Probing the Influence of Linker Length and Flexibility in the Design and Synthesis of New Trehalase Inhibitors

 ,
,  ,
,  , and
, and 
Abstract
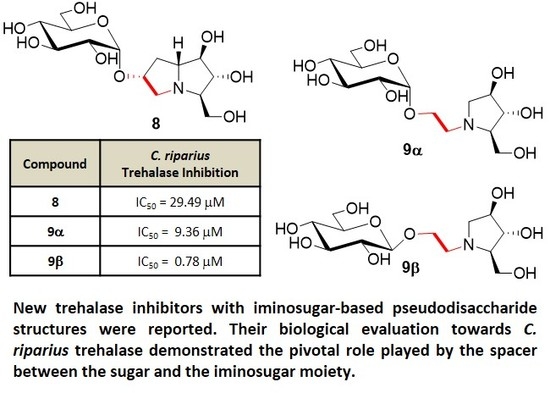
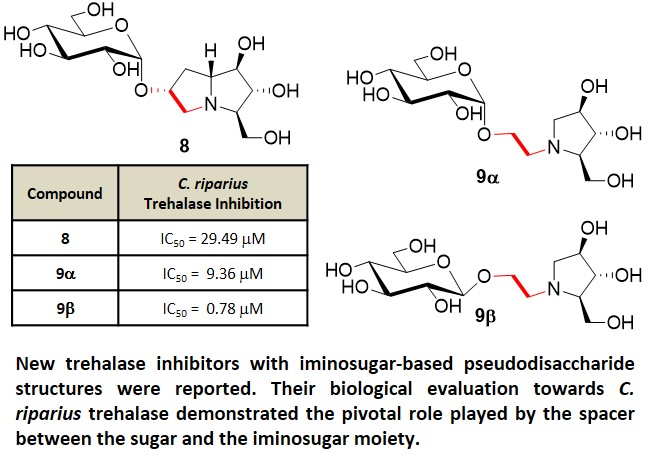
:
1. Introduction
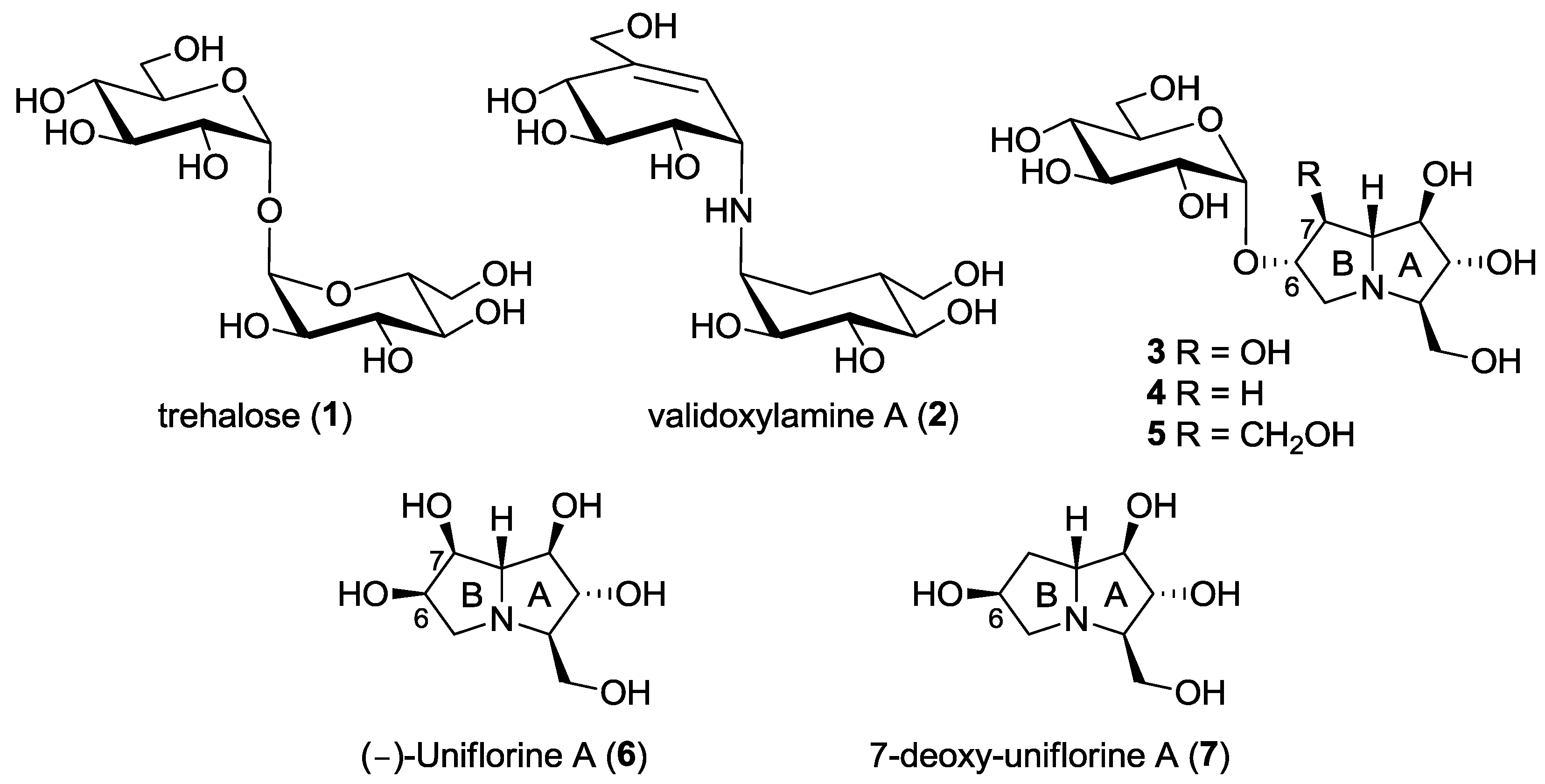
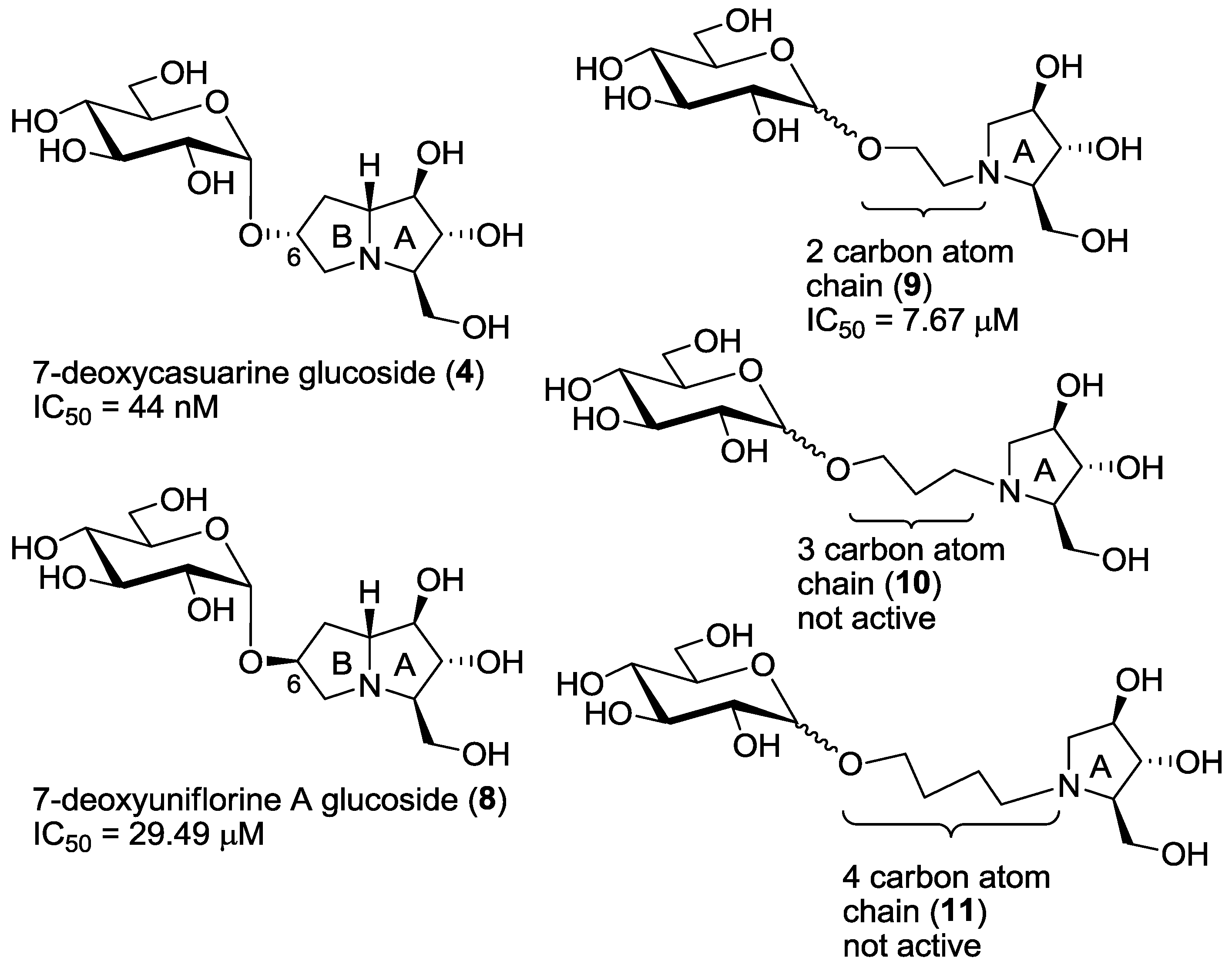
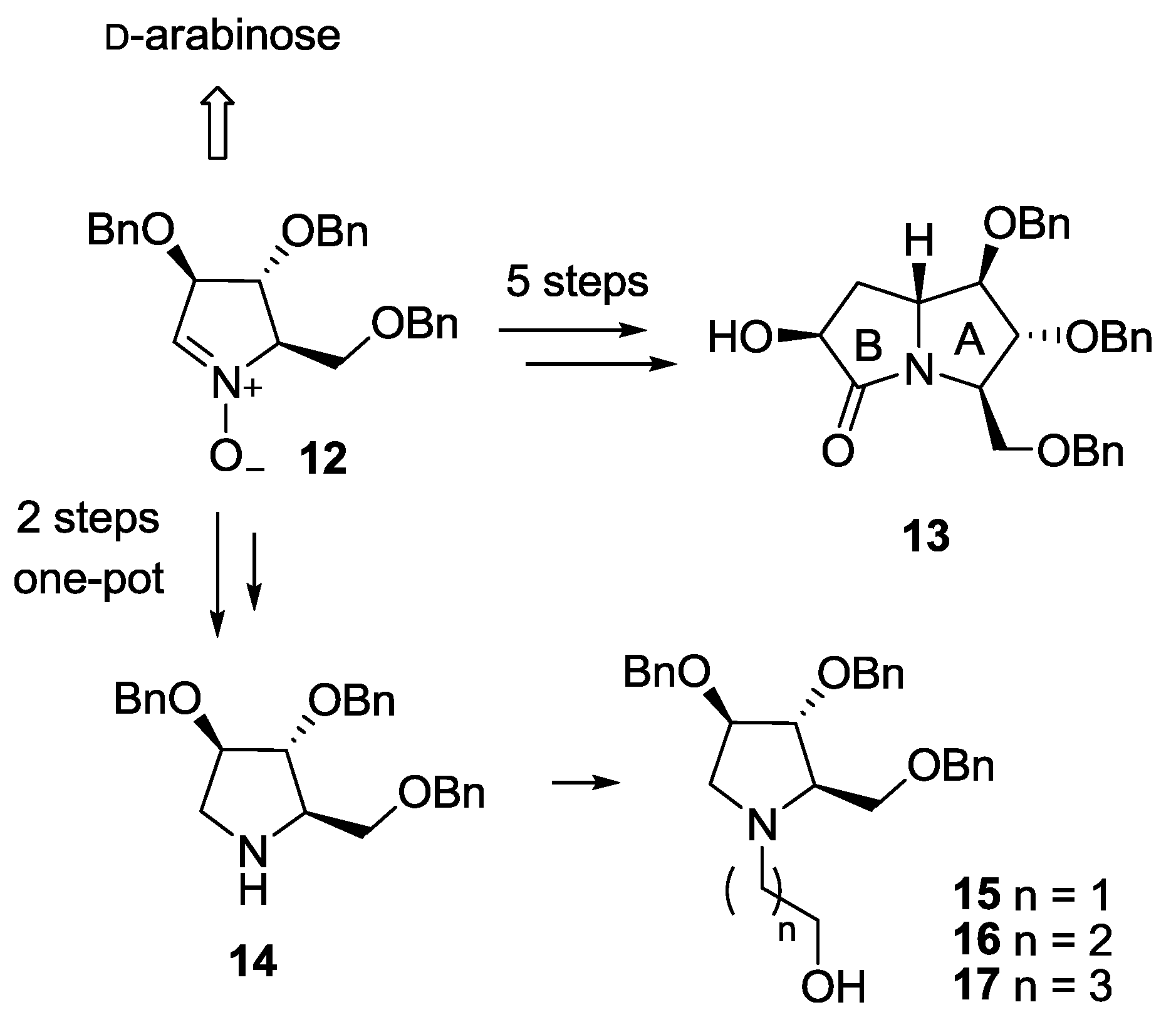
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
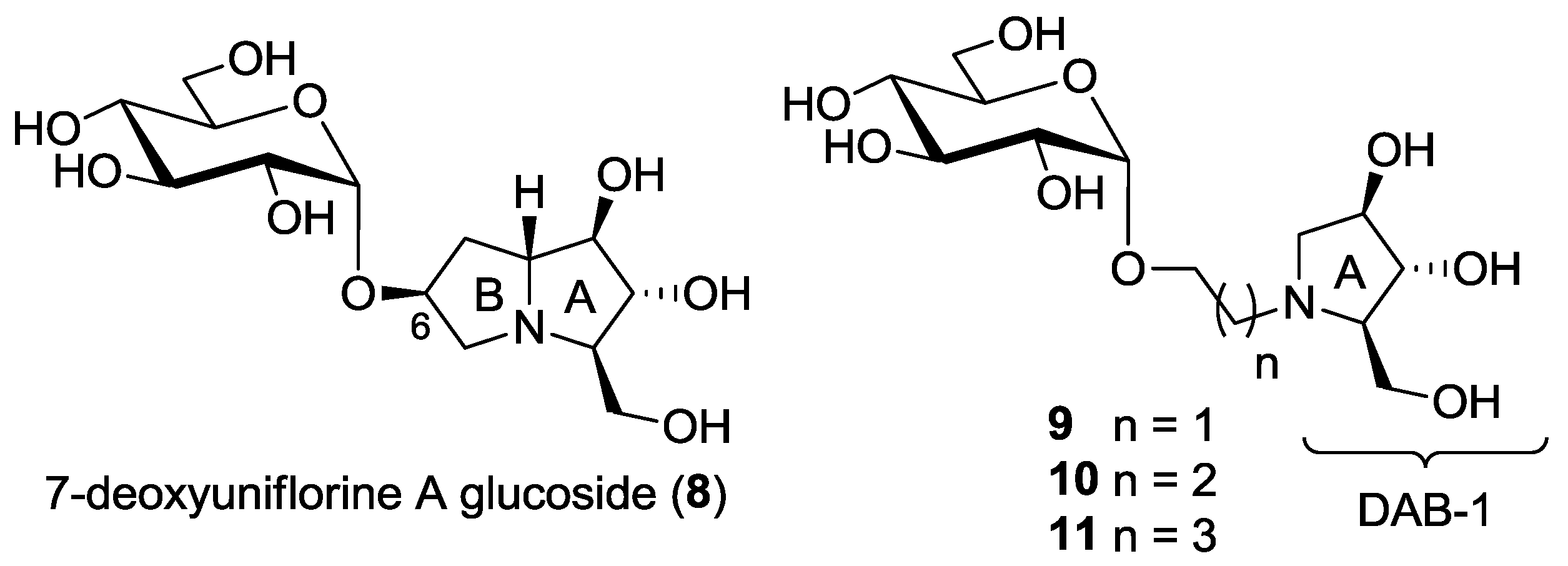
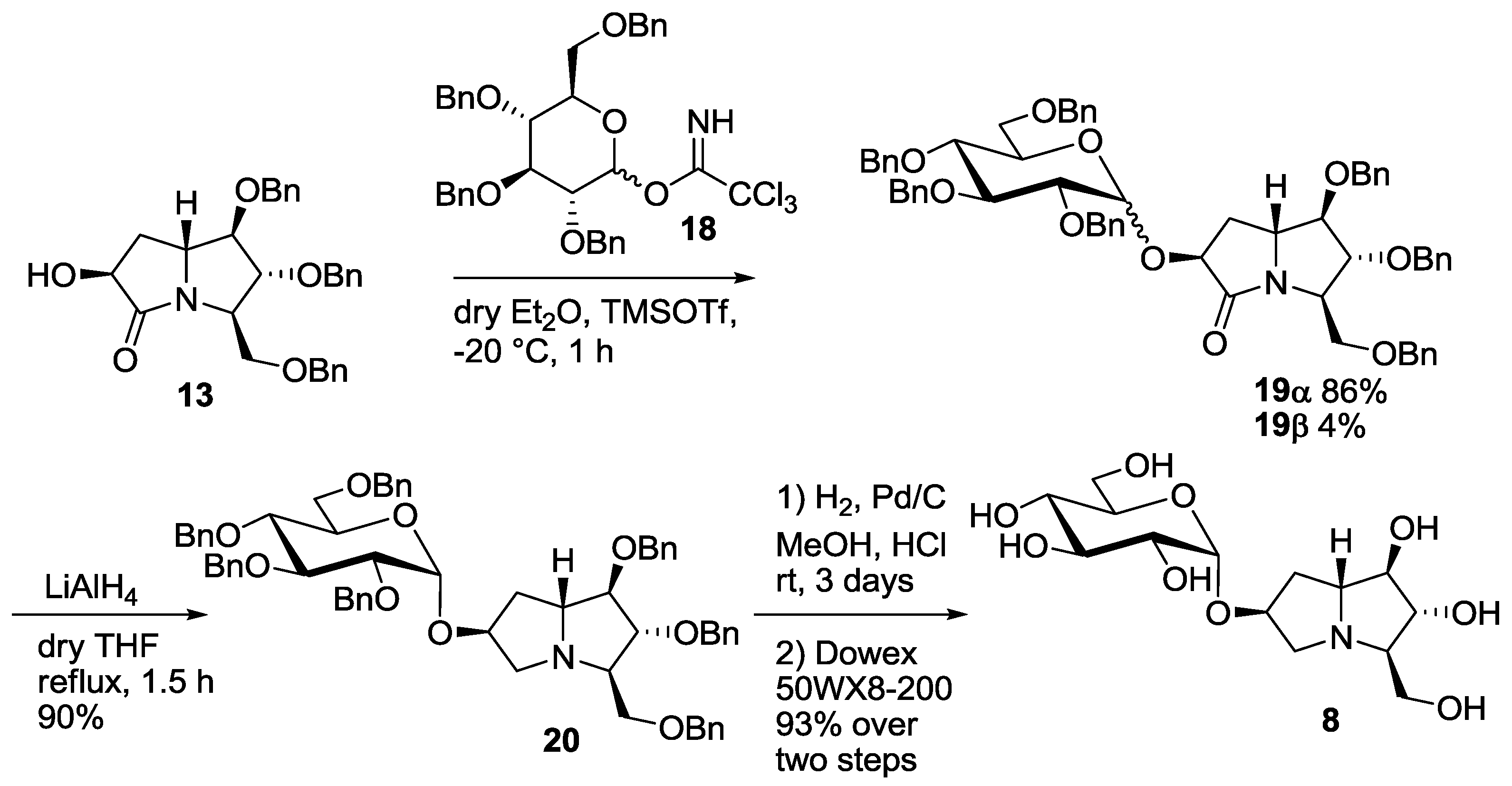
3.2. Synthesis and Purification of 7-Deoxyuniflorine A Glucoside (8)
3.2.1. Synthesis of Compounds 18
3.2.2. Synthesis of Compounds 19
3.2.3. Synthesis of Compound 20
3.2.4. Synthesis of Compound 8
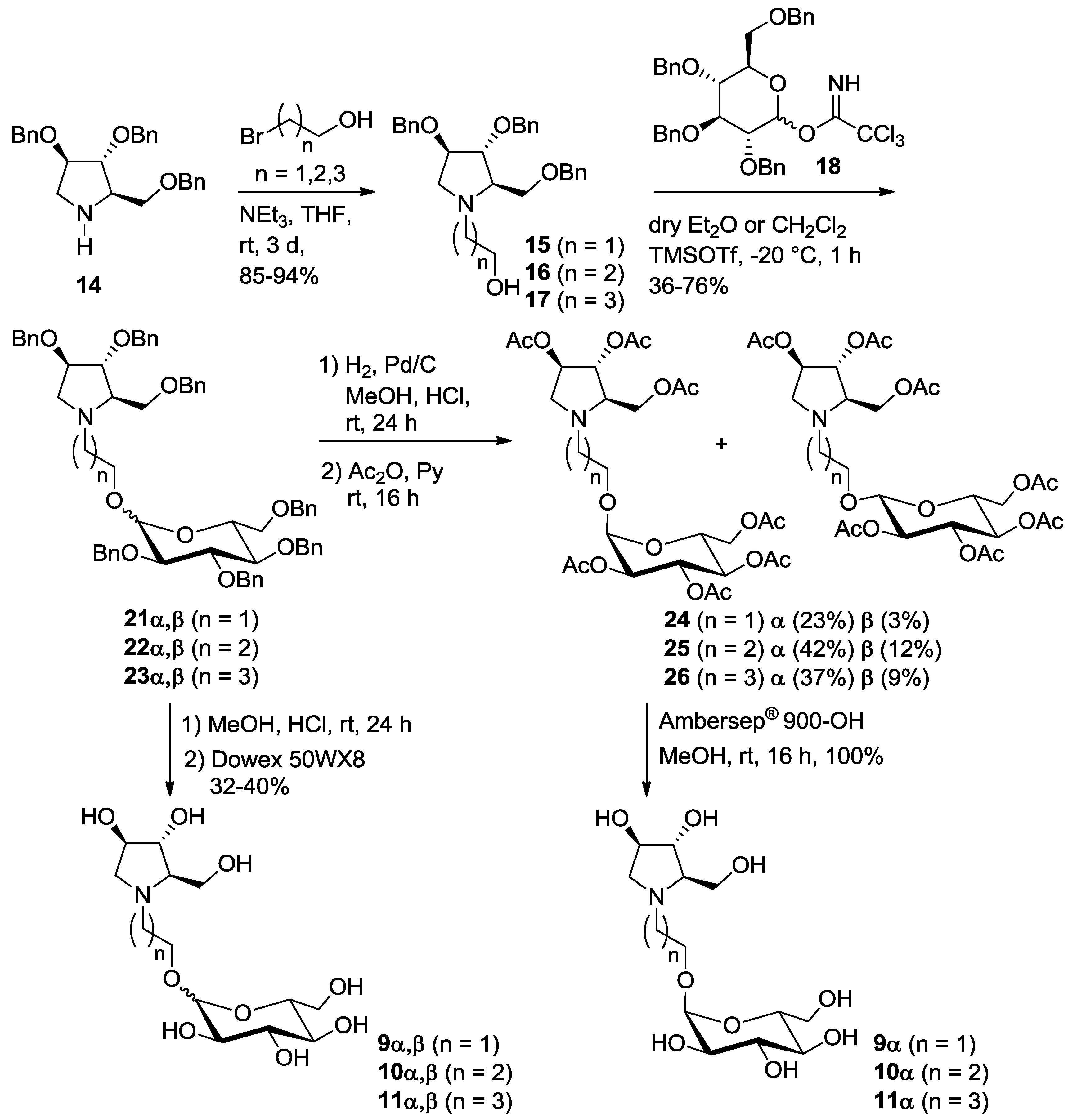
3.3. Synthesis and Purification of Pyrrolidine-based Pseudodisacharides 9α, 10α and 11α
3.3.1. Synthesis of Compound 15
3.3.2. Synthesis of Compound 16
3.3.3. Synthesis of Compound 17
3.3.4. Synthesis of Compounds 2123α/β
3.3.5. Synthesis of Peracetylated Compounds 24–26
3.3.6. Synthesis of Compounds 9–11
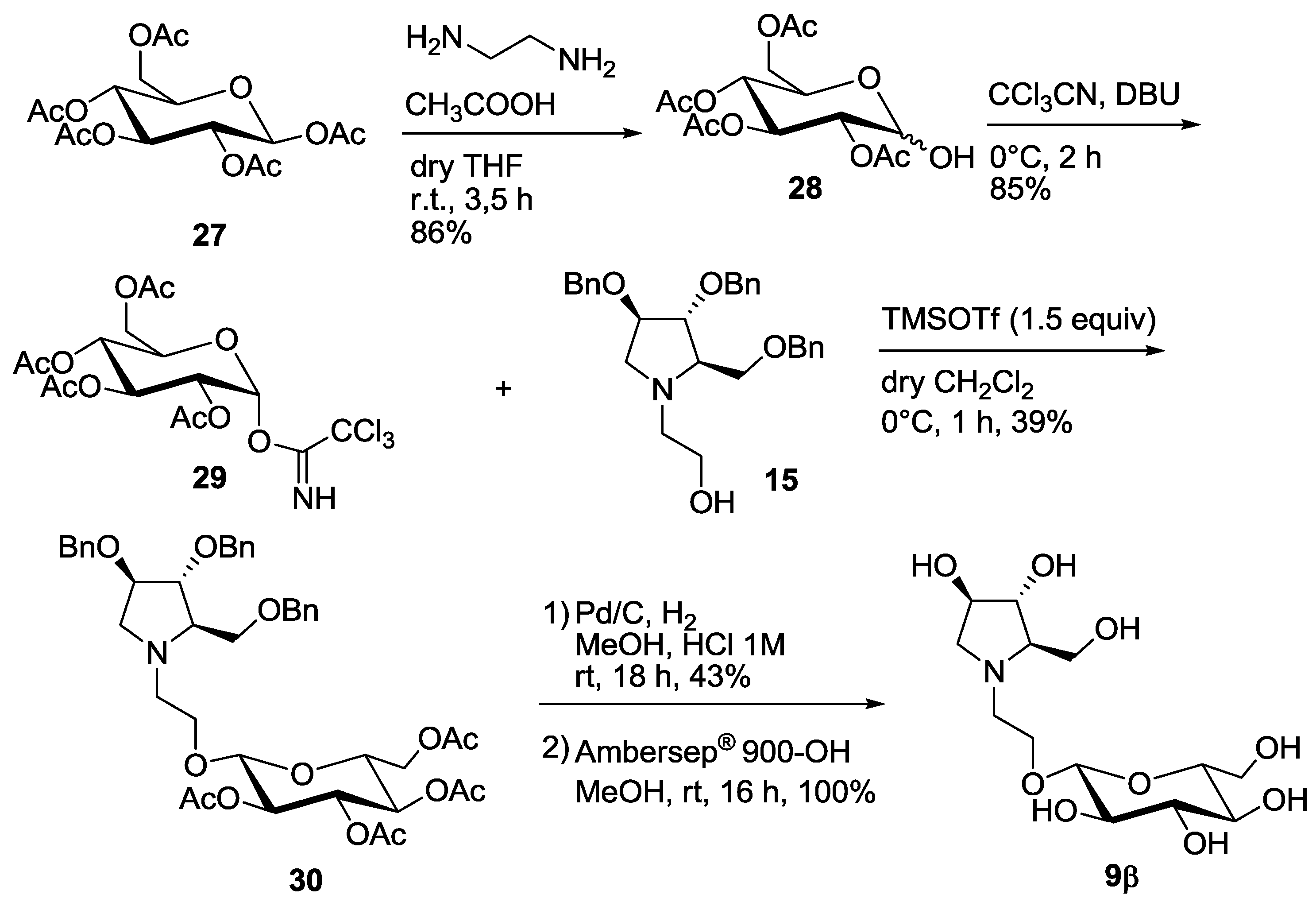
