
Adverse Drug Reaction Predictions Using Stacking Deep Heterogeneous Information Network Embedding Approach


Abstract
:1. Introduction
2. Datasets and Method
2.1. Datasets
- Drug-Drug interaction information (DDI): Tatonetti [12] mined side effects induced by DDIs from the FDA Adverse Event Reporting System (FAERS, http://www.fda.gov/cder/aers/default.htm) and developed a database called ”TWOSIDES”. The database contains 645 drugs and ADRs caused by 63,473 combinations of different drugs.
- Protein-Protein interaction data (PPI): We downloaded the PPI network data from the Human Protein Reference Database (HPRD, http://www.hprd.org). The dataset contains 9519 proteins and 37,062 protein-protein interactions.
- Other drug information: We also obtained other drug information from four online drug information databases (DrugBank [4], the PubChem Compound database [6], the SIDER database [5], and the OFFSIDES database [12]). DrugBank is a widely-used public drug information database. From the DrugBank database, we collected drug target protein and disease treatment information. The PubChem system generates a binary substructure fingerprint for chemical structures. From the PubChem database, we searched every drug’s chemical substructure. We also extracted drug side effect information from the SIDER and OFFSIDES databases. These two databases include most associations between drugs and side effects, and we integrated the drug-side effect data obtained from the two databases.
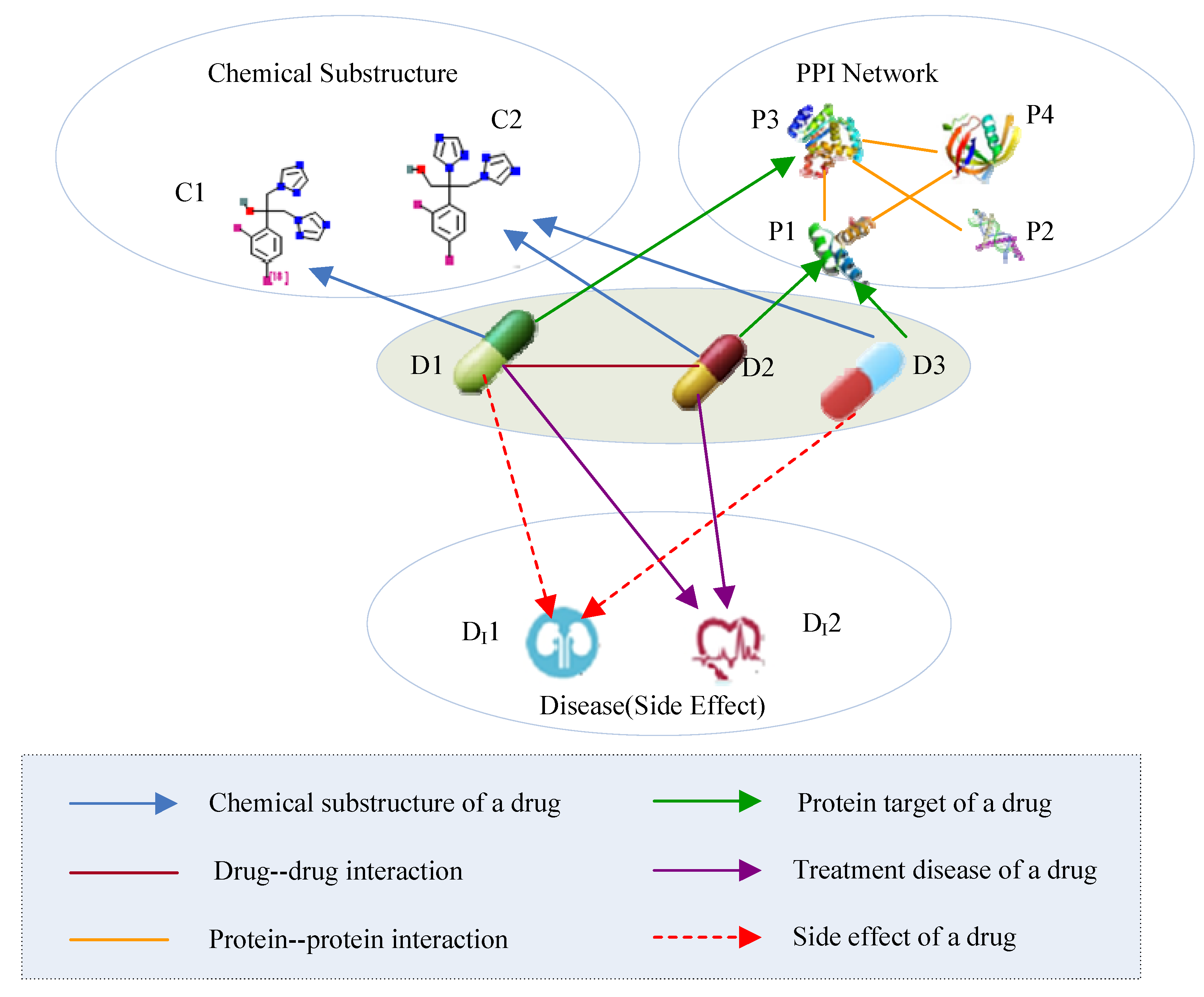
