
A Perspective on Reversibility in Controlled Polymerization Systems: Recent Progress and New Opportunities




Abstract
:
1. Introduction
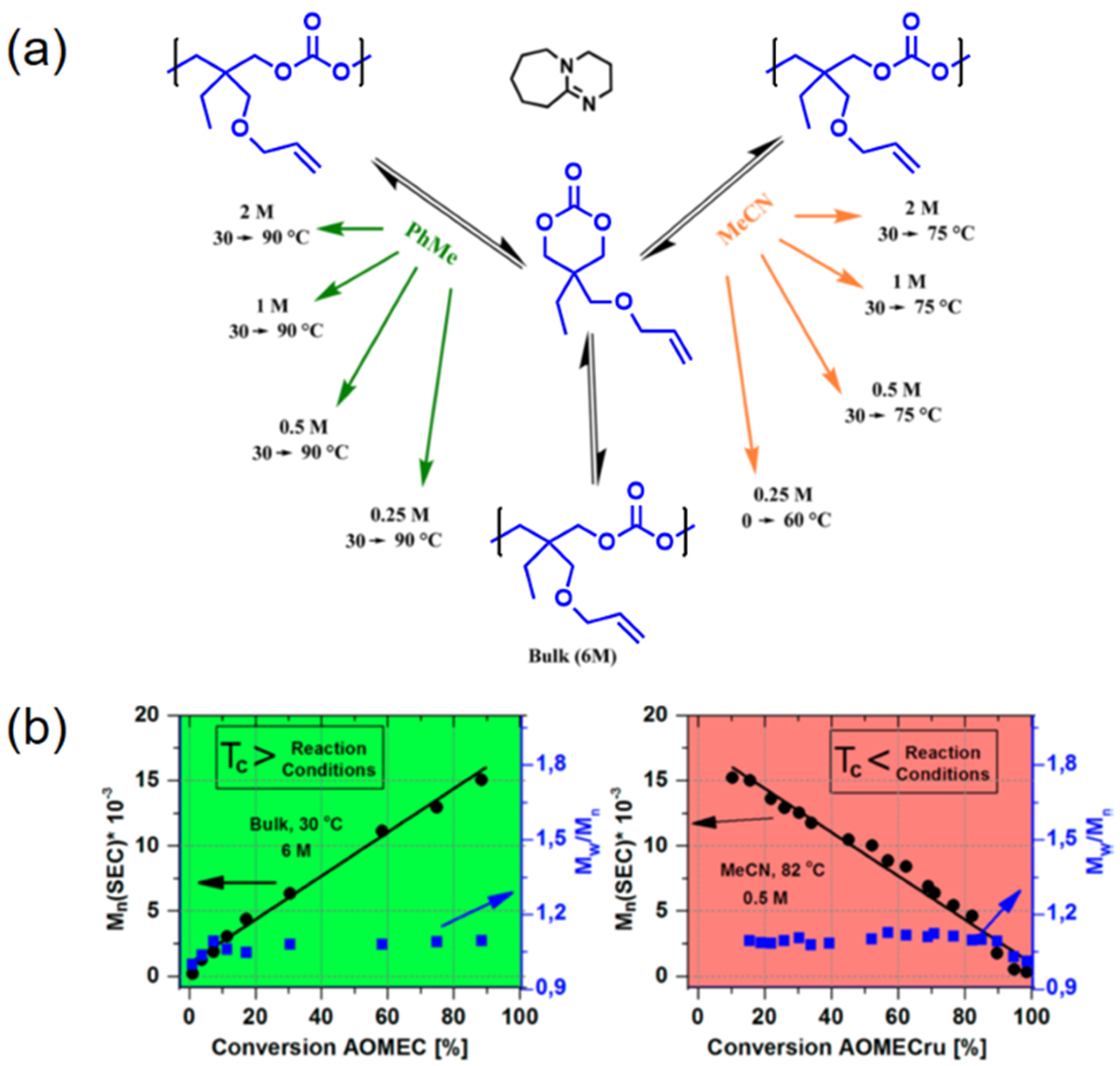
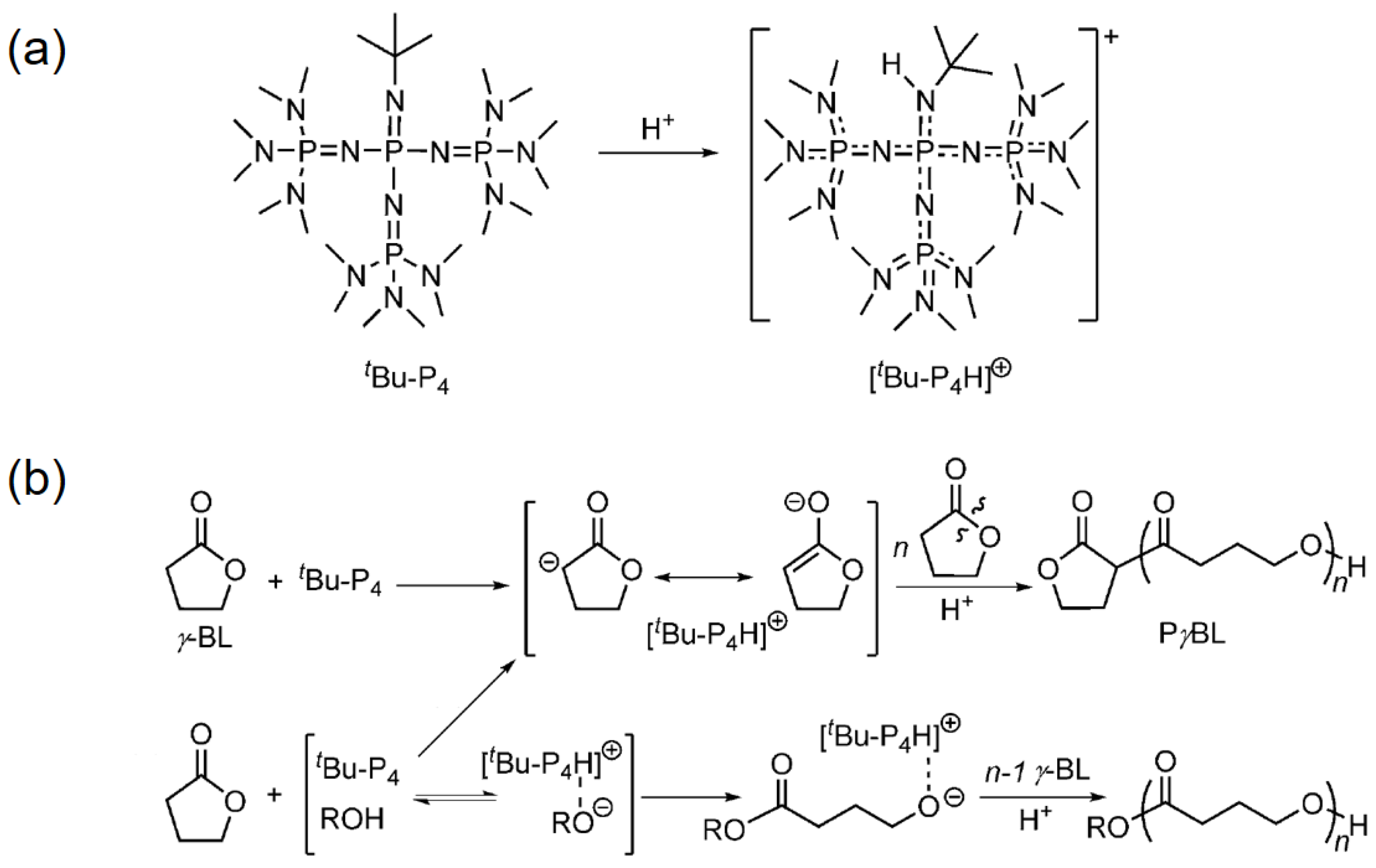
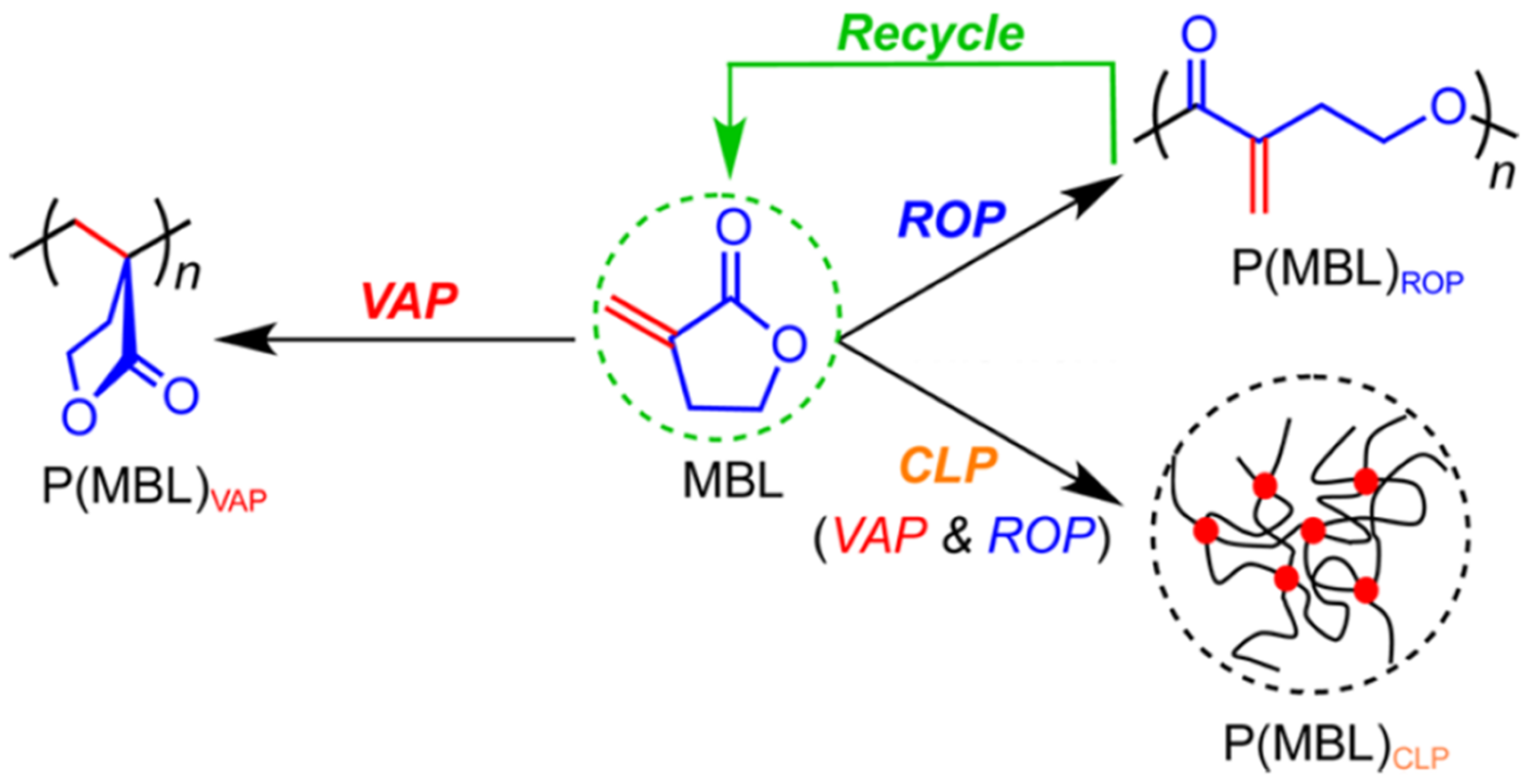
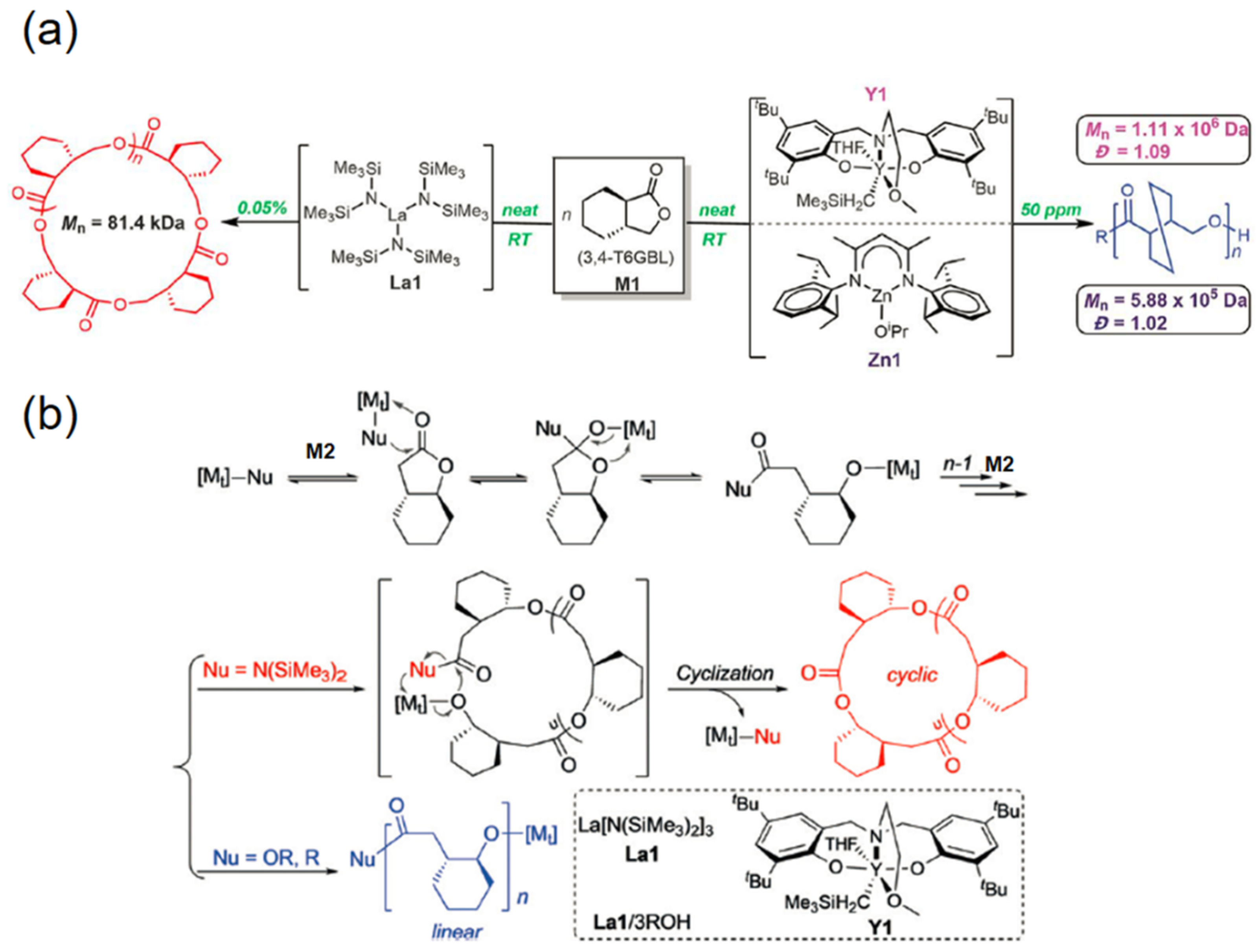
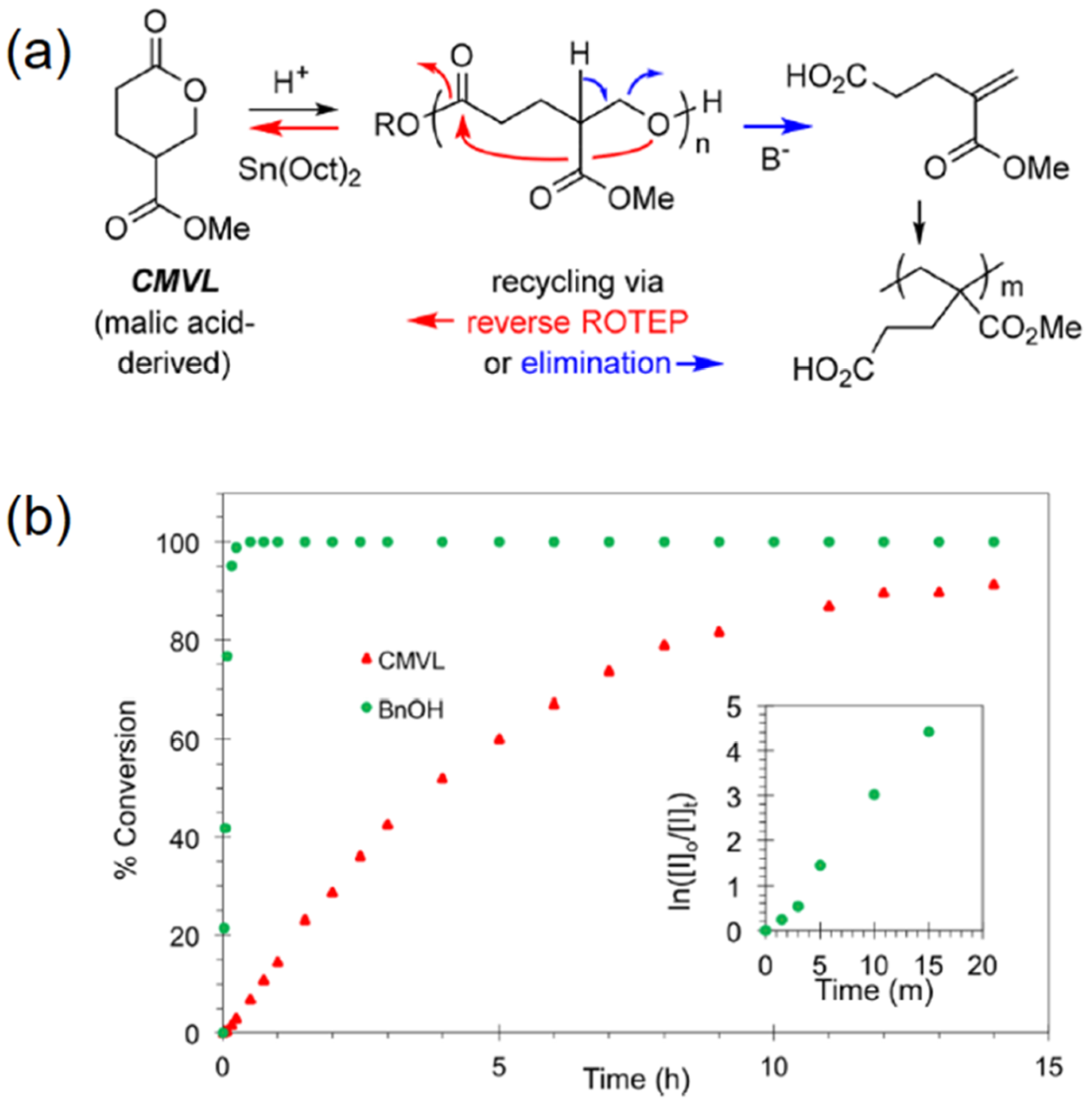
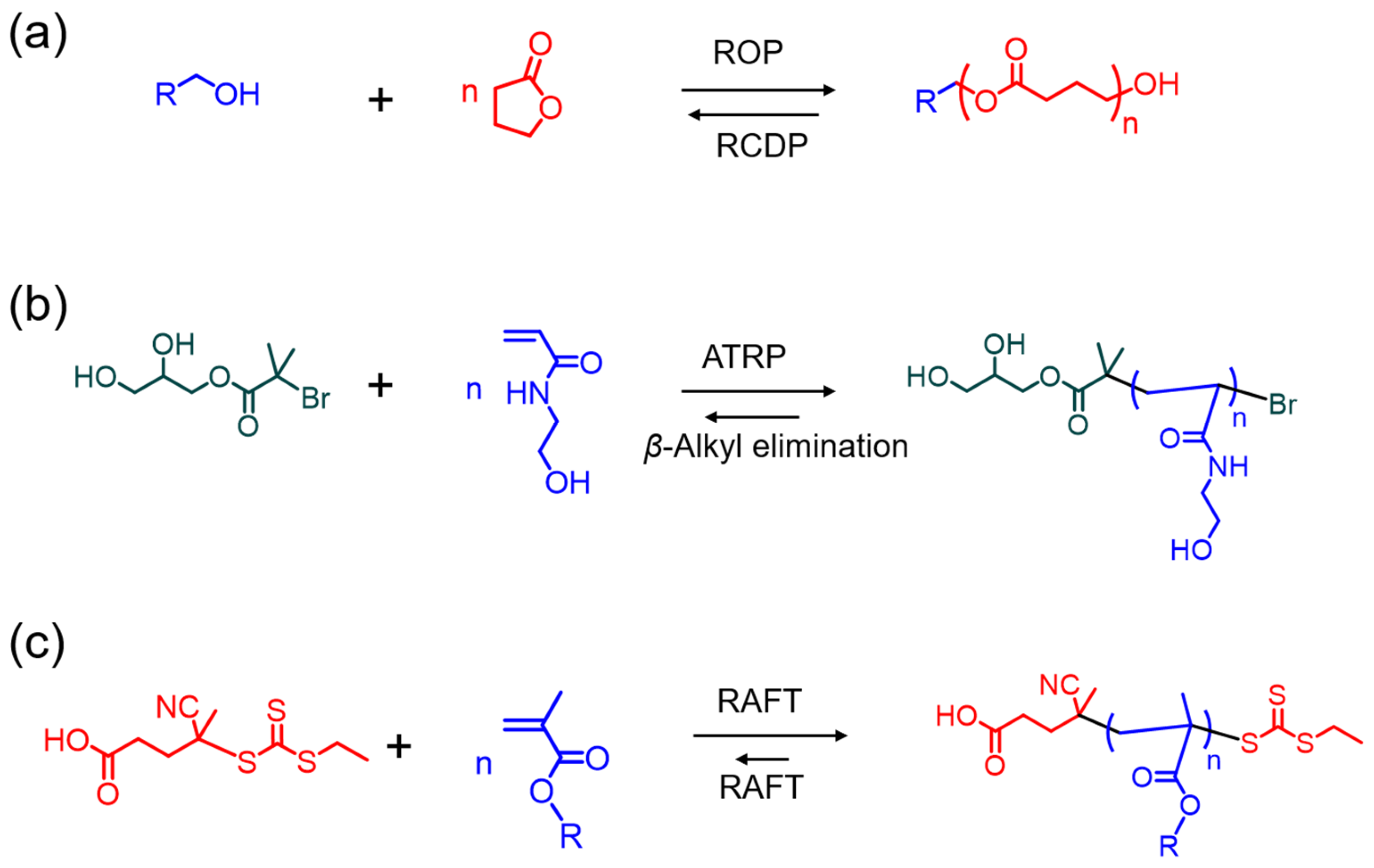
2. Reversible Polymerization in Ring-Opening Polymerization Systems
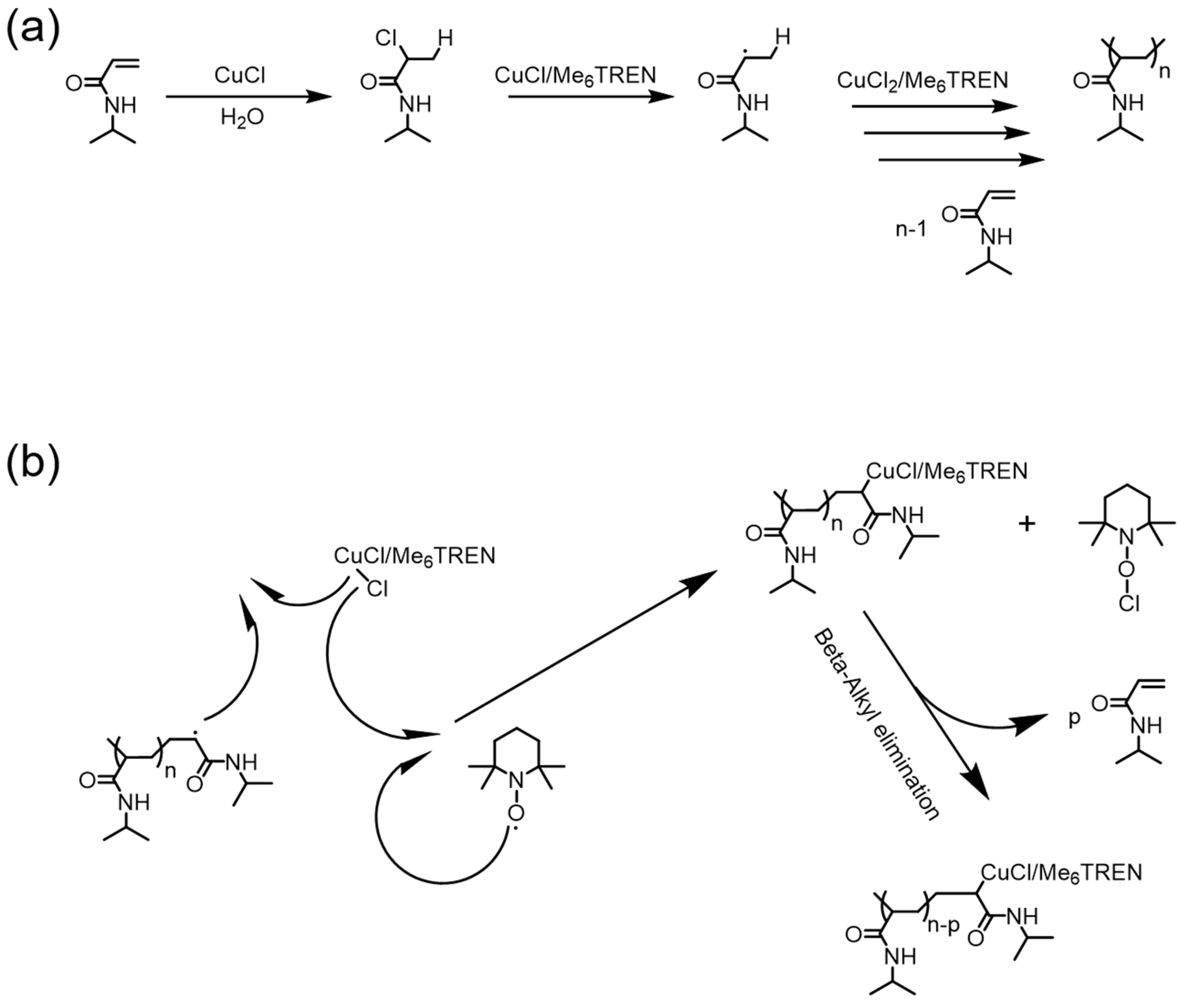
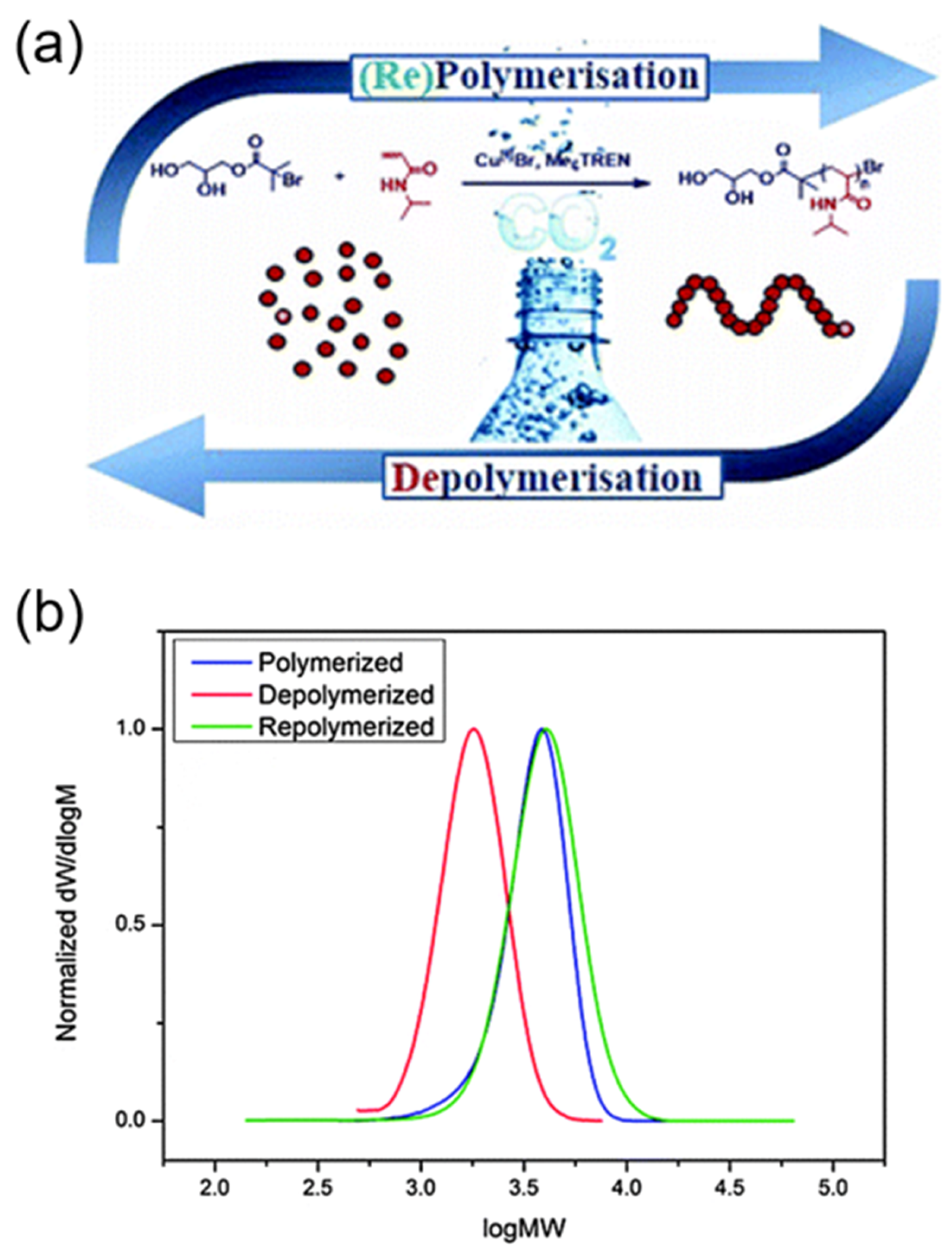
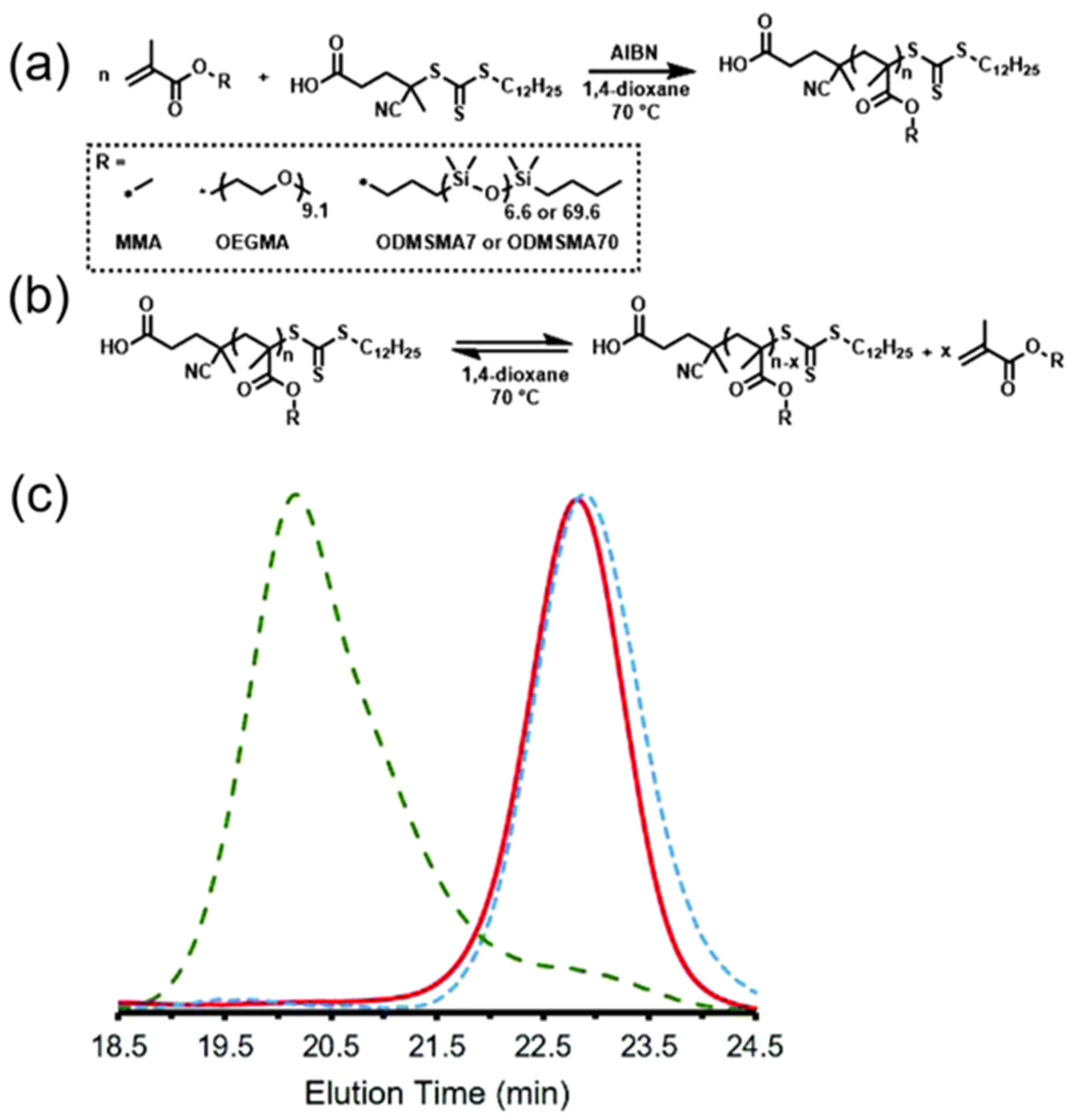
3. Reversible Polymerization in Reversible-Deactivation Radical Polymerization Systems
4. Closing Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sun, H.; Kabb, C.P.; Dai, Y.Q.; Hill, M.R.; Ghiviriga, I.; Bapat, A.P.; Sumerlin, B.S. Macromolecular metamorphosis via stimulus-induced transformations of polymer architecture. Nat. Chem. 2017, 9, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Stuart, M.A.C.; Huck, W.T.S.; Genzer, J.; Muller, M.; Ober, C.; Stamm, M.; Sukhorukov, G.B.; Szleifer, I.; Tsukruk, V.V.; Urban, M.; et al. Emerging applications of stimuli-responsive polymer materials. Nat. Mater. 2010, 9, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Urban, M.W. Self-healing polymeric materials. Chem. Soc. Rev. 2013, 42, 7446–7467. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Kabb, C.P.; Sumerlin, B.S. Thermally-labile segmented hyperbranched copolymers: Using reversible-covalent chemistry to investigate the mechanism of self-condensing vinyl copolymerization. Chem. Sci. 2014, 5, 4646–4655. [Google Scholar] [CrossRef]
- Chen, X.X.; Dam, M.A.; Ono, K.; Mal, A.; Shen, H.B.; Nutt, S.R.; Sheran, K.; Wudl, F. A thermally re-mendable cross-linked polymeric material. Science 2002, 295, 1698–1702. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Tang, H.L.; Sun, H. Progress in Photo-Responsive Polypeptide Derived Nano-Assemblies. Micromachines 2018, 9, 296. [Google Scholar] [CrossRef]
- Tang, H.; Tsarevsky, N.V. Lipoates as building blocks of sulfur-containing branched macromolecules. Polym Chem.-Uk 2015, 6, 6936–6945. [Google Scholar] [CrossRef]
- Tang, H.; Tsarevsky, N.V. Preparation and Functionalization of Linear and Reductively Degradable Highly Branched Cyanoacrylate-Based Polymers. J. Polym. Sci. Pol. Chem. 2016, 54, 3683–3693. [Google Scholar] [CrossRef]
