Diabetes Drug Discovery: hIAPP1–37 Polymorphic Amyloid Structures as Novel Therapeutic Targets


Abstract
:1. Introduction
2. Results
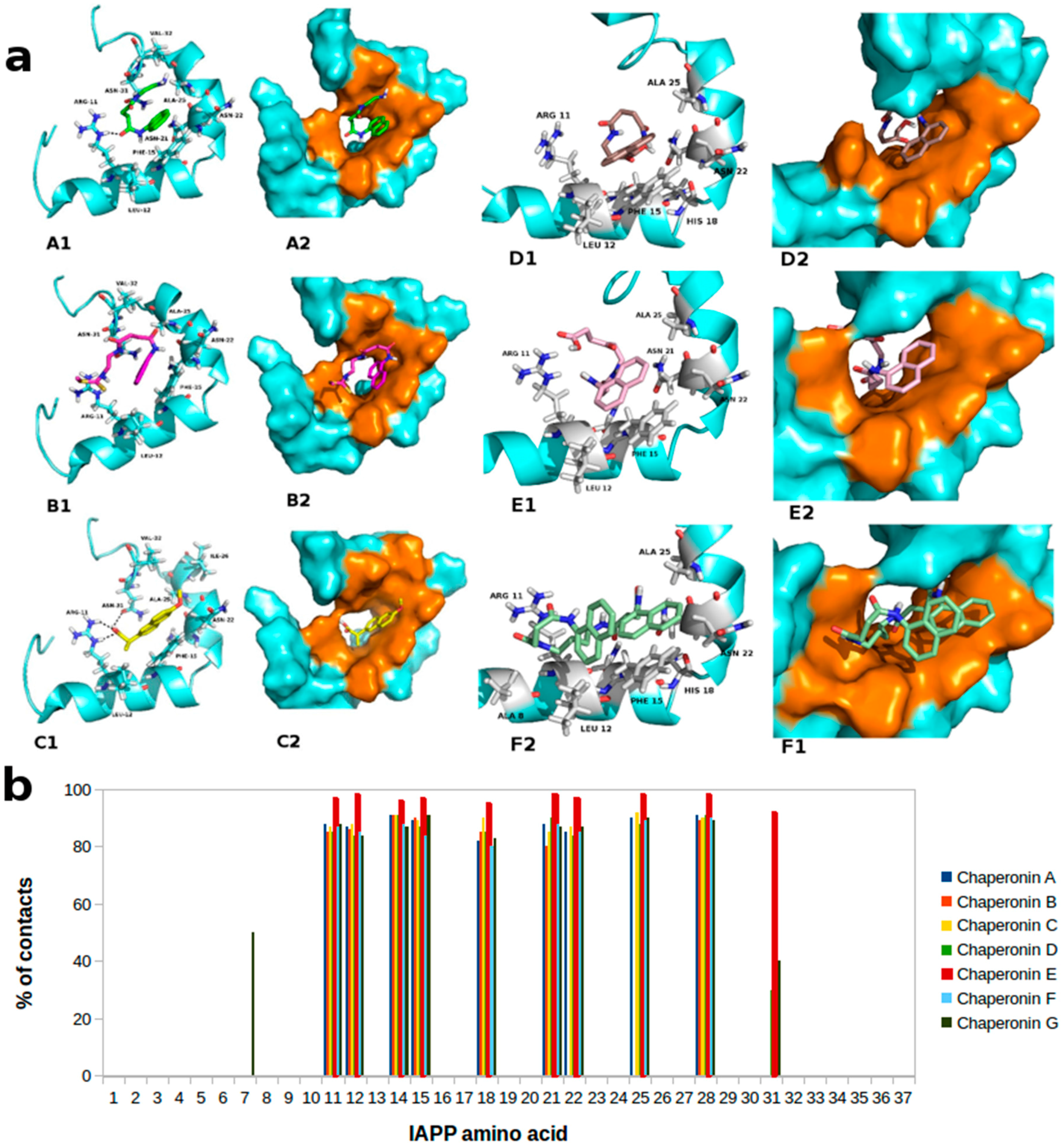
