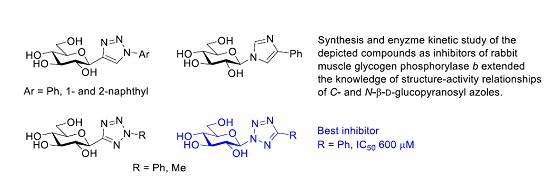
Synthesis of New C- and N-β-d-Glucopyranosyl Derivatives of Imidazole, 1,2,3-Triazole and Tetrazole, and Their Evaluation as Inhibitors of Glycogen Phosphorylase



Abstract
:
1. Introduction
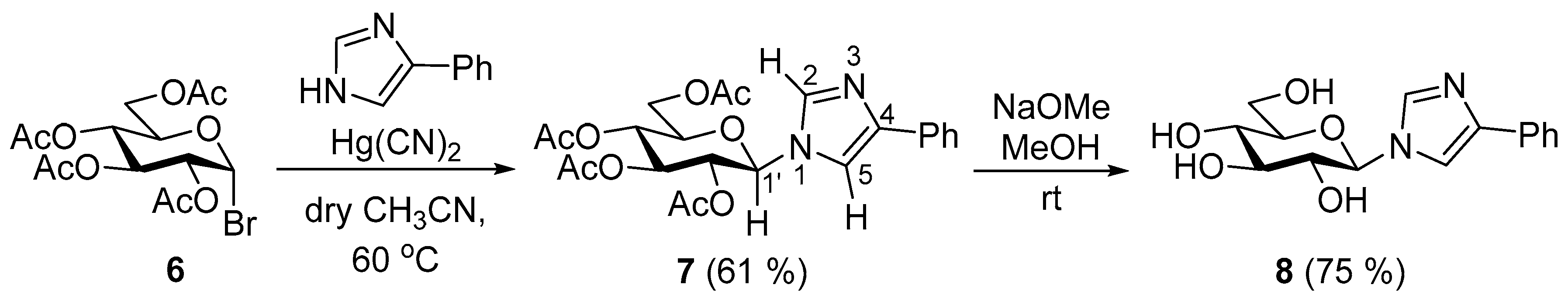
2. Results and Discussion
3. Experimental
3.1. General Methods
3.1.1. General Procedure 1 for the Synthesis of O-Peracetylated or O-Perbenzylated 1-aryl-4-β-d-Glucopyranosyl-1,2,3-triazoles from Azido-Arenes
3.1.2. General Procedure 2 for the Synthesis of O-Peracetylated or O-Perbenzylated 1-aryl-4-β-d-Glucopyranosyl-1,2,3-triazoles from Arylboronic Acids by Using CuSO4/L-Ascorbic Acid Catalytic System
3.1.3. General Procedure 3 for Removal of the O-Acetyl Protecting Groups
3.1.4. General Procedure 4 for the Synthesis of O-Peracetylated N-(β-d-Glucopyranosyl)tetrazoles
3.2. Characterization of the Comounds
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Praly, J.P.; Vidal, S. Inhibition of Glycogen Phosphorylase in the Context of Type 2 Diabetes, with Focus on Recent Inhibitors Bound at the Active Site. Mini-Rev. Med. Chem. 2010, 10, 1102–1126. [Google Scholar] [CrossRef]
- Somsák, L. Glucose derived inhibitors of glycogen phosphorylase. Comptes Rendus Chim. 2011, 14, 211–223. [Google Scholar] [CrossRef]
- Hayes, J.M.; Kantsadi, A.L.; Leonidas, D.D. Natural products and their derivatives as inhibitors of glycogen phosphorylase: Potential treatment for type 2 diabetes. Phytochem. Rev. 2014, 13, 471–498. [Google Scholar] [CrossRef]
- Henke, B.R. Inhibition of glycogen phosphorylase as a strategy for the treatment of type 2 diabetes. RSC Drug Discov. Ser. 2012, 27, 324–365. [Google Scholar] [CrossRef]
- Gaboriaud-Kolar, N.; Skaltsounis, A.L. Glycogen phosphorylase inhibitors: a patent review (2008–2012). Expert Opin. Ther. Patents 2013, 23, 1017–1032. [Google Scholar] [CrossRef] [PubMed]
- Donnier-Maréchal, M.; Vidal, S. Glycogen phosphorylase inhibitors: A patent review (2013–2015). Expert Opin. Ther. Patents 2016, 26, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Tracey, W.; Treadway, J.; Magee, W.; McPherson, R.; Levy, C.; Wilder, D.; Li, Y.; Yue, C.; Zavadoski, W.; Gibbs, E.; et al. A novel glycogen phosphorylase inhibitor, CP-368296, reduces myocardial ischemic injury. Diabetes 2003, 52, A135. [Google Scholar]
- Tracey, W.R.; Treadway, J.L.; Magee, W.P.; Sutt, J.C.; McPherson, R.K.; Levy, C.B.; Wilder, D.E.; Yu, L.J.; Chen, Y.; Shanker, R.M.; et al. Cardioprotective effects of ingliforib, a novel glycogen phosphorylase inhibitor. Am. J. Physiol.-Heart Circul. Physiol. 2004, 286, H1177–H1184. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Sun, H. Pharmacological manipulation of brain glycogenolysis as a therapeutic approach to cerebral ischemia. Mini-Rev. Med. Chem. 2010, 10, 1188–1193. [Google Scholar] [CrossRef] [PubMed]