- Snyder, R.L.; Fortman, D.J.; De Hoe, G.X.; Hillmyer, M.A.; Dichtel, W.R. Reprocessable Acid-Degradable Polycarbonate Vitrimers. Macromolecules 2018, 51, 389–397. [Google Scholar] [CrossRef]
- Denissen, W.; Winne, J.M.; Du Prez, F.E. Vitrimers: Permanent organic networks with glass-like fluidity. Chem. Sci. 2016, 7, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Rottger, M.; Domenech, T.; van der Weegen, R.; Nicolay, A.B.R.; Leibler, L. High-performance vitrimers from commodity thermoplastics through dioxaborolane metathesis. Science 2017, 356, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Honda, S.; Toyota, T. Photo-triggered solvent-free metamorphosis of polymeric materials. Nat. Commun. 2017, 8, 502. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.W.; Xue, C.G.; Weis, P.; Suzuki, Y.; Huang, S.L.; Koynov, K.; Auernhammer, G.K.; Berger, R.; Butt, H.J.; Wu, S. Photoswitching of glass transition temperatures of azobenzene-containing polymers induces reversible solid-to-liquid transitions. Nat. Chem. 2017, 9, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Alfurhood, J.A.; Sun, H.; Bachler, P.R.; Sumerlin, B.S. Hyperbranched poly(N-(2-hydroxypropyl)methacrylamide) via RAFT self-condensing vinyl polymerization. Polym Chem. 2016, 7, 2099–2104. [Google Scholar] [CrossRef]
- Sun, H.; Kabb, C.P.; Sims, M.B.; Sumerlin, B.S. Architecture-transformable polymers: Reshaping the future of stimuli-responsive polymers. Prog. Polym. Sci. 2018. [Google Scholar] [CrossRef]
- Diercks, C.S.; Yaghi, O.M. The atom, the molecule, and the covalent organic framework. Science 2017, 355, eaal1585. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.M.; Parent, L.R.; Flanders, N.C.; Bisbey, R.P.; Vitaku, E.; Kirschner, M.S.; Schaller, R.D.; Chen, L.X.; Gianneschi, N.C.; Dichtel, W.R. Seeded growth of single-crystal two-dimensional covalent organic frameworks. Science 2018, 361, 53. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Li, X.J.; Tang, H.L.; Wang, W.Y.; Ramella, D.; Luan, Y. A facile 2H-chromene dimerization through an ortho-quinone methide intermediate catalyzed by a sulfonyl derived MIL-101 MOF. New J. Chem. 2018, 42, 12722–12728. [Google Scholar] [CrossRef]
- Miao, Z.C.; Zhou, Z.H.; Tang, H.L.; Yu, M.D.; Ramella, D.; Du, X.; Luan, Y. Homodimerization of 2H-chromenes catalyzed by BrOnsted-acid derived UiO-66 MOFs. Catal. Sci. Technol. 2018, 8, 3406–3413. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, W.Y.; Tang, H.L.; Ramella, D.; Luan, Y. Modification of Cu2+into Zr-based metal-organic framework (MOF) with carboxylic units as an efficient heterogeneous catalyst for aerobic epoxidation of olefins. Mol. Catal. 2018, 456, 57–64. [Google Scholar] [CrossRef]
