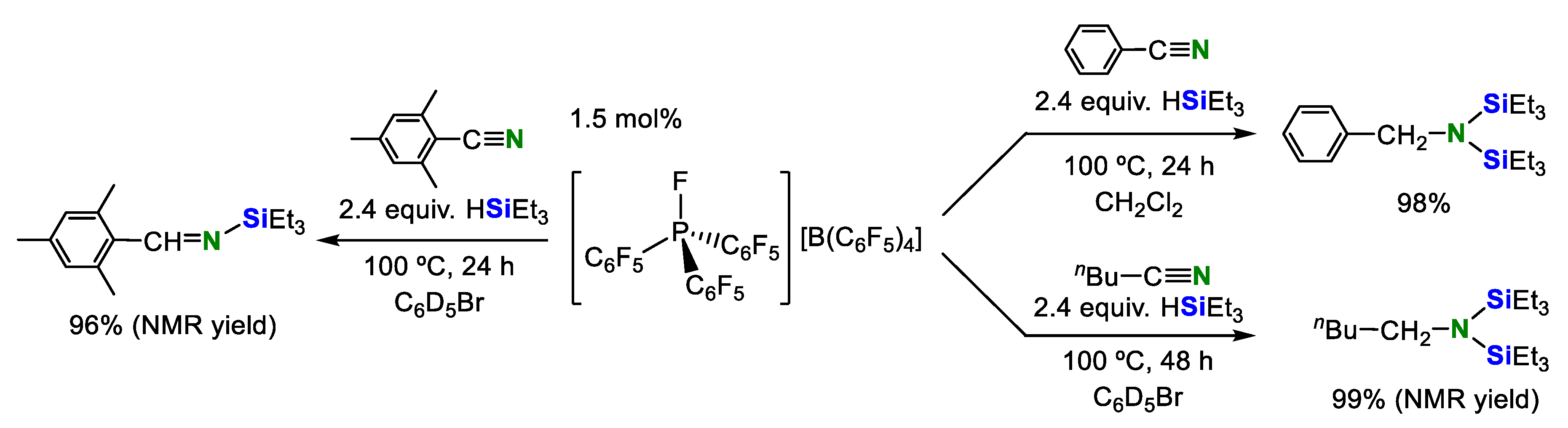
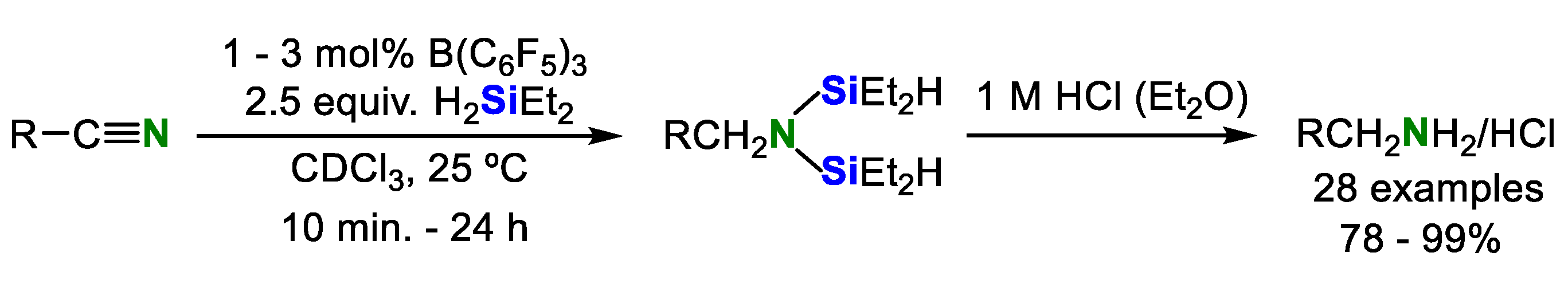
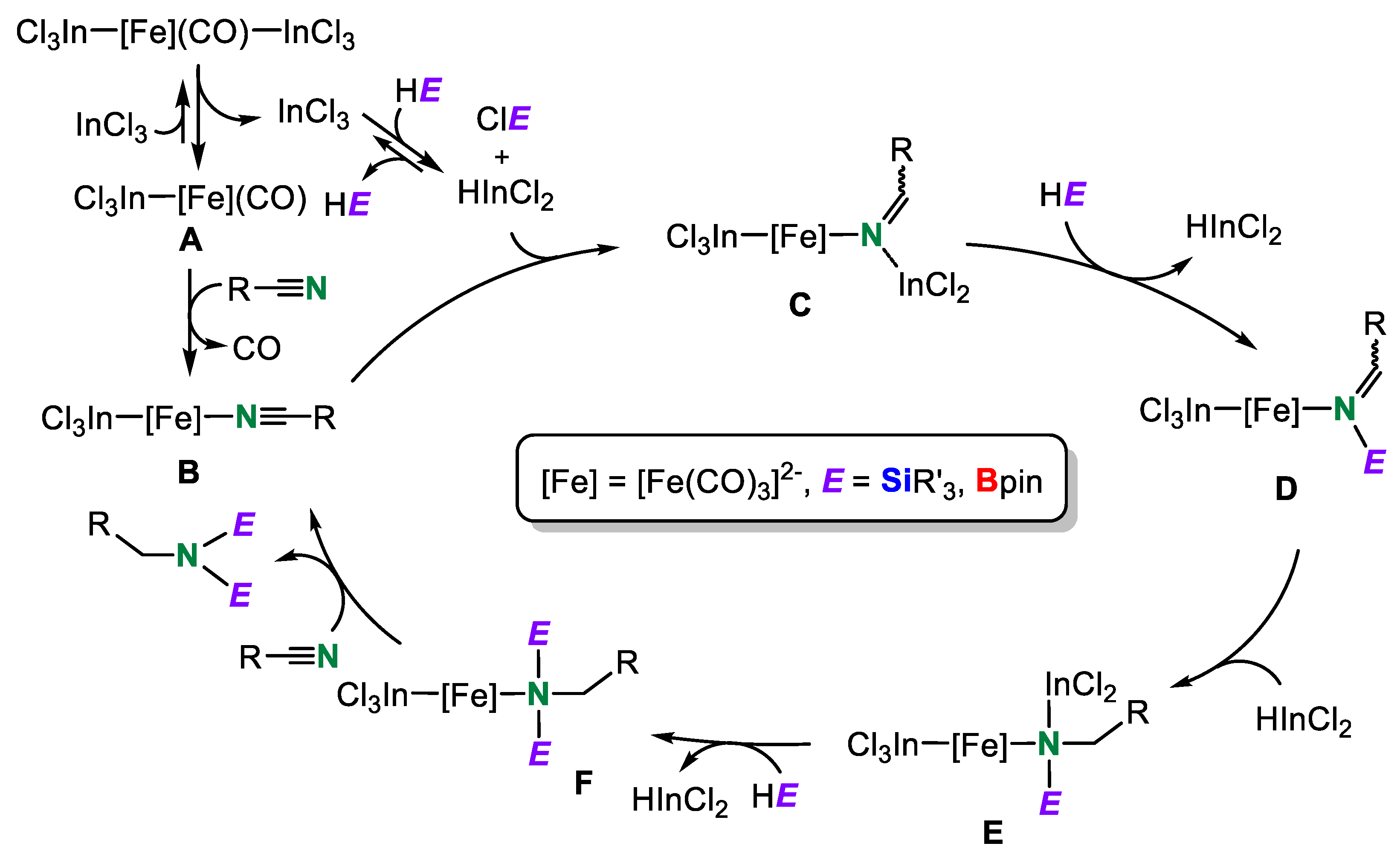
2. Double Hydrosilylation of Organonitriles





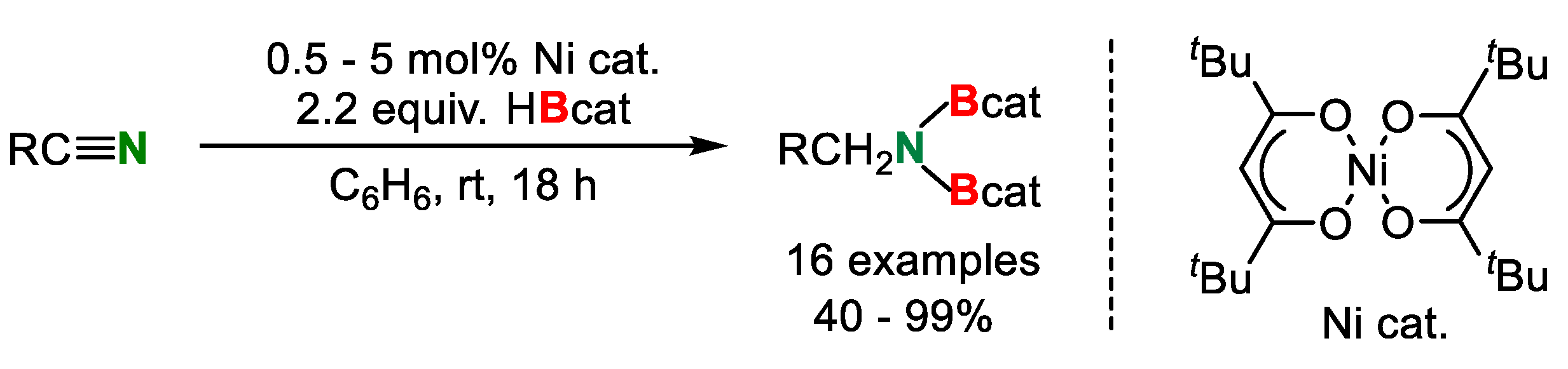
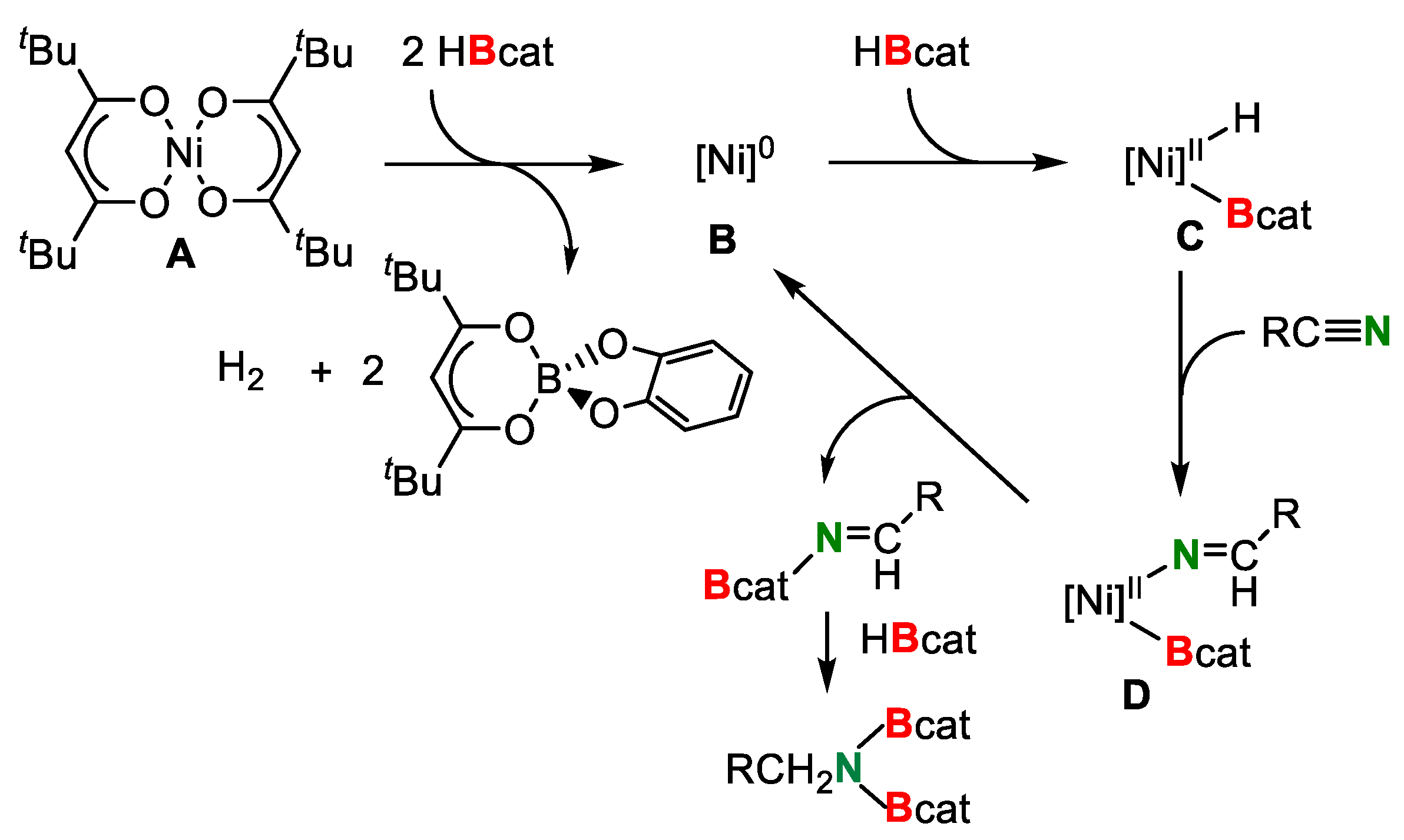
3. Double Hydroborylation of Organonitriles
4. Dihydroborylsilylation of Acetonitrile




5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Birot, M.; Pillot, J.-P.; Dunogues, J. Comprehensive Chemistry of Polycarbosilanes, Polysilazanes, and Polycarbosilazanes as Precursors of Ceramics. Chem. Rev. 1995, 95, 1443–1477. [Google Scholar] [CrossRef]
- Colombo, P.; Mera, G.; Riedel, R.; Sorarù, G.D. Polymer-Derived Ceramics: 40 Years of Research and Innovation in Advanced Ceramics. J. Am. Ceram. Soc. 2010, 93, 1805–1837. [Google Scholar] [CrossRef]
- Meng, L.; Zhang, X.; Tang, Y.; Su, K.; Kong, J. Hierarchically porous silicon−carbon−nitrogen hybrid materials towards highly efficient and selective adsorption of organic dyes. Sci. Rep. 2015, 5, 7910. [Google Scholar] [CrossRef] [PubMed]
- Viard, A.; Miele, P.; Bernard, S. Polymer-derived ceramics route toward SiCN and SiBCN fibers: From chemistry of polycarbosilazanes to the design and characterization of ceramic fibers. J. Ceram. Soc. Jpn. 2016, 124, 967–980. [Google Scholar] [CrossRef]
- Jansen, M.; Jäschke, T. Crystal Structure and Spectroscopic Characterisation of Hexamethyldisilazane-trichloroaluminum [(H3C)3Si]2NH·AlCl3. Z. Naturforsch. B J. Chem. Sci. 2000, 55, 763–767. [Google Scholar] [CrossRef]
- Ayed, T.; Barthelat, J.-C.; Tangour, B.; Pradère, C.; Donnadieu, B.; Grellier, M.; Sabo-Etienne, S. Structure and Bonding in a Disilazane Ruthenium Complex. Catalytic Selective Deuteration of Disilazane. Organometallics 2005, 24, 3824–3826. [Google Scholar] [CrossRef]