- Guan, T.; Qian, Y.S.; Tang, X.Z.; Huang, M.H.; Huang, L.F.; Li, Y.M.; Sun, H.B. Maslinic Acid, a Natural Inhibitor of Glycogen Phosphorylase, Reduces Cerebral Ischemic Injury in Hyperglycemic Rats by GLT-1 Up-Regulation. J. Neurosci. Res. 2011, 89, 1829–1839. [Google Scholar] [CrossRef] [PubMed]
- Schnier, J.B.; Nishi, K.; Monks, A.; Gorin, F.A.; Bradbury, E.M. Inhibition of glycogen phosphorylase (GP) by CP-91,149 induces growth inhibition correlating with brain GP expression. Biochem. Biophys. Res. Commun. 2003, 309, 126–134. [Google Scholar] [CrossRef]
- Geschwind, J.F.; Georgiades, C.S.; Ko, Y.H.; Pedersen, P.L. Recently elucidated energy catabolism pathways provide opportunities for novel treatments in hepatocellular carcinoma. Expert Rev. Anticancer Ther. 2004, 4, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Boros, L.G.; Go, V.L.W.; Lee, W.N.P. Glycogen phosphorylase inhibitor CP-320626 inhibits pancreatic cancer cell proliferation via inhibiting pentose cycle metabolism and glycolysis. Pancreas 2003, 27, 368–420. [Google Scholar]
- Favaro, E.; Bensaad, K.; Chong, M.G.; Tennant, D.A.; Ferguson, D.J.P.; Snell, C.; Steers, G.; Turley, H.; Li, J.L.; Günther, U.L.; et al. Glucose Utilization via Glycogen Phosphorylase Sustains Proliferation and Prevents Premature Senescence in Cancer Cells. Cell Metab. 2012, 16, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Zois, C.E.; Favaro, E.; Harris, A.L. Glycogen metabolism in cancer. Biochem. Pharmacol. 2014, 92, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Zois, C.E.; Harris, A.L. Glycogen metabolism has a key role in the cancer microenvironment and provides new targets for cancer therapy. J. Mol. Med. 2016, 94, 137–154. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.H.; Hoover, D.J.; Armento, S.J.; Stock, I.A.; McPherson, R.K.; Danley, D.E.; Stevenson, R.W.; Barrett, E.J.; Treadway, J.L. Discovery of a human liver glycogen phosphorylase inhibitor that lowers blood glucose in vivo. Proc. Natl. Acad. Sci. USA 1998, 95, 1776–1781. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Murakami, K.; Nishikawa, M.; Nakayama, O.; Hino, M. FR258900, a novel glycogen phosphorylase inhibitor isolated from Fungus No. 138354 - II. Anti-hyperglycemic effects in diabetic animal models. J. Antibiot. 2005, 58, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Docsa, T.; Czifrák, K.; Hüse, C.; Somsák, L.; Gergely, P. The effect of glucopyranosylidene-spiro-thiohydantoin on the glycogen metabolism in liver tissues of streptozotocin-induced and obese diabetic rats. Mol. Med. Rep. 2011, 4, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Docsa, T.; Marics, B.; Németh, J.; Hüse, C.; Somsák, L.; Gergely, P.; Peitl, B. Insulin sensitivity is modified by a glycogen phosphorylase inhibitor: glucopyranosylidene-spiro-thiohydantoin in streptozotocin-induced diabetic rats. Curr. Top. Med. Chem. 2015, 15, 2390–2394. [Google Scholar] [CrossRef] [PubMed]