3.4. Synthesis of Compound 9β
3.4.1. Synthesis of 2,3,4,6-Tetra-O-acetyl-α/β-d-glucopyranose 28 [18]
3.4.2. Synthesis of 2,3,4,6-Tetra-O-acetyl-α-d-glucopyranosyltrichloroacetimidate 29 [19]
3.4.3. Synthesis of Compound 30
3.4.4. Synthesis of Compound 9β
3.5. Biological Evaluation of Compounds 8, 9α, 9β, 10α, 11α and the α/β Mixture of 9,10 and 11
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Elbein, A.D.; Pan, Y.T.; Pastuszak, I.; Carroll, D. New insights on trehalose: A multifunctional molecule. Glycobiology 2003, 13, 17R–27R. [Google Scholar] [CrossRef] [PubMed]
- Defaye, J.; Driguez, H.; Henrissat, B.; Bar-Guilloux, E. Stereochemistry of the hydrolysis of α,α-trehalose by trehalase, determined by using a labelled substrate. Carbohydr. Res. 1983, 124, 265–273. [Google Scholar] [CrossRef]
- Bini, D.; Cardona, F.; Gabrielli, L.; Russo, L.; Cipolla, L. Trehalose mimetics as inhibitors of trehalose processing enzymes. In Carbohydrate Chemis Try: Volume 37; Rauter, A.P., Lindhorst, T., Eds.; Royal Society of Chemistry: Cambridge, UK, 2012; pp. 259–302. ISBN 978-1-84973-154-6. [Google Scholar]