- Tanabe, Y.; Misaki, T.; Kurihara, M.; Iida, A.; Nishii, Y. Silazanes/catalytic bases: Mild, powerful and chemoselective agents for the preparation of enol silyl ethers from ketones and aldehydes. Chem. Commun. 2002, 0, 1628–1629. [Google Scholar] [CrossRef]
- Shimizu, K.; Minami, Y.; Goto, O.; Ikehira, H.; Hiyama, T. Silicon-based C–N Cross-coupling Reaction. Chem. Lett. 2014, 43, 438–440. [Google Scholar]
- Suginome, M.; Uehlin, L.; Murakami, M. Aminoboranes as “Compatible” Iminium Ion Generators in Aminative C−C Bond Formations. J. Am. Chem. Soc. 2004, 126, 13196–13197. [Google Scholar] [CrossRef] [PubMed]
- Nixon, T.D.; Whittlesey, M.K.; Williams, J.M.J. Ruthenium-catalysed transfer hydrogenation reactions with dimethylamine borane. Tetrahedron Lett. 2011, 52, 6652–6654. [Google Scholar] [CrossRef]
- Laval, S.; Dayoub, W.; Favre-Reguillon, A.; Berthod, M.; Demonchaux, P.; Mignani, G.; Lemaire, M. A mild and efficient method for the reduction of nitriles. Tetrahedron Lett. 2009, 50, 7005–7007. [Google Scholar] [CrossRef]
- Das, S.; Wendt, B.; Möller, K.; Junge, K.; Beller, M. Two Iron Catalysts are Better than One: A General and Convenient Reduction of Aromatic and Aliphatic Primary Amides. Angew. Chem. Int. Ed. 2012, 51, 1662–1666. [Google Scholar] [CrossRef] [PubMed]
- Bornschein, C.; Werkmeister, S.; Junge, K.; Beller, M. TBAF-catalyzed hydrosilylation for the reduction of aromatic nitriles. New J. Chem. 2013, 37, 2061–2065. [Google Scholar] [CrossRef]
- Marciniec, B.; Guliński, J.; Urbaniak, W.; Kornetka, Z.W. Comprehensive Handbook on Hydrosilylation; Pergamon: Oxford, UK, 1992. [Google Scholar]
- Dhillon, R.S. Hydroboration and Organic Synthesis. 9-Borabicyclo[3.3.1]Nonane (9-BBN); Springer: Berlin, Germany, 2007. [Google Scholar]