- Deng, H.X.; Grunder, S.; Cordova, K.E.; Valente, C.; Furukawa, H.; Hmadeh, M.; Gandara, F.; Whalley, A.C.; Liu, Z.; Asahina, S.; et al. Large-Pore Apertures in a Series of Metal-Organic Frameworks. Science 2012, 336, 1018–1023. [Google Scholar] [CrossRef] [PubMed]
- Peterson, G.I.; Larsen, M.B.; Boydston, A.J. Controlled Depolymerization: Stimuli-Responsive Self-Immolative Polymers. Macromolecules 2012, 45, 7317–7328. [Google Scholar] [CrossRef]
- Zhou, J.W.; Guimard, N.K.; Inglis, A.J.; Namazian, M.; Lin, C.Y.; Coote, M.L.; Spyrou, E.; Hilf, S.; Schmidt, F.G.; Barner-Kowollik, C. Thermally reversible Diels-Alder-based polymerization: An experimental and theoretical assessment. Polym Chem.-UK 2012, 3, 628–639. [Google Scholar] [CrossRef]
- Sun, H.; Dobbins, D.J.; Dai, Y.Q.; Kabb, C.P.; Wu, S.J.; Alfurhood, J.A.; Rinaldi, C.; Sumerlin, B.S. Radical Departure: Thermally-Triggered Degradation of Azo-Containing Poly(beta-thioester)s. ACS Macro Lett. 2016, 5, 688–693. [Google Scholar] [CrossRef]
- Yokozawa, T.; Ohta, Y. Transformation of Step-Growth Polymerization into Living Chain-Growth Polymerization. Chem. Rev. 2016, 116, 1950–1968. [Google Scholar] [CrossRef] [PubMed]
- Li, X.W.; Figg, C.A.; Wang, R.W.; Jiang, Y.; Lyu, Y.F.; Sun, H.; Liu, Y.; Wang, Y.Y.; Teng, I.T.; Hou, W.J.; et al. Cross-Linked Aptamer-Lipid Micelles for Excellent Stability and Specificity in Target-Cell Recognition. Angew Chem. Int. Ed. 2018, 57, 11589–11593. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Wu, C.C.; Chen, T.; Sun, H.; Cansiz, S.; Zhang, L.Q.; Cui, C.; Hou, W.J.; Wu, Y.; Wan, S.; et al. DNA micelle flares: A study of the basic properties that contribute to enhanced stability and binding affinity in complex biological systems. Chem. Sci. 2016, 7, 6041–6049. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hou, W.J.; Sun, H.; Cui, C.; Zhang, L.Q.; Jiang, Y.; Wu, Y.X.; Wang, Y.Y.; Li, J.; Sumerlin, B.S.; et al. Thiol-ene click chemistry: A biocompatible way for orthogonal bioconjugation of colloidal nanoparticles. Chem. Sci. 2017, 8, 6182–6187. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Zhang, L.Q.; Wang, S.; Liu, Y.; Wu, C.C.; Cui, C.; Sun, H.; Shi, M.L.; Jiang, Y.; Li, L.; et al. Molecular Recognition-Based DNA Nanoassemblies on the Surfaces of Nanosized Exosomes. J. Am. Chem. Soc. 2017, 139, 5289–5292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubbs, R.B.; Grubbs, R.H. 50th Anniversary Perspective: Living Polymerization-Emphasizing the Molecule in Macromolecules. Macromolecules 2017, 50, 6979–6997. [Google Scholar] [CrossRef]