- Gibson, R.P.; Gloster, T.M.; Roberts, S.; Warren, R.A.; de Gracia, S.; García, Á.; Chiara, J.L.; Davies, G.J. Molecular basis for trehalase inhibition revealed by the structure of trehalase in complex with potent inhibitors. Angew. Chem. Int. Ed. 2007, 46, 4115–4119. [Google Scholar] [CrossRef] [PubMed]
- Cardona, F.; Parmeggiani, C.; Faggi, E.; Bonaccini, C.; Gratteri, P.; Sim, L.; Gloster, T.M.; Roberts, S.; Davies, G.J.; Rose, D.R.; et al. Total syntheses of casuarine and its 6-O-α-glucoside: Complementary inhibition towards glycoside hydrolases of families GH31 and GH37. Chem. Eur. J. 2009, 15, 1627–1636. [Google Scholar] [CrossRef] [PubMed]
- Cardona, F.; Goti, A.; Parmeggiani, C.; Parenti, P.; Forcella, M.; Fusi, P.; Cipolla, L.; Roberts, S.M.; Davies, G.J.; Gloster, T.M. Casuarine-6-O-α-d-glucoside and its analogues are tight binding inhibitors of insect and bacterial trehalases. Chem. Commun. 2010, 46, 2629–2631. [Google Scholar] [CrossRef]