- Luo, Y.-R. Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Reddy, N.P.; Uchimaru, Y.; Lautenschlager, H.-J.; Tanaka, M. Platinum-Catalyzed Novel Reactions of Nitriles and an Azirine with o-Bis(dimethylsilyl)benzene. Chem. Lett. 1992, 21, 45–48. [Google Scholar] [CrossRef]
- Hamdaoui, M.; Desrousseaux, C.; Habbita, H.; Djukic, J.-P. Iridacycles as Catalysts for the Autotandem Conversion of Nitriles into Amines by Hydrosilylation: Experimental Investigation and Scope. Organometallics 2017, 36, 4864–4882. [Google Scholar] [CrossRef]
- Gutsulyak, D.V.; Nikonov, G.I. Chemoselective Catalytic Hydrosilylation of Nitriles. Angew. Chem. Int. Ed. 2010, 49, 7553–7556. [Google Scholar] [CrossRef] [PubMed]
- Murai, T.; Sakane, T.; Kato, S. Cobalt carbonyl catalyzed reduction of aromatic nitriles with a hydrosilane leading to N,N-disilylamines. Tetrahedron Lett. 1985, 26, 5145–5148. [Google Scholar] [CrossRef]
- Murai, T.; Sakane, T.; Kato, S. Cobalt carbonyl catalyzed hydrosilylation of nitriles: A new preparation of N,N-disilylamines. J. Org. Chem. 1990, 55, 449–453. [Google Scholar] [CrossRef]
- Caporusso, A.M.; Panziera, N.; Pertici, P.; Pitzalis, E.; Salvadori, P.; Vitulli, G.; Martra, G. Hydrosilylation of aromatic nitriles promoted by solvated rhodium atom-derived catalysts. J. Mol. Catal. A 1999, 150, 275–285. [Google Scholar] [CrossRef]
- Corriu, R.J.P.; Moreau, J.J.E.; Pataud-Sat, M. Reactions de l’ortho-bis(dimethylsilyl)benzene avec les nitriles catalysees par des complexes du rhodium. J. Organomet. Chem. 1982, 228, 301–308. [Google Scholar] [CrossRef]
- Campos, J.; Rubio, M.; Esqueda, A.C.; Carmona, E. Large-scale preparation and labelling reactions of deuterated silanes. J. Label. Compd. Radiopharm. 2012, 55, 29–38. [Google Scholar] [CrossRef]