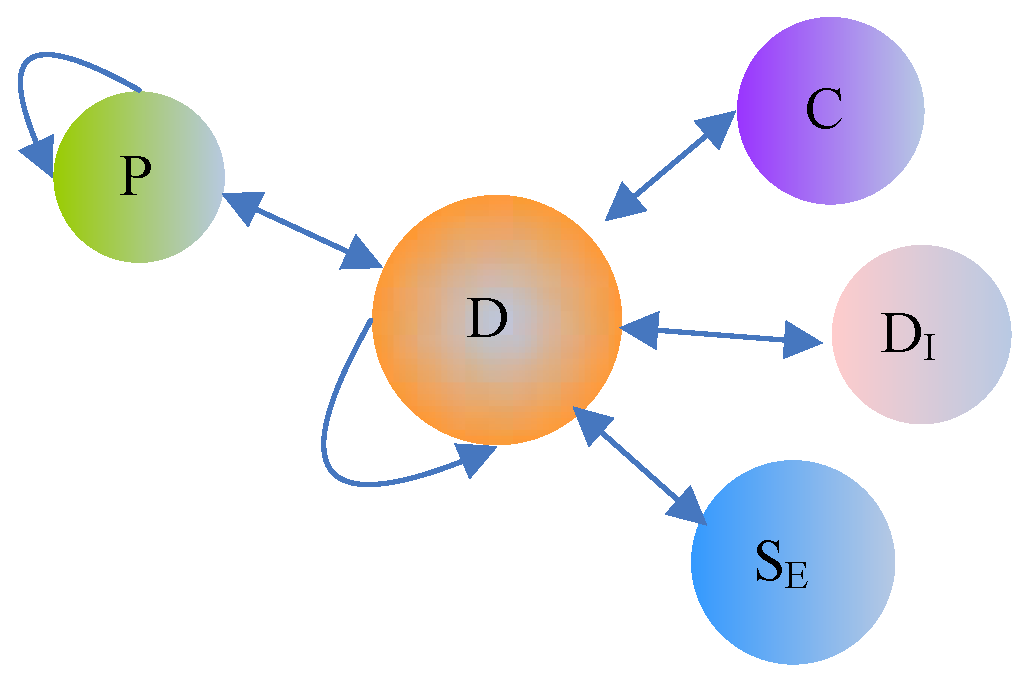
2.2. Drug HIN
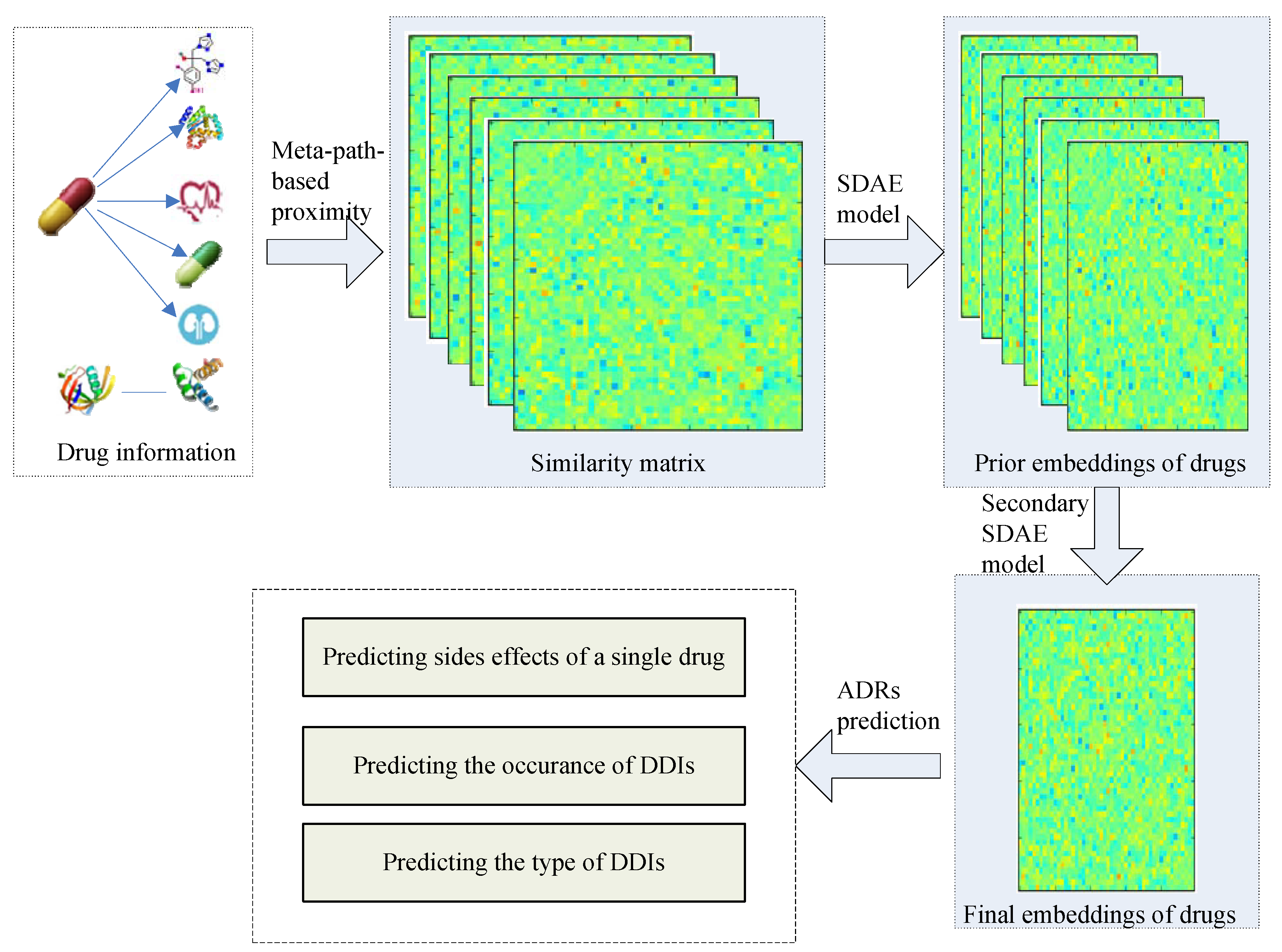
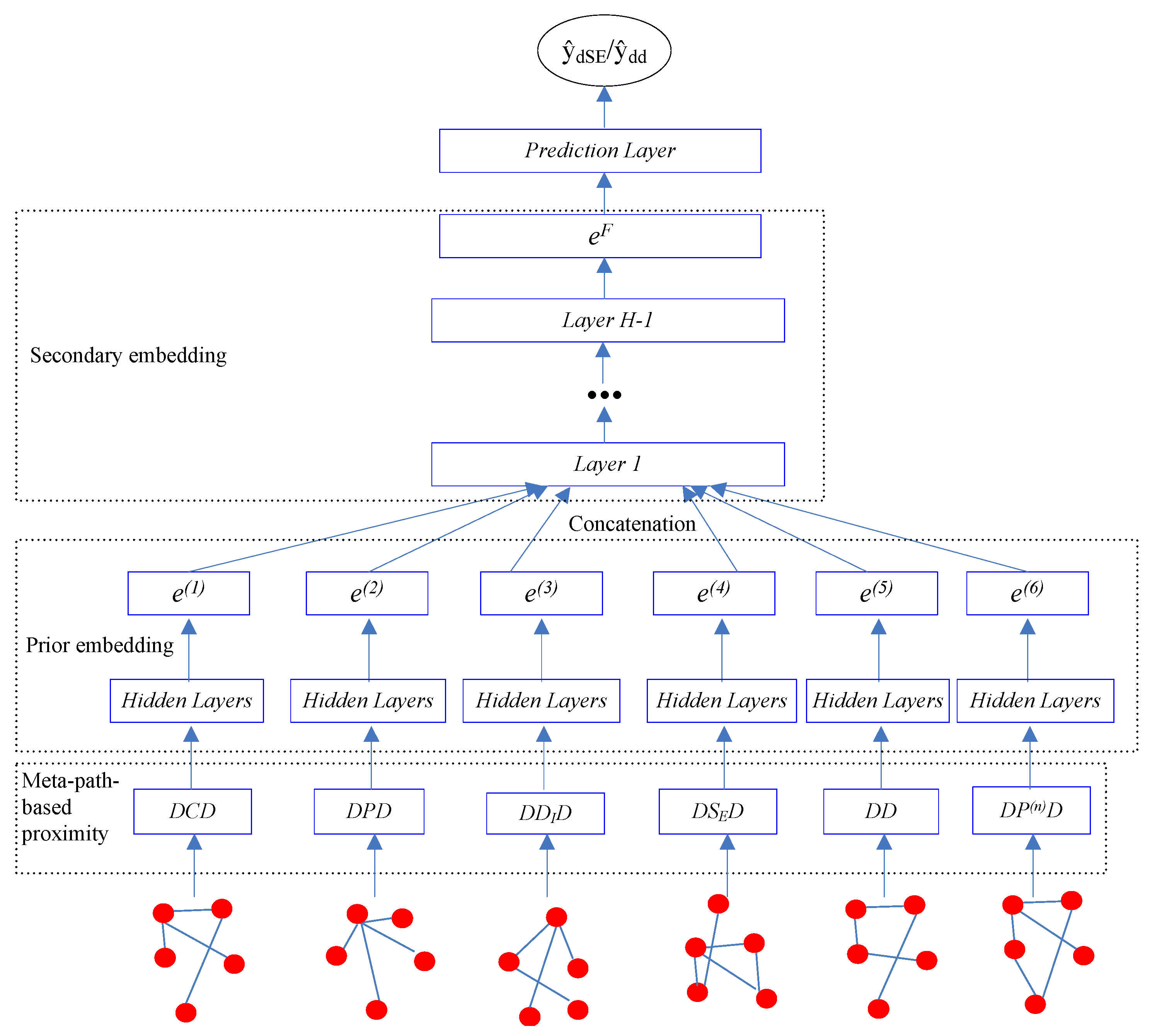
2.3. Stacking Deep HIN Embedding
2.3.1. Meta-Path-Based Proximity
- Constructing drug-drug interaction sub-network.
- Constructing sub-networks using PathSim proximity.
- Reconstructing the drug-target sub-network using the target propagation method.
2.3.2. Prior Drug Embedding
2.3.3. Secondary Drug Embedding
2.4. Prediction Formulation
3. Experiment
3.1. Implementation and Evaluation Strategy
- Concatenate drug features: This method is a simple original HIN embedding method [28]. The approach constructs a feature vector for each drug by concatenating the PCA representation of each correlation matrix, which represents one aspect of the drug character.