- Tan, J.B.; Sun, H.; Yu, M.G.; Sumerlin, B.S.; Zhang, L. Photo-PISA: Shedding Light on Polymerization-Induced Self-Assembly. ACS Macro Lett. 2015, 4, 1249–1253. [Google Scholar] [CrossRef]
- Hill, M.R.; Carmean, R.N.; Sumerlin, B.S. Expanding the Scope of RAFT Polymerization: Recent Advances and New Horizons. Macromolecules 2015, 48, 5459–5469. [Google Scholar] [CrossRef]
- Matyjaszewski, K. Atom Transfer Radical Polymerization (ATRP): Current Status and Future Perspectives. Macromolecules 2012, 45, 4015–4039. [Google Scholar] [CrossRef]
- Zheng, Y.; Abbas, Z.M.; Sarkar, A.; Marsh, Z.; Stefik, M.; Benicewicz, B.C. Surface-initiated reversible addition-fragmentation chain transfer polymerization of chloroprene and mechanical properties of matrix-free polychloroprene nanocomposites. Polymer 2018, 135, 193–199. [Google Scholar] [CrossRef]
- Zheng, Y.; Huang, Y.C.; Benicewicz, B.C. A Useful Method for Preparing Mixed Brush Polymer Grafted Nanoparticles by Polymerizing Block Copolymers from Surfaces with Reversed Monomer Addition Sequence. Macromol. Rapid Comm. 2017, 38, 1700300. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Sun, H.; Quan, D.P. Synthesis of poly(l-lactide-co-5-amino-5-methyl-1,3-dioxan-2-ones) [P(L-LA-co-TAc)] containing amino groups via organocatalysis and post-polymerization functionalization. Polymer 2016, 97, 614–622. [Google Scholar] [CrossRef]
- Dai, Y.Q.; Sun, H.; Pal, S.; Zhang, Y.L.; Park, S.; Kabb, C.P.; Wei, W.D.; Sumerlin, B.S. Near-IR-induced dissociation of thermally-sensitive star polymers. Chem. Sci. 2017, 8, 1815–1821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matyjaszewski, K. Architecturally Complex Polymers with Controlled Heterogeneity. Science 2011, 333, 1104–1105. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Tang, H.L.; Feng, A.C.; Thang, S.H. Synthesis of Zwitterionic Polymers by Living/Controlled Radical Polymerization and Its Applications. Prog. Chem. 2018, 30, 1097–1111. [Google Scholar] [CrossRef]
- Zheng, Y.; Huang, Y.C.; Abbas, Z.M.; Benicewicz, B.C. Surface-initiated polymerization-induced self-assembly of bimodal polymer-grafted silica nanoparticles towards hybrid assemblies in one step. Polym Chem.-UK 2016, 7, 5347–5350. [Google Scholar] [CrossRef]