- Huckaba, A.J.; Hollis, T.K.; Reilly, S.W. Homobimetallic Rhodium NHC Complexes as Versatile Catalysts for Hydrosilylation of a Multitude of Substrates in the Presence of Ambient Air. Organometallics 2013, 32, 6248–6256. [Google Scholar] [CrossRef]
- Pérez, M.; Qu, Z.-W.; Caputo, C.B.; Podgorny, V.; Hounjet, L.J.; Hansen, A.; Dobrovetsky, R.; Grimme, S.; Stephan, D.W. Hydrosilylation of Ketones, Imines and Nitriles Catalysed by Electrophilic Phosphonium Cations: Functional Group Selectivity and Mechanistic Considerations. Chem. Eur. J. 2015, 21, 6491–6500. [Google Scholar] [CrossRef] [PubMed]
- Gandhamsetty, N.; Jeong, J.; Park, J.; Park, S.-W.; Chang, S. Boron-Catalyzed Silylative Reduction of Nitriles in Accessing Primary Amines and Imines. J. Org. Chem. 2015, 80, 7281–7287. [Google Scholar] [CrossRef] [PubMed]
- Gandhamsetty, N.; Park, J.; Jeong, J.; Park, S.-W.; Park, S.; Chang, S. Chemoselective Silylative Reduction of Conjugated Nitriles under Metal-Free Catalytic Conditions: β-Silyl Amines and Enamines. Angew. Chem. Int. Ed. 2015, 54, 6832–6836. [Google Scholar] [CrossRef] [PubMed]
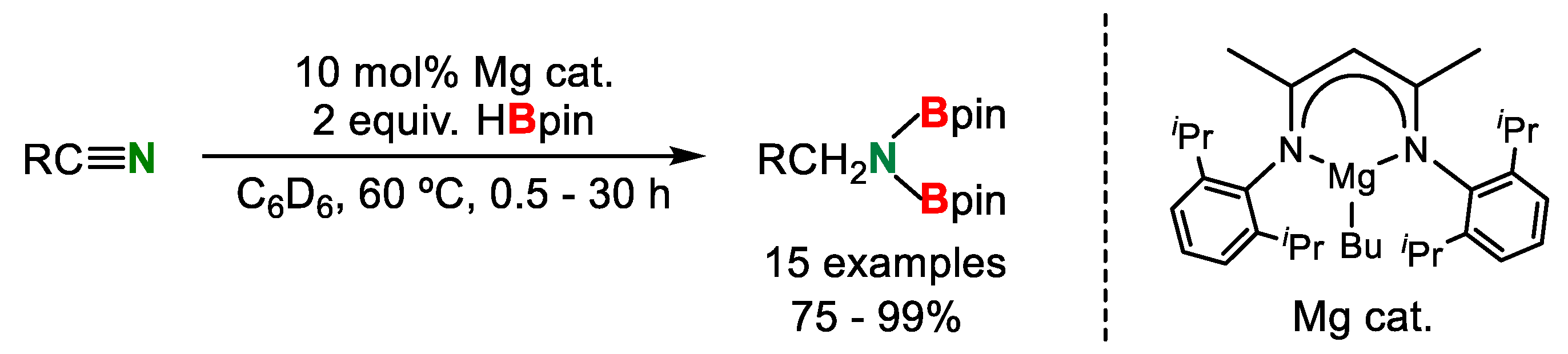
- Weetman, C.; Anker, M.D.; Arrowsmith, M.; Hill, M.S.; Kociok-Kohn, G.; Liptrot, D.J.; Mahon, M.F. Magnesium-catalysed nitrile hydroboration. Chem. Sci. 2016, 7, 628–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, A.D.; Entsminger, S.W.; Fout, A.R. Insights into a Chemoselective Cobalt Catalyst for the Hydroboration of Alkenes and Nitriles. ACS Catal. 2017, 7, 3730–3734. [Google Scholar] [CrossRef]
- Nakamura, G.; Nakajima, Y.; Matsumoto, K.; Srinivas, V.; Shimada, S. Nitrile hydroboration reactions catalysed by simple nickel salts, bis(acetylacetonato)nickel(II) and its derivatives. Catal. Sci. Technol. 2017, 7, 3196–3199. [Google Scholar] [CrossRef]