- GraphCNN [34]: GraphCNN is a recently-proposed network embedding method based on spectral convolutional operation and achieves state-of-the-art performance on important prediction problems in recommender systems. Here, first, we linearly integrated similarity matrices based on all meta-paths except the target propagation meta-path and then learned the drug embeddings using the same GraphCNN structure described in [35].
- metapath2vec++ [36]: metapath2vec++ is a heterogeneous information network embedding method based on a meta-path-guided random walk strategy.
3.2. Experimental Results
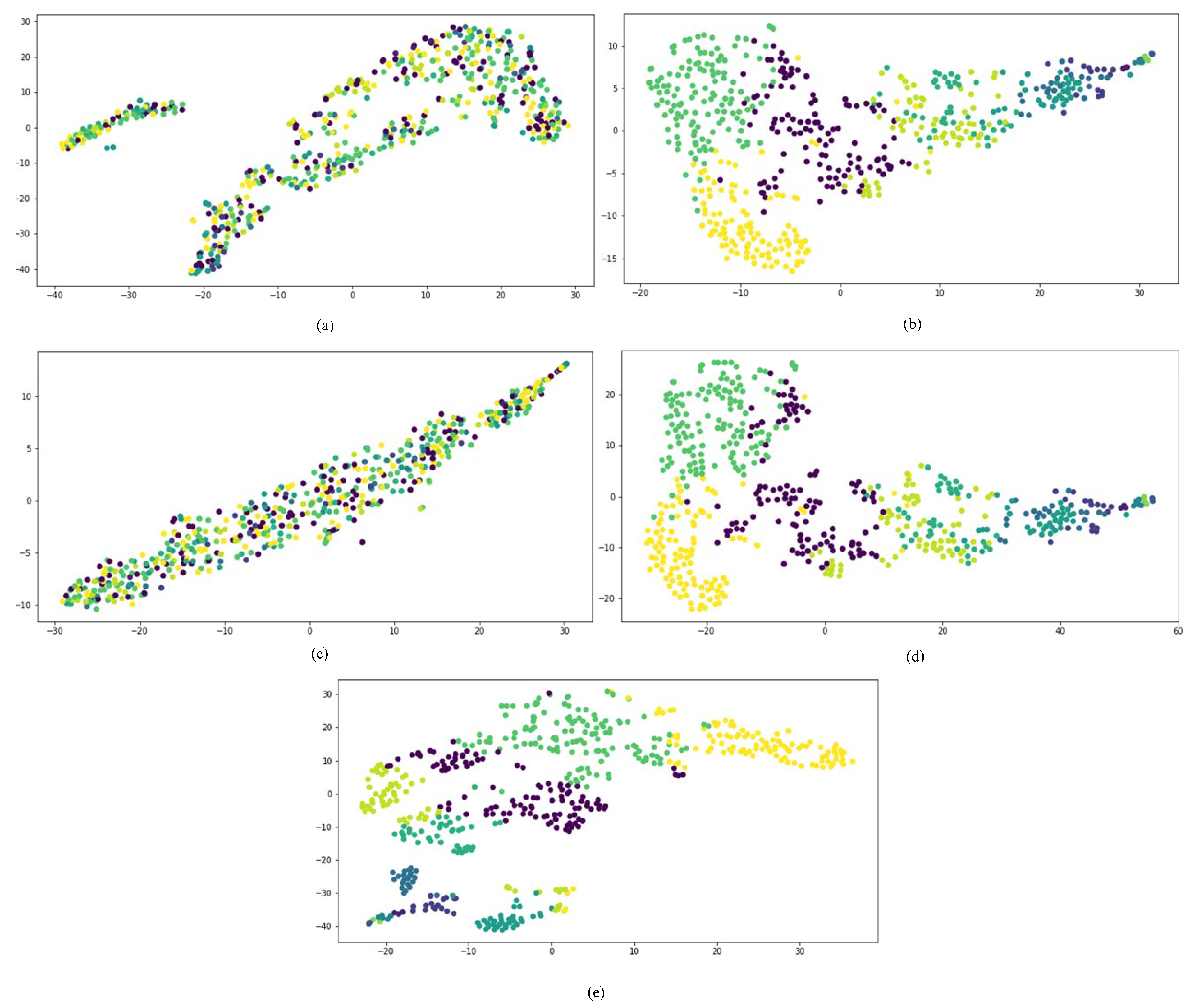
3.2.1. Visualization Results
3.2.2. Prediction Results
- Task 1: Predicting side effects of a single drug.
- Task 2: Binary prediction of the occurrence of DDIs.
- Task 3: Multi-label prediction of specific adverse DDI types.