- Goyard, D.; Kónya, B.; Chajistamatiou, A.S.; Chrysina, E.D.; Leroy, J.; Balzarin, S.; Tournier, M.; Tousch, D.; Petit, P.; Duret, C.; et al. Glucose-derived spiro-isoxazolines are anti-hyperglycemic agents against type 2 diabetes through glycogen phosphorylase inhibition. Eur. J. Med. Chem. 2016, 108, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Nagy, L.; Docsa, T.; Brunyánszki, A.; Szántó, M.; Hegedűs, C.; Márton, J.; Kónya, B.; Virág, L.; Somsák, L.; Gergely, P.; et al. Glycogen phosphorylase inhibitor N-(3,5-dimethyl-benzoyl)-N’-(β-d-glucopyranosyl) urea improves glucose tolerance under normoglycemic and diabetic conditions through rearranging hepatic metabolism. PLoS ONE 2013, 8, e69420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, L.; Márton, J.; Vida, A.; Kis, G.; Bokor, É.; Kun, S.; Gönczi, M.; Docsa, T.; Tóth, A.; Antal, M.; et al. Glycogen phosphorylase inhibition improves β-cell function. Br. J. Pharmacol. 2018, 175, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Somsák, L.; Czifrák, K.; Tóth, M.; Bokor, É.; Chrysina, E.D.; Alexacou, K.M.; Hayes, J.M.; Tiraidis, C.; Lazoura, E.; Leonidas, D.D.; et al. New inhibitors of glycogen phosphorylase as potential antidiabetic agents. Curr. Med. Chem. 2008, 15, 2933–2983. [Google Scholar] [CrossRef] [PubMed]
- Stravodimos, G.A.; Chetter, B.A.; Kyriakis, E.; Kantsadi, A.L.; Chatzileontiadou, D.S.M.; Skamnaki, V.T.; Kato, A.; Hayes, J.M.; Leonidas, D.D. Phytogenic Polyphenols as Glycogen Phosphorylase Inhibitors: The Potential of Triterpenes and Flavonoids for Glycaemic Control in Type 2 Diabetes. Curr. Med. Chem. 2017, 24, 384–403. [Google Scholar] [CrossRef]
- Watson, K.A.; Mitchell, E.P.; Johnson, L.N.; Cruciani, G.; Son, J.C.; Bichard, C.J.F.; Fleet, G.W.J.; Oikonomakos, N.G.; Kontou, M.; Zographos, S.E. Glucose Analogue Inhibitors of Glycogen Phosphorylase: from Crystallographic Analysis to Drug Prediction using GRID Force-Field and GOLPE Variable Selection. Acta Cryst. 1995, D51, 458–472. [Google Scholar] [CrossRef] [PubMed]
- Somsák, L.; Kovács, L.; Tóth, M.; Ősz, E.; Szilágyi, L.; Györgydeák, Z.; Dinya, Z.; Docsa, T.; Tóth, B.; Gergely, P. Synthesis of and a Comparative Study on the Inhibition of Muscle and Liver Glycogen Phosphorylases by Epimeric Pairs of d-Gluco- and d-Xylopyranosylidene-spiro-(thio)hydantoins and N-(d-Glucopyranosyl) Amides. J. Med. Chem. 2001, 44, 2843–2848. [Google Scholar] [CrossRef] [PubMed]