- Kaithal, A.; Chatterjee, B.; Gunanathan, C. Ruthenium-Catalyzed Selective Hydroboration of Nitriles and Imines. J. Org. Chem. 2016, 81, 11153–11161. [Google Scholar] [CrossRef] [PubMed]
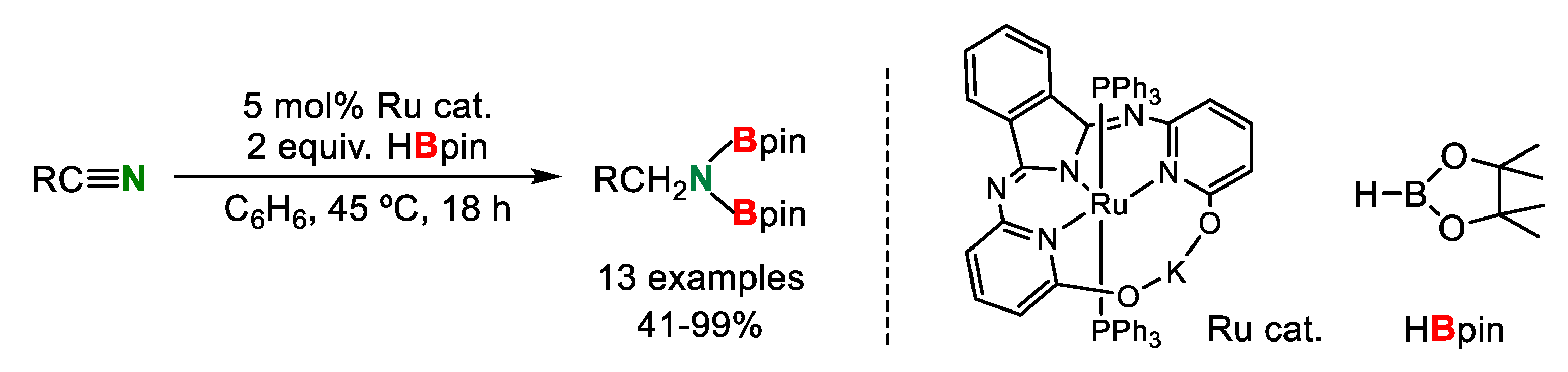
- Geri, J.B.; Szymczak, N.K. A Proton-Switchable Bifunctional Ruthenium Complex That Catalyzes Nitrile Hydroboration. J. Am. Chem. Soc. 2015, 137, 12808–12814. [Google Scholar] [CrossRef] [PubMed]
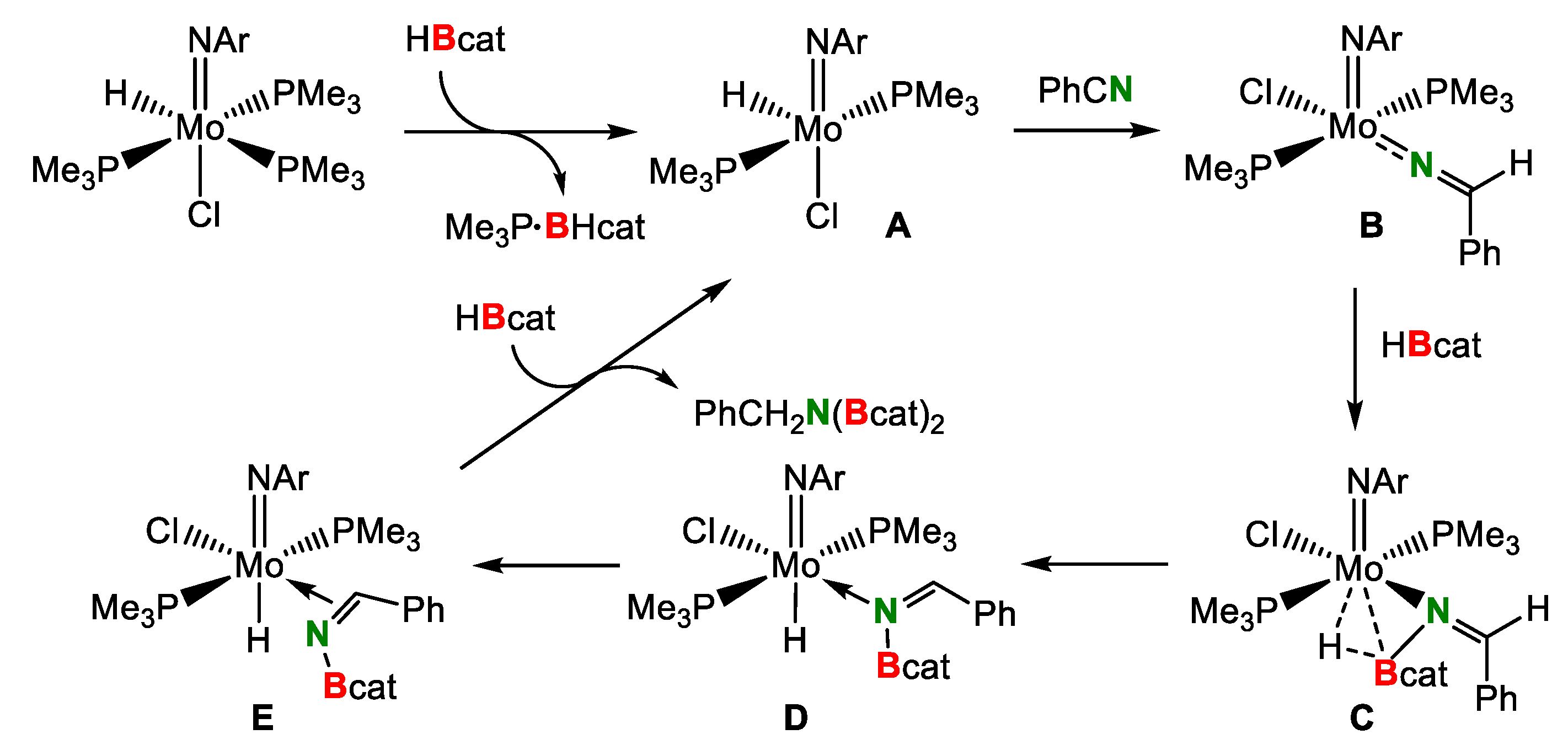
- Khalimon, A.Y.; Farha, P.; Kuzmina, L.G.; Nikonov, G.I. Catalytic hydroboration by an imido-hydrido complex of Mo(IV). Chem. Commun. 2012, 48, 455–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalimon, A.Y.; Farha, P.M.; Nikonov, G.I. Imido−hydrido complexes of Mo(IV): Catalysis and mechanistic aspects of hydroboration reactions. Dalton Trans. 2015, 44, 18945–18956. [Google Scholar] [CrossRef] [PubMed]
- Baldus, P.; Jansen, M.; Sporn, D. Ceramic Fibers for Matrix Composites in High-Temperature Engine Applications. Science 1999, 285, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Weinmann, M.; Kroschel, M.; Jäschke, T.; Nuss, J.; Jansen, M.; Kolios, G.; Morillo, A.; Tellaeche, C.; Nieken, U. Towards continuous processes for the synthesis of precursors of amorphous Si/B/N/C ceramics. J. Mater. Chem. 2008, 18, 1810–1818. [Google Scholar] [CrossRef]
- Shriver, D.F. Transition metal basicity. Acc. Chem. Res. 1970, 3, 231–238. [Google Scholar] [CrossRef]
- Parkin, G.A. Simple Description of the Bonding in Transition-Metal Borane Complexes. Organometallics 2006, 25, 4744–4747. [Google Scholar] [CrossRef]
- Hill, A.F. An Unambiguous Electron-Counting Notation for Metallaboratranes. Organometallics 2006, 25, 4741–4743. [Google Scholar] [CrossRef]
- Bouhadir, G.; Amgoune, A.; Bourissou, D. Chapter 1 phosphine-boranes and related ambiphilic compounds: Synthesis, structure, and coordination to transition metals. In Advances in Organometallic Chemstry; Hill, A.F., Fink, M.J., Eds.; Elsevier: London, UK, 2010; Volume 58, pp. 1–107. ISBN 978-0-12-374784-6. [Google Scholar]