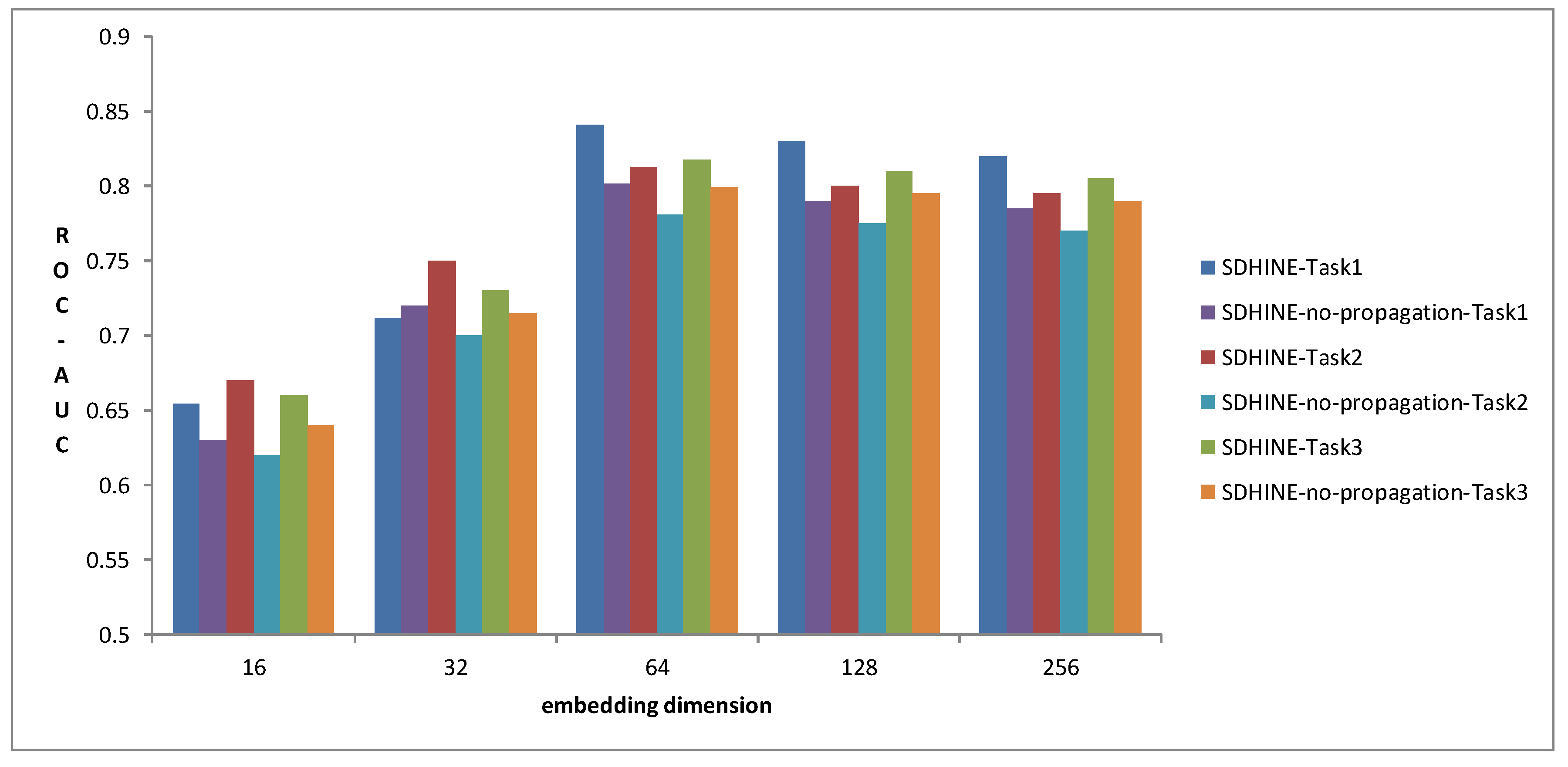
3.3. Performance Comparison of Different Embedding Dimensions
3.4. Case Studies
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Giacomini, K.M.; Krauss, R.M.; Dan, M.R.; Eichelbaum, M.; Hayden, M.R.; Nakamura, Y. When good drugs go bad. Nature 2007, 446, 975–977. [Google Scholar] [CrossRef] [PubMed]
- Whitebread, S.; Hamon, J.; Bojanic, D.; Urban, L. Keynote review: In vitro safety pharmacology profiling: An essential tool for successful drug development. Drug Discov. Today 2005, 10, 1421–1433. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Furumichi, M.; Tanabe, M.; Hirakawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010, 38, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Knox, C.; Law, V.; Jewison, T.; Liu, P.; Ly, S.; Frolkis, A.; Pon, A.; Banco, K.; Mak, C.; Neveu, V. DrugBank 3.0: A comprehensive resource for ’Omics’ research on drugs. Nucleic Acids Res. 2011, 39, D1035. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Campillos, M.; Letunic, I.; Jensen, L.J.; Bork, P. A side effect resource to capture phenotypic effects of drugs. Mol. Syst. Biol. 2010, 6, 343. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cheng, T.; Wang, Y.; Bryant, S.H. PubChem as a public resource for drug discovery. Drug Discovery Today 2010, 15, 1052–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanishi, Y.; Pauwels, E.; Kotera, M. Drug side effect prediction based on the integration of chemical and biological spaces. J. Chem. Inf. Model. 2012, 52, 3284–3292. [Google Scholar] [CrossRef]
- Li, J.; Zheng, S.; Chen, B.; Butte, A.J.; Swamidass, S.J.; Lu, Z. A survey of current trends in computational drug repositioning. Brief. Bioinf. 2016, 17, 2–12. [Google Scholar] [CrossRef]
- Xu, B.; Shi, X.F.; Zhao, Z.H.; Zheng, W. Leveraging Biomedical Resources in Bi-LSTM for Drug Drug Interaction Extraction. IEEE Access 2018, 17, 33432–33439. [Google Scholar] [CrossRef]
- Vilar, S.; Tatonetti, N.P.; Hripcsak, G. 3D Pharmacophoric Similarity improves Multi Adverse Drug Event Identification in Pharmacovigilance. Sci. Rep. 2015, 5, 8809. [Google Scholar] [CrossRef] [Green Version]
- Labute, M.X.; Zhang, X.; Lenderman, J.; Bennion, B.J.; Wong, S.E.; Lightstone, F.C. Adverse drug reaction prediction using scores produced by large-scale drug-protein target docking on high-performance computing machines. PLoS ONE 2014, 9, e106298. [Google Scholar] [CrossRef] [PubMed]
- Tatonetti, N.P.; Ye, P.P.; Daneshjou, R.; Altman, R.B. Data-Driven Prediction of Drug Effects and Interactions. Sci. Transl. Med. 2012, 4, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Ping, Z.; Fei, W.; Hu, J. Towards Drug Repositioning: A Unified Computational Framework for Integrating Multiple Aspects of Drug Similarity and Disease Similarity. AMIA Annu. Symp. Proc. 2014, 2014, 1258–1267. [Google Scholar]
- Zhang, W.; Chen, Y.; Tu, S.; Liu, F.; Qu, Q. Drug side effect prediction through linear neighborhoods and multiple data source integration. In Proceedings of the IEEE International Conference on Bioinformatics and Biomedicine, Shenzhen, China, 15–18 December 2016; pp. 427–434. [Google Scholar]
- Segura-Bedmar, I. Using a shallow linguistic kernel for drug–drug interaction extraction. J. Biomed. Inf. 2011, 44, 789–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, B.; Yang, H.; Xiao, C.; Zhang, P.; Wei, X.; Wang, F. Multitask Dyadic Prediction and Its Application in Prediction of Adverse Drug-Drug Interaction. In Proceedings of the Thirty-First AAAI Conference on Artificial Intelligence, San Francisco, CA, USA, 4–9 February 2017; pp. 1367–1373. [Google Scholar]