- Györgydeák, Z.; Hadady, Z.; Felföldi, N.; Krakomperger, A.; Nagy, V.; Tóth, M.; Brunyánszky, A.; Docsa, T.; Gergely, P.; Somsák, L. Synthesis of N-(β-d-glucopyranosyl)- and N-(2-acetamido-2-deoxy-β-d-glucopyranosyl) amides as inhibitors of glycogen phosphorylase. Bioorg. Med. Chem. 2004, 12, 4861–4870. [Google Scholar] [CrossRef] [PubMed]
- Chrysina, E.D.; Bokor, É.; Alexacou, K.-M.; Charavgi, M.-D.; Oikonomakos, G.N.; Zographos, S.E.; Leonidas, D.D.; Oikonomakos, N.G.; Somsák, L. Amide-1,2,3-triazole bioisosterism: The glycogen phosphorylase case. Tetrahedron: Asymmetry 2009, 20, 733–740. [Google Scholar] [CrossRef]
- Chrysina, E.D. The Prototype of Glycogen Phosphorylase. Mini-Rev. Med. Chem. 2010, 10, 1093–1101. [Google Scholar] [CrossRef]
- Patani, G.A.; LaVoie, E.J. Bioisosterism: A rational approach in drug design. Chem. Rev. 1996, 96, 3147–3176. [Google Scholar] [CrossRef] [PubMed]
- Lima, L.M.A.; Barreiro, E.J. Bioisosterism: A useful strategy for molecular modification and drug design. Curr. Med. Chem. 2005, 12, 23–49. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A. Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design. J. Med. Chem. 2011, 54, 2529–2591. [Google Scholar] [CrossRef] [PubMed]
- Bokor, É.; Docsa, T.; Gergely, P.; Somsák, L. Synthesis of 1-(d-glucopyranosyl)-1,2,3-triazoles and their evaluation as glycogen phosphorylase inhibitors. Bioorg. Med. Chem. 2010, 18, 1171–1180. [Google Scholar] [CrossRef] [PubMed]
- Hadady, Z.; Tóth, M.; Somsák, L. C-(β-d-glucopyranosyl) heterocycles as potential glycogen phosphorylase inhibitors. Arkivoc 2004, 140–149. [Google Scholar] [CrossRef]
- Chrysina, E.D.; Kosmopolou, M.N.; Tiraidis, C.; Kardarakis, R.; Bischler, N.; Leonidas, D.D.; Hadady, Z.; Somsák, L.; Docsa, T.; Gergely, P.; et al. Kinetic and crystallographic studies on 2-(β-d-glucopyranosyl)-5-methyl-1,3,4-oxadiazole, -benzothiazole, and -benzimidazole, inhibitors of muscle glycogen phosphorylase b. Evidence for a new binding site. Protein Sci. 2005, 14, 873–888. [Google Scholar] [CrossRef] [PubMed]
- Tóth, M.; Kun, S.; Bokor, É.; Benltifa, M.; Tallec, G.; Vidal, S.; Docsa, T.; Gergely, P.; Somsák, L.; Praly, J.P. Synthesis and structure-activity relationships of C-glycosylated oxadiazoles as inhibitors of glycogen phosphorylase. Bioorg. Med. Chem. 2009, 17, 4773–4785. [Google Scholar] [CrossRef] [PubMed]
- Benltifa, M.; Vidal, S.; Fenet, B.; Msaddek, M.; Goekjian, P.G.; Praly, J.P.; Brunyánszki, A.; Docsa, T.; Gergely, P. In the Search of Glycogen Phosphorylase Inhibitors: 5-Substituted 3-C-Glucopyranosyl-1,2,4-Oxadiazoles from β-d-Glucopyranosyl Cyanides upon Cyclization of O-Acyl-amidoxime Intermediates. Eur. J. Org. Chem. 2006, 4242–4256. [Google Scholar] [CrossRef]