- Zheng, Y.; Huang, Y.C.; Abbas, Z.M.; Benicewicz, B.C. One-pot synthesis of inorganic nanoparticle vesicles via surface-initiated polymerization-induced self-assembly. Polym. Chem.-UK 2017, 8, 370–374. [Google Scholar] [CrossRef]
- Kamber, N.E.; Jeong, W.; Waymouth, R.M.; Pratt, R.C.; Lohmeijer, B.G.G.; Hedrick, J.L. Organocatalytic ring-opening polymerization. Chem. Rev. 2007, 107, 5813–5840. [Google Scholar] [CrossRef] [PubMed]
- Nuyken, O.; Pask, S.D. Ring-Opening Polymerization-An Introductory Review. Polymers 2013, 5, 361–403. [Google Scholar] [CrossRef]
- Liu, H.; Cheng, Y.L.; Chen, J.J.; Chang, F.; Wang, J.C.; Ding, J.X.; Chen, X.S. Component effect of stem cell-loaded thermosensitive polypeptide hydrogels on cartilage repair. Acta. Biomater. 2018, 73, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Xu, W.G.; Li, S.X.; Qiu, H.P.; Li, Z.B.; Wang, C.X.; Wang, X.Q.; Ding, J.X. Polylactide-Cholesterol Stereocomplex Micelle Encapsulating Chemotherapeutic Agent for Improved Antitumor Efficacy and Safety. J. Biomed. Nanotechnol. 2018, 14, 2102–2113. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.X.; Xiao, C.S.; He, C.L.; Li, M.Q.; Li, D.; Zhuang, X.L.; Chen, X.S. Facile preparation of a cationic poly(amino acid) vesicle for potential drug and gene co-delivery. Nanotechnology 2011, 22, 494012. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.X.; Xiao, C.S.; Zhao, L.; Cheng, Y.L.; Ma, L.L.; Tang, Z.H.; Zhuang, X.L.; Chen, X.S. Poly(L-glutamic acid) Grafted with Oligo(2-(2-(2-methoxyethoxy)ethoxy) ethyl methacrylate): Thermal Phase Transition, Secondary Structure, and Self-Assembly. J. Polym. Sci. Pol. Chem. 2011, 49, 2665–2676. [Google Scholar] [CrossRef]
- Braunecker, W.A.; Matyjaszewski, K. Controlled/living radical polymerization: Features, developments, and perspectives. Prog. Polym. Sci. 2007, 32, 93–146. [Google Scholar] [CrossRef]
- Yeow, J.; Chapman, R.; Gormley, A.J.; Boyer, C. Up in the air: Oxygen tolerance in controlled/living radical polymerisation. Chem. Soc. Rev. 2018, 47, 4357–4387. [Google Scholar] [CrossRef] [PubMed]
- Zetterlund, P.B.; Thickett, S.C.; Perrier, S.; Bourgeat-Lami, E.; Lansalot, M. Controlled/Living Radical Polymerization in Dispersed Systems: An Update. Chem. Rev. 2015, 115, 9745–9800. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Wang, L.; Lu, L.; Wang, Q.; Benicewicz, B.C. pH and Thermal Dual-Responsive Nanoparticles for Controlled Drug Delivery with High Loading Content. Acs Omega. 2017, 2, 3399–3405. [Google Scholar] [CrossRef] [PubMed]