- D’Adamio, G.; Sgambato, A.; Forcella, M.; Caccia, S.; Parmeggiani, C.; Casartelli, M.; Parenti, P.; Bini, D.; Cipolla, L.; Fusi, P.; et al. New synthetic and biological evaluation of uniflorine A derivatives: Towards specific insect trehalase inhibitors. Org. Biomol. Chem. 2015, 13, 886–892. [Google Scholar] [CrossRef] [PubMed]
- Bini, D.; Forcella, M.; Cipolla, L.; Fusi, P.; Matassini, C.; Cardona, F. Synthesis of novel iminosugar-based trehalase inhibitors by Cross-Metathesis reactions. Eur. J. Org. Chem. 2011, 3995–4000. [Google Scholar] [CrossRef]
- Bini, D.; Cardona, F.; Forcella, M.; Parmeggiani, C.; Parenti, P.; Nicotra, F.; Cipolla, L. Synthesis and biological evaluation of nojirimycin- and pyrrolidine-based trehalase inhibitors. Beilstein J. Org. Chem. 2012, 8, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Cardona, F.; Faggi, E.; Liguori, F.; Cacciarini, M.; Goti, A. Total syntheses of hyacinthacine A2 and 7-deoxycasuarine by cycloaddition to a carbohydrate derived nitrone. Tetrahedron Lett. 2003, 44, 2315–2318. [Google Scholar] [CrossRef]
- Overkleeft, H.S.; van Wiltenburg, J.; Pandit, U.K. A facile transformation of sugar lactones to azasugars. Tetrahedron 1994, 50, 4215–4224. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, W.