- Benltifa, M.; Vidal, S.; Gueyrard, D.; Goekjian, P.G.; Msaddek, M.; Praly, J.P. 1,3-Dipolar cycloaddition reactions on carbohydrate-based templates: synthesis of spiro-isoxazolines and 1,2,4-oxadiazoles as glycogen phosphorylase inhibitors. Tetrahedron Lett. 2006, 47, 6143–6147. [Google Scholar] [CrossRef]
- Bokor, É.; Docsa, T.; Gergely, P.; Somsák, L. C-Glucopyranosyl-1,2,4-triazoles as new potent inhibitors of glycogen phosphorylase. ACS Med. Chem. Lett. 2013, 4, 612–615. [Google Scholar] [CrossRef] [PubMed]
- Kun, S.; Bokor, É.; Varga, G.; Szőcs, B.; Páhi, A.; Czifrák, K.; Tóth, M.; Juhász, L.; Docsa, T.; Gergely, P.; et al. New synthesis of 3-(β-d-glucopyranosyl)-5-substituted-1,2,4-triazoles, nanomolar inhibitors of glycogen phosphorylase. Eur. J. Med. Chem. 2014, 76, 567–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokor, É.; Kun, S.; Docsa, T.; Gergely, P.; Somsák, L. 4(5)-Aryl-2-C-glucopyranosyl-imidazoles as new nanomolar glucose analog inhibitors of glycogen phosphorylase. ACS Med. Chem. Lett. 2015, 6, 1215–1219. [Google Scholar] [CrossRef] [PubMed]
- Kantsadi, A.L.; Bokor, É.; Kun, S.; Stravodimos, G.A.; Chatzileontiadou, D.S.M.; Leonidas, D.D.; Juhász-Tóth, É.; Szakács, A.; Batta, G.; Docsa, T.; et al. Synthetic, enzyme kinetic, and protein crystallographic studies of C-β-d-glucopyranosyl pyrroles and imidazoles reveal and explain low nanomolar inhibition of human liver glycogen phosphorylase. Eur. J. Med. Chem. 2016, 123, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Bokor, É.; Kun, S.; Goyard, D.; Tóth, M.; Praly, J.P.; Vidal, S.; Somsák, L. C-Glycopyranosyl arenes and hetarenes: Synthetic methods and bioactivity focused on antidiabetic potential. Chem. Rev. 2017, 117, 1687–1764. [Google Scholar] [CrossRef] [PubMed]
- Dondoni, A.; Mariotti, G.; Marra, A. Synthesis of α- and β-Glycosyl Asparagine Ethylene Isosteres (C-Glycosyl Asparagines) via Sugar Acetylenes and Garner Aldehyde Coupling. J. Org. Chem. 2002, 67, 4475–4486. [Google Scholar] [CrossRef] [PubMed]
- Meldal, M.; Tornoe, C.W. Cu-catalyzed azide-alkyne cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef] [PubMed]
- Bokor, É.; Koppány, C.; Gonda, Z.; Novák, Z.; Somsák, L. Evaluation of bis-triphenylphosphano-copper(I)-butyrate (C3H7COOCu(PPh3)2) as catalyst for the synthesis of 1-(d-glycopyranosyl)-4-substituted-1,2,3-triazoles. Carbohydr. Res. 2012, 351, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Tao, C.Z.; Cui, X.; Li, J.; Liu, A.X.; Liu, L.; Guo, Q.X. Copper-catalyzed synthesis of aryl azides and 1-aryl-1,2,3-triazoles from boronic acids. Tetrahedron Lett. 2007, 48, 3525–3529. [Google Scholar] [CrossRef]
- Kun, S.; Nagy, G.Z.; Tóth, M.; Czecze, L.; Nguyen van Nhien, A.; Docsa, T.; Gergely, P.; Charavgi, M.D.; Skourti, P.V.; Chrysina, E.D.; et al. Synthesis of variously coupled conjugates of d-glucose, 1,3,4-oxadiazole, and 1,2,3-triazole for inhibition of glycogen phosphorylase. Carbohydr. Res. 2011, 346, 1427–1438. [Google Scholar] [CrossRef] [PubMed]