- Amgoune, A.; Bourissou, D. σ-Acceptor, Z-type ligands for transition metals. Chem. Commun. 2011, 47, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Braunschweig, H.; Dewhurst, R.D. Transition metals as Lewis bases: “Z-type” boron ligands and metal-to-boron dative bonding. Dalton Trans. 2011, 40, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Kameo, H.; Nakazawa, H. Recent Developments in the Coordination Chemistry of Multidentate Ligands Featuring a Boron Moiety. Chem. Asian J. 2013, 8, 1720–1734. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.S.; Gabbaï, F.P. Coordination- and Redox-Noninnocent Behavior of Ambiphilic Ligands Containing Antimony. Acc. Chem. Res. 2016, 49, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.S.; Gabbaï, F.P. Coordination and Redox Non-innocent Behavior of Hybrid Ligands Containing Tellurium. Chem. Lett. 2016, 45, 376–384. [Google Scholar] [CrossRef] [Green Version]
- Bouhadir, G.; Bourissou, D. Complexes of ambiphilic ligands: Reactivity and catalytic applications. Chem. Soc. Rev. 2016, 45, 1065–1079. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, M.V.; Xie, J.; Lu, C.C. Stable Dihydrogen Complexes of Cobalt(−I) Suggest an Inverse trans-Influence of Lewis Acidic Group 13 Metalloligands. J. Am. Chem. Soc. 2017, 139, 6570–6573. [Google Scholar] [CrossRef] [PubMed]
- Kameo, H.; Kawamoto, T.; Sakaki, S.; Bourissou, D.; Nakazawa, H. Transition-Metal-Mediated Cleavage of Fluoro-Silanes under Mild Conditions. Chem. Eur. J. 2016, 22, 2370–2375. [Google Scholar] [CrossRef] [PubMed]