-J.; Ye, J.-L.; Huang, P.-Q. A versatile approach to pyrrolidine azasugars and homoazasugars based on a highly diastereoselective reductive benzyloxymethylation of protected tartarimide. Tetrahedron 2007, 63, 6346–6357. [Google Scholar] [CrossRef]
- D’Adamio, G.; Matassini, C.; Parmeggiani, C.; Catarzi, S.; Morrone, A.; Goti, A.; Paoli, P.; Cardona, F. Evidence for a Multivalent Effect in Inhibition of Sulfatases Involved in Lysosomal Storage Disorders (LSDs). RSC Adv. 2016, 6, 64847–64851. [Google Scholar] [CrossRef]
- Damager, I.; Olsen, C.E.; Møller, B.L.; Motawia, M.S. Chemical synthesis of 6‴-α-maltotriosyl-maltohexaose as substrate for enzymes in starch biosynthesis and degradation. Carbohydr. Res. 1999, 320, 19–30. [Google Scholar] [CrossRef]
- Stick, R.V.; Williams, S.J. Formation of the glycosidic linkage. In Carbohydrates the Essential Molecules of Life, 2nd ed.; Stick, R.V., Williams, S.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2009; pp. 1133–1202. ISBN 978-0-240-52118-3. [Google Scholar]