- Matyjaszewski, K. Atom Transfer Radical Polymerization: From Mechanisms to Applications. Isr. J. Chem. 2012, 52, 206–220. [Google Scholar] [CrossRef]
- Siegwart, D.J.; Oh, J.K.; Matyjaszewski, K. ATRP in the design of functional materials for biomedical applications. Prog. Polym. Sci. 2012, 37, 18–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrier, S. 50th Anniversary Perspective: RAFT Polymerization-A User Guide. Macromolecules 2017, 50, 7433–7447. [Google Scholar] [CrossRef]
- Alfurhood, J.A.; Sun, H.; Kabb, C.P.; Tucker, B.S.; Matthews, J.H.; Luesch, H.; Sumerlin, B.S. Poly(N-(2-hydroxypropyl)-methacrylamide)-valproic acid conjugates as block copolymer nanocarriers. Polym Chem. 2017, 8, 4983–4987. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.Y.; Ding, J.J.; Zhang, B.; Qiu, F.; Zhuang, X.D.; Chen, Y. Recent Advances in RAFT Polymerization: Novel Initiation Mechanisms and Optoelectronic Applications. Polymers 2018, 10, 318. [Google Scholar] [CrossRef]
- Almeida, C.; Costa, H.; Kadhirvel, P.; Queiroz, A.M.; Dias, R.C.S.; Costa, M.R.P.F.N. Electrochemical activity of sulfur networks synthesized through RAFT polymerization. J. Appl. Polym. Sci. 2016, 133, 43993. [Google Scholar] [CrossRef]
- Nicolas, J.; Guillaneuf, Y.; Lefay, C.; Bertin, D.; Gigmes, D.; Charleux, B. Nitroxide-mediated polymerization. Prog Polym Sci 2013, 38, 63–235. [Google Scholar] [CrossRef]
- Yang, L.; Sun, H.; Liu, Y.; Hou, W.; Yang, Y.; Cai, R.; Cui, C.; Zhang, P.; Pan, X.; Li, X.; et al. Self-assembled aptamer-hyperbranched polymer nanocarrier for targeted and photoresponsive drug delivery. Angew Chem. Int. Ed. 2018. [Google Scholar] [CrossRef]
- Flanders, M.J.; Gramlich, W.M. Reversible-addition fragmentation chain transfer (RAFT) mediated depolymerization of brush polymers. Polym. Chem.-UK 2018, 9, 2328–2335. [Google Scholar] [CrossRef]
- Miller, S.A. Sustainable Polymers: Opportunities for the Next Decade. ACS Macro. Lett. 2013, 2, 550–554. [Google Scholar] [CrossRef]
- Li, L.J.; Shu, X.; Zhu, J. Low temperature depolymerization from a copper-based aqueous vinyl polymerization system. Polymer 2012, 53, 5010–5015. [Google Scholar] [CrossRef]
- Olsen, P.; Odelius, K.; Albertsson, A.C. Ring-Closing Depolymerization: A Powerful Tool for Synthesizing the Allyloxy-Functionalized Six-Membered Aliphatic Carbonate Monomer 2-Allyloxymethyl-2-ethyltrimethylene Carbonate. Macromolecules 2014, 47, 6189–6195. [Google Scholar] [CrossRef]