- Kameo, H.; Ikeda, K.; Bourissou, D.; Sakaki, S.; Takemoto, S.; Nakazawa, H.; Matsuzaka, H. Transition-Metal-Mediated Germanium–Fluorine Activation: Inverse Electron Flow in σ-Bond Metathesis. Organometallics 2016, 35, 713–719. [Google Scholar] [CrossRef]
- Cammarota, R.C.; Lu, C.C. Tuning Nickel with Lewis Acidic Group 13 Metalloligands for Catalytic Olefin Hydrogenation. J. Am. Chem. Soc. 2015, 137, 12486–12489. [Google Scholar] [CrossRef] [PubMed]
- Schindler, T.; Lux, M.; Peters, M.; Scharf, L.T.; Osseili, H.; Maron, L.; Tauchert, M.E. Synthesis and Reactivity of Palladium Complexes Featuring a Diphosphinoborane Ligand. Organometallics 2015, 34, 1978–1984. [Google Scholar] [CrossRef]
- Fong, H.; Moret, M.-E.; Lee, Y.; Peters, J.C. Heterolytic H2 Cleavage and Catalytic Hydrogenation by an Iron Metallaboratrane. Organometallics 2013, 32, 3053–3062. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.S.; Rittle, J.; Peters, J.C. Catalytic conversion of nitrogen to ammonia by an iron model complex. Nature 2013, 501, 84–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, W.-C.; Gu, W.; MacInnis, M.C.; Timpa, S.D.; Bhuvanesh, N.; Zhou, J.; Ozerov, O.V. Facile Insertion of Rh and Ir into a Boron–Phenyl Bond, Leading to Boryl/Bis(phosphine) PBP Pincer Complexes. J. Am. Chem. Soc. 2016, 138, 2086–2089. [Google Scholar] [CrossRef] [PubMed]
- You, D.; Yang, H.; Sen, S.; Gabbaï, F.P. Modulating the σ-Accepting Properties of an Antimony Z-type Ligand via Anion Abstraction: Remote-Controlled Reactivity of the Coordinated Platinum Atom. J. Am. Chem. Soc. 2018, 140, 9644–9651. [Google Scholar] [CrossRef] [PubMed]
- Itazaki, M.; Ito, M.; Nakazawa, H. Synthesis, Structure and Reactivity of Ruthenium(0) Indane Complex fac-[Ru(NCMe)3(CO)2(InX3)] (X = Cl, Br). Eur. J. Inorg. Chem. 2015, 2015, 2033–2036. [Google Scholar] [CrossRef]