- Kantsadi, A.L.; Stravodimos, G.A.; Kyriakis, E.; Chatzileontiadou, D.S.; Solovou, T.G.; Kun, S.; Bokor, É.; Somsák, L.; Leonidas, D.D. van der Waals interactions govern C-β-d-glucopyranosyl triazoles’ nM inhibitory potency in human liver glycogen phosphorylase. J. Struct. Biol. 2017, 199, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Alzeer, J.; Vasella, A. Oligosaccharide Analogues of Polysaccharides. Part 2. Regioselective deprotection of monosaccharide-derived monomers and dimers. Helv. Chim. Acta 1995, 78, 177–193. [Google Scholar] [CrossRef]
- Patt, M.; Sorger, D.; Scheunemann, M.; Stocklin, G. Adduct of 2-[F18]FDG and 2-nitroimidazole as a putative radiotracer for the detection of hypoxia with PET: Synthesis, in vitro- and in vivo-characterisation. Appl. Radiat. Isot. 2002, 57, 705–712. [Google Scholar] [CrossRef]
- Onaka, T.; Umemoto, H.; Miki, Y.; Nakamura, A.; Maegawa, T. [Cu(OH)(TMEDA)]2Cl2-Catalyzed Regioselective 2-Arylation of 5-Substituted Tetrazoles with Boronic Acids under Mild Conditions. J. Org. Chem. 2014, 79, 6703–6707. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Tanaka, Y.; Kakehi, A.; Kondo, K. Facile synthesis of 2,5-disubstituted tetrazoles by reaction of phenylsulfonylhydrazones with arenediazonium salts. Bull. Chem. Soc. Jpn. 1976, 49, 1920–1923. [Google Scholar] [CrossRef]
- Tóth, M.; Kövér, K.E.; Bényei, A.; Somsák, L. C-Glycosylmethylene carbenes: synthesis of anhydro-aldose tosylhydrazones as precursors; generation and a new synthetic route to exo-glycals. Org. Biomol. Chem. 2003, 1, 4039–4046. [Google Scholar] [CrossRef] [PubMed]
- Tóth, M.; Somsák, L.; Goyard, D. Preparation of 2,6-Anhydro-aldose-tosylhydrazones. In Carbohydrate Chemistry: Proven Synthetic Methods; Kováč, P., Ed.; CRC Press: Boca Raton, FL, USA, 2012; Volume 1, pp. 355–365. [Google Scholar]
- Roe, A. Preparation of Aromatic Fluorine Compounds from Diazonium Fluoborates. In Organic Reactions; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 193–228. [Google Scholar]
- Flood, D.T. Fluorobenzene. Org. Synth. 1933, 13, 46–49. [Google Scholar] [CrossRef]
- Kobe, J.; Prhavc, M.; Hohnjec, M.; Townsend, L.B. Preparation and utility of 5-β-d-ribofuranosyl-1H-tetrazole as a key synthon for C-nucleoside synthesis. Nucleosides Nucleotides 1994, 13, 2209–2244. [Google Scholar] [CrossRef]
- Ostrovskii, V.A.; Koldobskii, G.I.; Trifonov, R.E. Tetrazoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A.R., Ramsden, C.A., Scriven, E.F.V., Taylor, R.J.K., Eds.; Elsevier: Oxford, UK, 2008; Volume 6, pp. 257–423. [Google Scholar]