- Wang, W.; Kong, F. New Synthetic Methodology for Regio- and Stereoselective Synthesis of Oligosaccharides via Sugar Ortho Ester Intermediates. J. Org. Chem. 1998, 63, 5744–5745. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Yang, Q.; Gong, S.; Fu, Q.; Xiao, Q. Synthesis and enzymatic evaluation of phosphoramidon and its β anomer: Anomerization of α-l-rhamnose triacetate upon phosphitylation. Bioorg. Med. Chem. 2013, 21, 6778–6787. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Lorente, G.; Palomo, J.M.; Cocca, J.; Mateo, C.; Moro, P.; Terreni, M.; Fernandez-Lafuenteb, R.; Guisanb, J.M. Regio-selective deprotection of peracetylated sugars via lipase hydrolysis. Tetrahedron 2003, 59, 5705–5711. [Google Scholar] [CrossRef]
- Koketsu, M.; Kuwahara, M. First Synthesis of a Trisaccharide of Glycosylkaemferide: A Resistance Factor in Carnations. Synth. Commun. 2004, 34, 239–245. [Google Scholar] [CrossRef]
- Forcella, M.; Mozzi, A.; Bigi, A.; Parenti, P.; Fusi, P. Molecular cloning of soluble trehalase from Chironomus riparius larvae, its heterologous expression in Escherichia coli and bioinformatic analysis. Arch. Insect Biochem. Physiol. 