- Olsen, P.; Undin, J.; Odelius, K.; Keul, H.; Albertsson, A.C. Switching from Controlled Ring-Opening Polymerization (cROP) to Controlled Ring-Closing Depolymerization (cRCDP) by Adjusting the Reaction Parameters That Determine the Ceiling Temperature. Biomacromolecules 2016, 17, 3995–4002. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, D.J.; Nikolaou, V.; Collins, J.; Waldron, C.; Anastasaki, A.; Bassett, S.P.; Howdle, S.M.; Blanazs, A.; Wilson, P.; Kempe, K.; et al. Controlled aqueous polymerization of acrylamides and acrylates and "in situ" depolymerization in the presence of dissolved CO2. Chem. Commun. 2016, 52, 6533–6536. [Google Scholar] [CrossRef] [PubMed]
- Fahnhorst, G.W.; Hoye, T.R. A Carbomethoxylated Polyvalerolactone from Malic Acid: Synthesis and Divergent Chemical Recycling. ACS Macro. Lett. 2018, 7, 143–147. [Google Scholar] [CrossRef]
- Zhu, J.B.; Chen, E.Y.X. Living Coordination Polymerization of a Six-Five Bicyclic Lactone to Produce Completely Recyclable Polyester. Angew. Chem. Int. Edit. 2018, 57, 12558–12562. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Chen, E.Y.X. Completely recyclable biopolymers with linear and cyclic topologies via ring-opening polymerization of gamma-butyrolactone. Nat. Chem. 2016, 8, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.Y.; Hong, M.; Falivene, L.; Caporaso, L.; Cavallo, L.; Chen, E.Y.X. The Quest for Converting Biorenewable Bifunctional alpha-Methylene-gamma-butyrolactone into Degradable and Recyclable Polyester: Controlling Vinyl-Addition/Ring-Opening/Cross-Linking Pathways. J. Am. Chem. Soc. 2016, 138, 14326–14337. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.B.; Watson, E.M.; Tang, J.; Chen, E.Y.X. A synthetic polymer system with repeatable chemical recyclability. Science 2018, 360, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Chen, E.Y.X. Towards Truly Sustainable Polymers: A Metal-Free Recyclable Polyester from Biorenewable Non-Strained -Butyrolactone. Angew. Chem. Int. Edit. 2016, 55, 4188–4193. [Google Scholar] [CrossRef] [PubMed]
- Albertsson, A.C.; Varma, I.K. Recent developments in ring opening polymerization of lactones for biomedical applications. Biomacromolecules 2003, 4, 1466–1486. [Google Scholar] [CrossRef] [PubMed]