- Itazaki, M.; Ito, M.; Nakashima, S.; Nakazawa, H. Synthesis and Characterization of [Fe(NCCH3)6][cis-Fe(InX3)2(CO)4] (X = Cl, Br, I) Containing Two Terminal Indium Fragments. Dalton Trans. 2016, 45, 1327–1330. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Itazaki, M.; Nakazawa, H. Selective Double Hydrosilylation of Nitriles Catalyzed by an Iron Complex Containing Indium Trihalide. ChemCatChem 2016, 8, 3323–3325. [Google Scholar] [CrossRef]
- Ito, M.; Itazaki, M.; Nakazawa, H. Selective Double Hydroboration and Dihydrobrylsilylation of Organonitriles by an Iron-indium Cooperative Catalytic System. Inorg. Chem. 2017, 56, 13709–13714. [Google Scholar] [CrossRef] [PubMed]
- Baba, A.; Shibata, I. Dihaloindium hydride as a novel reducing agent. Chem. Rec. 2005, 5, 323–335. [Google Scholar] [CrossRef] [PubMed]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
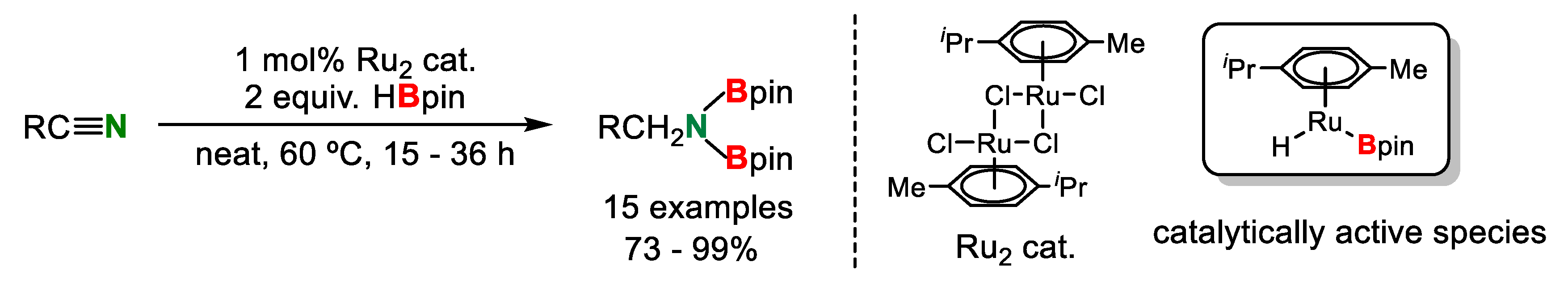{kind=link}
{kind=link}
{kind=link}
{kind=link}









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Itazaki, M.; Nakazawa, H. Selective Double Addition Reaction of an E‒H Bond (E = Si, B) to a C≡N Triple Bond of Organonitriles. Molecules 2018, 23, 2769. https://doi.org/10.3390/molecules23112769
Itazaki M, Nakazawa H. Selective Double Addition Reaction of an E‒H Bond (E = Si, B) to a C≡N Triple Bond of Organonitriles. Molecules. 2018; 23(11):2769. https://doi.org/10.3390/molecules23112769
Chicago/Turabian StyleItazaki, Masumi, and Hiroshi Nakazawa. 2018. "Selective Double Addition Reaction of an E‒H Bond (E = Si, B) to a C≡N Triple Bond of Organonitriles" Molecules 23, no. 11: 2769. https://doi.org/10.3390/molecules23112769
APA StyleItazaki, M., & Nakazawa, H. (2018). Selective Double Addition Reaction of an E‒H Bond (E = Si, B) to a C≡N Triple Bond of Organonitriles. Molecules, 23(11), 2769. https://doi.org/10.3390/molecules23112769