2012, 81, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Wegener, G.; Tschiedel, V.; Schlöder, P.; Ando, O.J. The toxic and lethal effects of the trehalase inhibitor trehazolin in locusts are caused by hypoglycaemia. J. Exp. Biol. 2003, 206, 1233–1240. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Compound | C. riparius Trehalase | Porcine Trehalase | Selectivity 2 |
|---|---|---|---|---|
| entry 1 | 4 | 44 ± 1 nM 1 | 479 ± 45 nM 1 | 10 |
| entry 2 | 6 | 177 ± 18 nM 1 | >1 mM 1 | >5649 |
| entry 3 | 7 | 175 ± 12 nM 1 | >1 mM 1 | >5714 |
| entry 4 | 8 | 29.49 ± 7.26 μM | 190.60± 34.15 μM | 6 |
| entry 5 | 9α,β | 2.30 ± 0.13 μM | 7.67 ± 3.91 μM | 3 |
| entry 6 | 9α | 9.36 ± 1.49 μM | 27.64 ± 5.35 μM | 3 |
| entry 7 | 9β | 0.784 ± 0.059 μM | 5.84 ± 0.26 μM | 7 |
| entry 8 | 10α,β | >1000 μM | n.d. 3 | - |
| entry 9 | 10α | >1000 μM | n.d. 3 | - |
| entry 10 | 11α,β | >1000 μM | n.d. 3 | - |
| entry 11 | 11α | >1000 μM | n.d. 3 | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Adamio, G.; Forcella, M.; Fusi, P.; Parenti, P.; Matassini, C.; Ferhati, X.; Vanni, C.; Cardona, F. Probing