- Couri, M.R.; Luduvico, I.; Santos, L.; Alves, R.; Prado, M.A.; Gil, R.F. Microwave-assisted efficient preparation of novel carbohydrate tetrazole derivatives. Carbohydr. Res. 2007, 342, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Wittenberger, S.J.; Donner, B.G. Dialkyltin Oxide-Mediated Addition of Trimethylsilyl Azide to Nitriles—A Novel Preparation of 5-Substituted Tetrazoles. J. Org. Chem. 1993, 58, 4139–4141. [Google Scholar] [CrossRef]
- Valverde, I.E.; Mindt, T.L. 1,2,3-Triazoles as Amide-bond Surrogates in Peptidomimetics. Chimia 2013, 67, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Bonandi, E.; Fumagalli, G.; Perdicchia, D.; Christodoulou, M.S.; Rastelli, G.; Passarella, D. The 1,2,3-triazole ring as a bioisostere in medicinal chemistry. Drug Discov. Today 2017, 22, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Watson, K.A.; Mitchell, E.P.; Johnson, L.N.; Son, J.C.; Bichard, C.J.F.; Orchard, M.G.; Fleet, G.W.J.; Oikonomakos, N.G.; Leonidas, D.D.; Kontou, M.; et al. Design of Inhibitors of Glycogen Phosphorylase: A Study of α- and β-C-Glucosides and 1-Thio-β-d-glucose Compounds. Biochemistry 1994, 33, 5745–5758. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.C.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef] [PubMed]
- Udodong, U.E.; Fraser-Reid, B. Formal total synthesis of 1-β-methylcarbapenem via a novel route to deoxyamino sugars. J. Org. Chem. 1989, 54, 2103–2112. [Google Scholar] [CrossRef]
Sample Availability: Not available. |


{kind=link}
{kind=link}
| R | CH3 |  |  | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| a | b | c | ||||||||
| I |  | 32 [26] | 81 [26] 144 [27] | 10 [28] 13 [29] | ||||||
| II |  | - | 151 [29] 162 [34] | 16 [29] 36 [34] | ||||||
| III |  | 212 [35] 145 [36] | 10% at 625 μM [37] | 10% at 625 μM [37] | ||||||
| IV |  | No inh. at 625 μM [38] | 10% at 625 μM [38] | 38 [38] | ||||||
| V |  | - | 27 [39] 64 [37] | 12 * [37] | ||||||
| VI |  | 499 [41] | 7 [40,41] | 0.41 [40,41] | ||||||
| VII |  | - | 0.28 [42,43] | 0.031 [42,43] | ||||||
| VIII |  | No inh. at 625 μM [35] | ||||||||
| * A Ki value of 2.4 μM was measured by N.G. Oikonomakos et al. (unpublished results in ref. [37]) | ||||||||||
| Target compounds in this study: |  | |||||||||
| IX |  | X |  | XI |  | XII |  | |||
 | |||||||
| Reagents and conditions: (a) ArN3, CuO(CO)C3H7(PPh3)2, dry CH2Cl2, r.t.; (b) і. ArB(OH)2, NaN3, CuSO4·5H2O, MeOH, r.t., іі. 1 or 3, L-ascorbic acid, CH2Cl2-H2O (1:1), 50 °C; (c) H2, Pd(C), dry EtOH, dry EtOAc, r.t.; (d) TMSOTf, Ac2O, −40 °C; (e) і. H2, Pd(C), dry EtOAc, dry MeOH, 40 °C, іі. Ac2O, pyridine, 90 °C; (f) ~1M NaOMe in MeOH, r.t. | |||||||
| Ar | Conditions and Yields (%) | ||||||
| 2 | 4 | 5 | |||||
| a |  | a | 78 (from 1) | - | - | c | 92 (from 2a) |
| b |  | b | 79 (from 1) | d | 68 (from 2b) | f | 96 (from 4b) |
| b | 80 (from 3) | ||||||