- Zhang, W.; Chen, Y.; Liu, F.; Luo, F.; Tian, G.; Li, X. Predicting potential drug–drug interactions by integrating chemical, biological, phenotypic and network data. BMC Bioinf. 2017, 18, 18. [Google Scholar] [CrossRef] [PubMed]
- Zitnik, M.; Agrawal, M.; Leskovec, J. Modeling polypharmacy side effects with graph convolutional networks. Bioinformatics 2018, 34, i457–i466. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Xu, D.; Zhang, B.; Zhang, H.J.; Yang, Q.; Lin, S. Graph Embedding and Extensions: A General Framework for Dimensionality Reduction. IEEE Trans. Pattern Anal. Mach. Intell. 2007, 29, 40–51. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.; Lu, W.; Xu, Q. Deep neural networks for learning graph representations. In Proceedings of the Thirtieth AAAI Conference on Artificial Intelligence, Phoenix, AZ, USA, 12–17 February 2016; pp. 1145–1152. [Google Scholar]
- Huang, Z.; Mamoulis, N. Heterogeneous Information Network Embedding for Meta Path based Proximity. Available online: https://arxiv.org/abs/1701.05291 (accessed on 19 January 2017).
- Li, R.; Dong, Y.; Kuang, Q.; Wu, Y.; Li, Y.; Zhu, M.; Li, M. Inductive matrix completion for predicting adverse drug reactions (ADRs) integrating drug–target interactions. Chemom. Intell. Lab. Syst. 2015, 144, 71–79. [Google Scholar] [CrossRef]
- Ma, T.; Xiao, C.; Zhou, J.; Wang, F. Drug Similarity Integration Through Attentive Multi-view Graph Auto-Encoders. Available online: https://arxiv.org/abs/1804.10850 (accessed on 28 April 2018).
- Kelley, B.P.; Sharan, R.; Karp, R.M.; Sittler, T.; Root, D.E.; Stockwell, B.R.; Ideker, T. Conserved pathways within bacteria and yeast as revealed by global protein network alignment. Proc. Natl. Acad. Sci. USA 2003, 100, 11394–11399. [Google Scholar] [CrossRef] [Green Version]
- Yeh, C.Y.; Yeh, H.Y.; Arias, C.R.; Soo, V.W. Pathway Detection from Protein Interaction Networks and Gene Expression Data Using Color-Coding Methods and A* Search Algorithms. Sci. World J. 2012, 2012, 315797. [Google Scholar] [CrossRef]
- Codling, E.A.; Plank, M.J.; Benhamou, S. Random walk models in biology. J. R. Soc. Interface 2008, 5, 813–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Q.; Li, J.; Song, L.; Zeng, X.; Wang, G. Similarity computation strategies in the microRNA-disease network: A Survey. Brief. Funct. Genom. 2016, 15, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Li, Y.; Zhang, J.; Sun, Y.; Yu, P.S. A Survey of Heterogeneous Information Network Analysis. IEEE Trans. Knowl. Data. Eng. 2016, 29, 17–37. [Google Scholar] [CrossRef]
- Shakibian, H.; Charkari, N.M. Mutual information model for link prediction in heterogeneous complex networks. Sci. Rep. 2017, 7, 44981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.; Han, W.; Tang, J.; Qi, G.J.; Aggarwal, C.C.; Huang, T.S. Heterogeneous Network Embedding via Deep Architectures. In Proceedings of the 21th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Sydney, New South Wales, Australia, 10–13 August 2015; pp. 119–128. [Google Scholar]
- Katz, L. A new status index derived from sociometric analysis. Psychmetrika 1953, 18, 39–43. [Google Scholar] [CrossRef]
- Wang, D.; Cui, P.; Zhu, W. Structural Deep Network Embedding. In Proceedings of the 22nd ACM SIGKDD international conference on Knowledge discovery and data mining, San Francisco, CA, USA, 13–17 August 2016; pp. 1225–1234. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization. Available online: https://arxiv.org/abs/1412.6980 (accessed on 22 December 2014).