the Influence of Linker Length and Flexibility in the Design and Synthesis of New Trehalase Inhibitors. Molecules 2018, 23, 436. https://doi.org/10.3390/molecules23020436
D’Adamio G, Forcella M, Fusi P, Parenti P, Matassini C, Ferhati X, Vanni C, Cardona F. Probing the Influence of Linker Length and Flexibility in the Design and Synthesis of New Trehalase Inhibitors. Molecules. 2018; 23(2):436. https://doi.org/10.3390/molecules23020436
Chicago/Turabian StyleD’Adamio, Giampiero, Matilde Forcella, Paola Fusi, Paolo Parenti, Camilla Matassini, Xhenti Ferhati, Costanza Vanni, and Francesca Cardona. 2018. "Probing the Influence of Linker Length and Flexibility in the Design and Synthesis of New Trehalase Inhibitors" Molecules 23, no. 2: 436. https://doi.org/10.3390/molecules23020436
APA StyleD’Adamio, G., Forcella, M., Fusi, P., Parenti, P., Matassini, C., Ferhati, X., Vanni, C., & Cardona, F. (2018). Probing the Influence of Linker Length and Flexibility in the Design and Synthesis of New Trehalase Inhibitors. Molecules, 23(2), 436. https://doi.org/10.3390/molecules23020436