| c |  | a | 85 (from 1) | e | 29 (from 2c) | f | 94 (from 4c) |
| a | 91 (from 3) | ||||||
| d |  | - | - | e | 3 (from 2c) | - | - |
 | |||||||||||
| Reagents and conditions: (a) R’B(OH)2, CuCl2, TMEDA, K2CO3, dry CH2Cl2, r.t.; (b) CH2N2 in Et2O, dry CH2Cl2, r.t.; (c) PhN2BF4, dry pyridine, −40 °C; (d) ~1M NaOMe in MeOH, r.t. | |||||||||||
| Conditions, Yields (%) and Chemical Shifts (ppm) for Tetrazole C-5 (Solvent) | |||||||||||
| R’ | 10 | 11 | 13 | 14 | |||||||
| a | Phenyl | a | 95 | 162.2 (CDCl3) | - | - | d | 94 | 164.8 (DMSO-d6) | - | - |
| c | 61 | - | |||||||||
| e | Methyl | b | 38 | 162.1 (CDCl3) | 38 | 149.9 (CDCl3) | d | 72 | 163.9 (D2O) | 97 | 153.9 (D2O) |
 | ||||||||||||
| Reagents and conditions: (a) K2CO3, 4 Å molecular sieves, dry acetone, reflux; (b) ~1 M NaOMe in MeOH, r.t. | ||||||||||||
| R’ | Conditions, Yields (%) and Chemical Shifts (ppm) for Tetrazole C-5 (Solvent) | |||||||||||
| 15 | 16 | 17 | 18 | 19 | ||||||||
| a | Phenyl | a | 79 | 165.8 (CDCl3) | 17 | 155.9 (CDCl3) | - | b | 85 | 165.9 (D2O) | 86 | 157.3 (D2O) |
| e | Methyl | a | 26 | 163.9 (CDCl3) | - | 45 | b | 84 | 164.7 (D2O) | - | ||
| Entry | Compound | Inhibition * (μM) | |
|---|---|---|---|
| 1. | Ib |  | Ki 81 [26] Ki 144 [27] |
| 2. | IIb |  | Ki 151 [29] Ki 162 [34] |
| 3. | 20 |  | Ki 5400 [65] |
| 4. | 5a |  | N.I. |
| 5. | 5b |  | N.I. |
| 6. | 5c |  | N.I. |
| 7. | 8 |  | N.I. |
| 8. | 13a |  | N.I. |
| 9. | 13e |  | N.I. |
| 10. | 14e |  | N.I. |
| 11. | 18a |  | IC50 600 (calculated ** Ki 327) |
| 12. | 18e |  | N.I. |
| 13. | 19a |  | N.I. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kun, S.; Bokor, É.; Sipos, Á.; Docsa, T.; Somsák, L. Synthesis of New C- and N-β-d-Glucopyranosyl Derivatives of Imidazole, 1,2,3-Triazole and Tetrazole, and Their Evaluation as Inhibitors of Glycogen Phosphorylase. Molecules 2018, 23, 666. https://doi.org/10.3390/molecules23030666
Kun S, Bokor É, Sipos Á, Docsa T, Somsák L. Synthesis of New C- and N-β-d-Glucopyranosyl Derivatives of Imidazole, 1,2,3-Triazole and Tetrazole, and Their Evaluation as Inhibitors of Glycogen Phosphorylase. Molecules. 2018; 23(3):666. https://doi.org/10.3390/molecules23030666
Chicago/Turabian StyleKun, Sándor, Éva Bokor, Ádám Sipos, Tibor Docsa, and László Somsák. 2018. "Synthesis of New C- and N-β-d-Glucopyranosyl Derivatives of Imidazole, 1,2,3-Triazole and Tetrazole, and Their Evaluation as Inhibitors of Glycogen Phosphorylase" Molecules 23, no. 3: 666. https://doi.org/10.3390/molecules23030666
APA StyleKun, S., Bokor, É., Sipos, Á., Docsa, T., & Somsák, L. (2018). Synthesis of New C- and N-β-d-Glucopyranosyl Derivatives of Imidazole, 1,2,3-Triazole and Tetrazole, and Their Evaluation as Inhibitors of Glycogen Phosphorylase. Molecules, 23(3), 666. https://doi.org/10.3390/molecules23030666