- Kipf, T.N.; Welling, M. Semi-Supervised Classification with Graph Convolutional Networks. Available online: https://arxiv.org/abs/1609.02907 (accessed on 9 September 2016).
- Kipf, T.N.; Welling, M. Variational Graph Auto-Encoders. Available online: https://arxiv.org/abs/1611.0730821 (accessed on 21 November 2016).
- Dong, Y.; Chawla, N.V.; Swami, A. In metapath2vec: Scalable Representation Learning for Heterogeneous Networks. In Proceedings of the ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, New York, NY, USA, 24–27 August 2017; pp. 135–144. [Google Scholar]
- Maaten, L.V.D.; Hinton, G. Viualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Hashizume, K.; Nabeshima, T.; Fujiwara, T.; Machida, S.; Kurosaka, D. A case of herpetic epithelial keratitis after triamcinolone acetonide subtenon injection. Cornea 2009, 28, 463–464. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Figueroa, M.; Contreras, I.; Noval, S. Side-effects of triamcinolone in young patients. Arch. Soc. Esp. Oftalmol. 2006, 81, 405–407. [Google Scholar] [PubMed]
- Chew, E.Y.; Glassman, A.R.; Beck, R.W. Ocular side effects associated with peribulbar injections of triamcinolone acetonide for diabetic macular edema. Retina 2011, 31, 284. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Type | Data | Data Source | Dimension |
|---|---|---|---|
| Chemical | Substructures | PubChem | 548 × 881 |
| Biological | Target protein | DrugBank | 548 × 695 |
| Phenotypic | Treatment disease | DrugBank | 548 × 718 |
| Phenotypic | Side effect | SIDER, OFFSIDES | 548 × 1318 (1318 ADR events) |
| Interaction | DDIs | TWOSIDES | 548 × 548 × 1318 (1318 ADR events) |
| Interaction | PPI | HPRD | 9519 × 9519 (37,062 interactions) |
| Link Types | Abbreviated Form | Semantics of Link Types |
|---|---|---|
| Drug-Drug | D-D | Drug-drug interactions |
| Drug-Chemical | D-C | The chemical substructure of a drug |
| Drug-Protein | D-P | The target protein of a drug |
| Protein-Protein | P-P | Protein-protein interactions |
| Drug-Disease | D- | The therapeutic effect between a drug and a disease |
| Drug-Side Effect | D- | The side effect between a drug and a disease |
| Meta-Paths | Abbreviated Form | Semantics of Meta-Paths |
|---|---|---|
| Drug-Drug | DD | Drug-Drug interactions (at the drug embedding stage, interaction types are not considered). |
| Drug-Chemical-Drug | DCD | Two drugs have a similar chemical substructure. |
| Drug-Protein-Drug | DPD | Two drugs have the same target protein. |
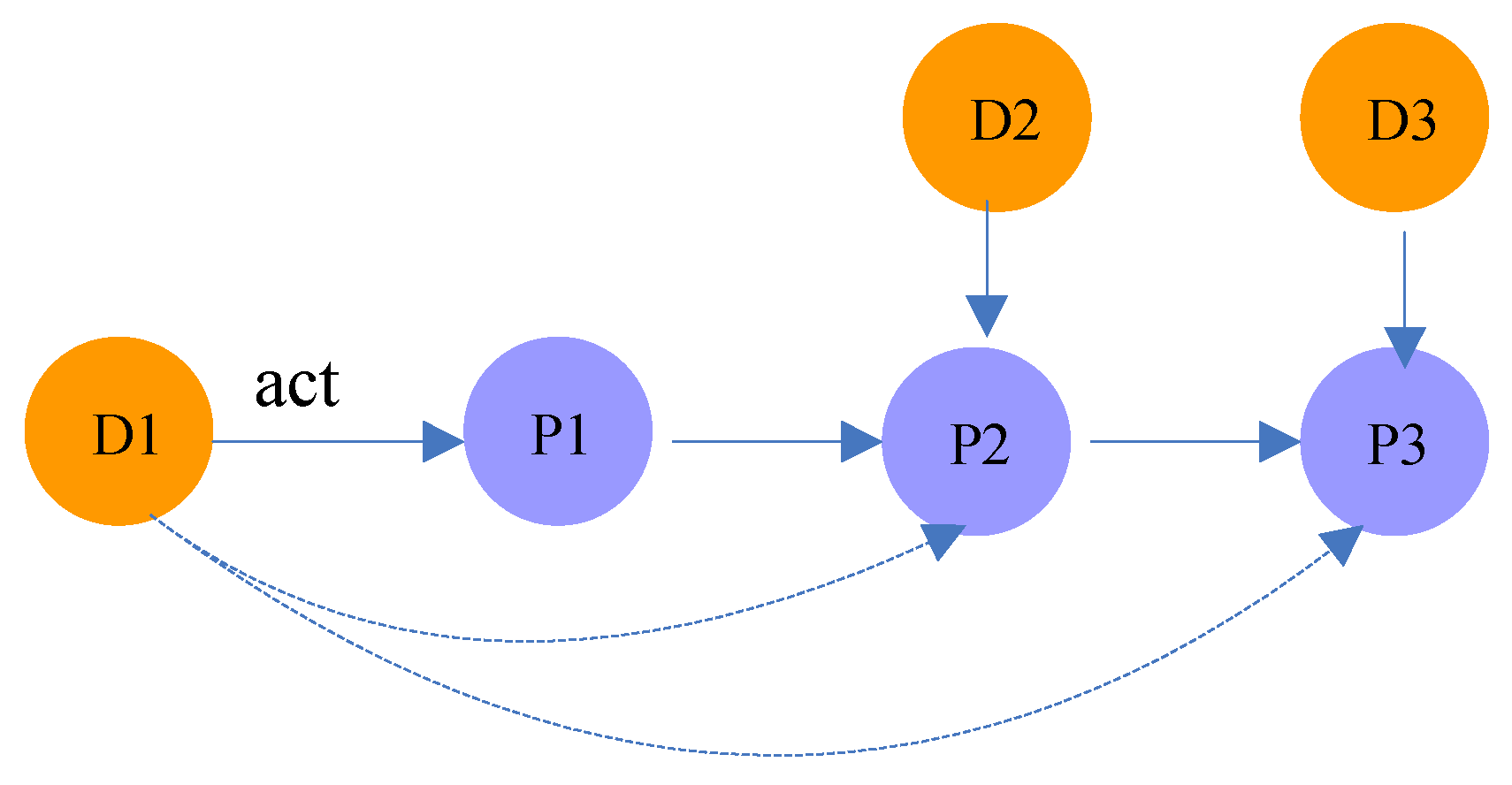