- Dechy-Cabaret, O.; Martin-Vaca, B.; Bourissou, D. Controlled ring-opening polymerization of lactide and glycolide. Chem. Rev. 2004, 104, 6147–6176. [Google Scholar] [CrossRef] [PubMed]
- Higaki, Y.; Okazaki, R.; Takahara, A. Semirigid Biobased Polymer Brush: Poly(alpha-methylene-gamma-butyrolactone) Brushes. ACS Macro. Lett. 2012, 1, 1124–1127. [Google Scholar] [CrossRef]
- Mosnacek, J.; Matyjaszewski, K. Atom transfer radical polymerization of tulipalin A: A naturally renewable monomer. Macromolecules 2008, 41, 5509–5511. [Google Scholar] [CrossRef]
- Al-Salem, S.M.; Lettieri, P.; Baeyens, J. Recycling and recovery routes of plastic solid waste (PSW): A review. Waste Manage. 2009, 29, 2625–2643. [Google Scholar] [CrossRef] [PubMed]
- Hungenburg, K.; Wulkow, B. Modeling and Simulation in Polymer Reaction Engineering: A Modular Approach; Wiley: Hoboken, NJ, USA, 2018; ISBN 978-3-527-33818-4. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Polymers | Polymerzation Mechanism | Depolymerization Mechanism | Depolymerization Conversion | References |
|---|---|---|---|---|
| Polyester | ROP | RCDP | ≥99% | Albertsson [64] |
| Polyester | ROP | RCDP | ≥99% | Chen [68] |
| Polyester | ROP | RCDP | ≥99% | Chen [71] |
| Polyester | ROP | RCDP | ≥99% | Chen [69] |
| Polyester | ROP | RCDP | ≥99% | Chen [70] |
| Polyester | ROP | RCDP | ≥99% | Chen [67] |
| Polyester | ROP | RCDP | ≥99% | Hoye [66] |
| Vinyl polymer | ATRP | β-alkyl elimination | 24–34% | Zhu [62] |
| Vinyl polymer | ATRP | Not identified | 43–71% | Haddleton [65] |
| Vinyl polymer | RAFT | RAFT | 28% | Gramlich [60] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, H.; Luan, Y.; Yang, L.; Sun, H. A Perspective on Reversibility in Controlled Polymerization Systems: Recent Progress and New Opportunities. Molecules 2018, 23, 2870. https://doi.org/10.3390/molecules23112870
Tang H, Luan Y, Yang L, Sun H. A Perspective on Reversibility in Controlled Polymerization Systems: Recent Progress and New Opportunities. Molecules. 2018; 23(11):2870. https://doi.org/10.3390/molecules23112870
Chicago/Turabian StyleTang, Houliang, Yi Luan, Lu Yang, and Hao Sun. 2018. "A Perspective on Reversibility in Controlled Polymerization Systems: Recent Progress and New Opportunities" Molecules 23, no. 11: 2870. https://doi.org/10.3390/molecules23112870
APA StyleTang, H., Luan, Y., Yang, L., & Sun, H. (2018). A Perspective on Reversibility in Controlled Polymerization Systems: Recent Progress and New Opportunities. Molecules, 23(11), 2870. https://doi.org/10.3390/molecules23112870