| Drug-Protein-…-Protein-Drug | (n ≥ 2) | There are protein-protein interactions between the targets of two drugs. For example, the path in Figure 4 indicates that the targets of and are and , respectively. Meanwhile, there is an interaction between and (in meta-path , there are protein-protein interactions). |
| Drug-Disease-Drug | Two drugs have the same therapeutic effect. | |
| Drug-Side Effect-Drug | Two drugs have the same side effect. |
| Models | MAP@20 | MAP@50 | MAP@100 | ROC-AUC |
|---|---|---|---|---|
| Concatenate drug features | 0.5590 | 0.5475 | 0.5310 | 0.7820 |
| GraphCNN | 0.6510 | 0.6493 | 0.6321 | 0.8190 |
| metapath2vec++ | 0.5835 | 0.5760 | 0.5628 | 0.7845 |
| SDHINE-no-target propagation | 0.6508 | 0.6416 | 0.6356 | 0.8021 |
| SDHINE | 0.6653 | 0.6479 | 0.6361 | 0.8407 |
| Models | MAP@20 | MAP@50 | MAP@100 | ROC-AUC |
|---|---|---|---|---|
| Concatenate drug features | 0.6122 | 0.5624 | 0.5432 | 0.7409 |
| GraphCNN | 0.6874 | 0.6715 | 0.6219 | 0.7918 |
| metapath2vec++ | 0.6542 | 0.6326 | 0.5986 | 0.7332 |
| SDHINE-no-target propagation | 0.6813 | 0.6718 | 0.6211 | 0.7814 |
| SDHINE | 0.7015 | 0.6854 | 0.6328 | 0.8124 |
| Models | MAP@20 | MAP@50 | MAP@100 | ROC-AUC |
|---|---|---|---|---|
| Concatenate drug features | 0.6596 | 0.6144 | 0.5045 | 0.74322 |
| GraphCNN | 0.6823 | 0.6681 | 0.6137 | 0.7851 |
| metapath2vec++ | 0.6766 | 0.6567 | 0.5118 | 0.7543 |
| SDHINE-no-target propagation | 0.6804 | 0.6622 | 0.6119 | 0.7996 |
| SDHINE | 0.6881 | 0.6745 | 0.6126 | 0.8175 |
| Top K | Side Effect | Confirmation |
|---|---|---|
| K = 1 | headache | yes |
| K = 2 | cough | yes |
| K = 3 | fever | yes |
| K = 4 | eye redness | no |
| K = 5 | sneezing | yes |
| K = 6 | nausea | yes |
| K = 7 | rash | yes |
| K = 8 | fatigue | yes |
| K = 9 | dry skin | no |
| K = 10 | conjunctivitis | yes |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, B.; Wang, H.; Wang, L.; Yuan, W. Adverse Drug Reaction Predictions Using Stacking Deep Heterogeneous Information Network Embedding Approach. Molecules 2018, 23, 3193. https://doi.org/10.3390/molecules23123193
Hu B, Wang H, Wang L, Yuan W. Adverse Drug Reaction Predictions Using Stacking Deep Heterogeneous Information Network Embedding Approach. Molecules. 2018; 23(12):3193. https://doi.org/10.3390/molecules23123193
Chicago/Turabian StyleHu, Baofang, Hong Wang, Lutong Wang, and Weihua Yuan. 2018. "Adverse Drug Reaction Predictions Using Stacking Deep Heterogeneous Information Network Embedding Approach" Molecules 23, no. 12: 3193. https://doi.org/10.3390/molecules23123193
APA StyleHu, B., Wang, H., Wang, L., & Yuan, W. (2018). Adverse Drug Reaction Predictions Using Stacking Deep Heterogeneous Information Network Embedding Approach. Molecules, 23(12), 3193. https://doi.org/10.3390/molecules23123193