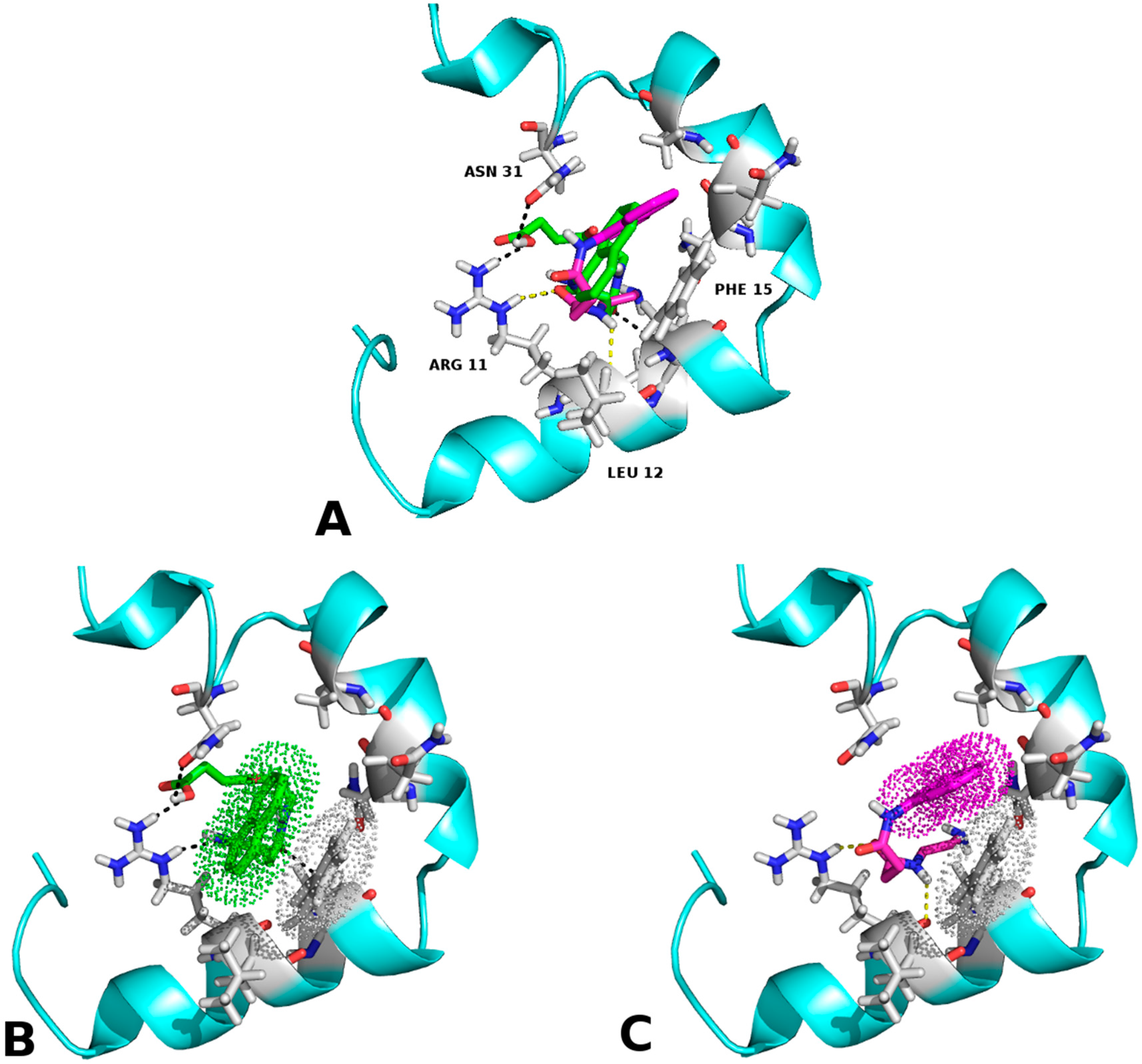
2.1. Molecular Framework of Interaction of Pharmacological Chaperones and the Human Islet Amyloid Polypeptide
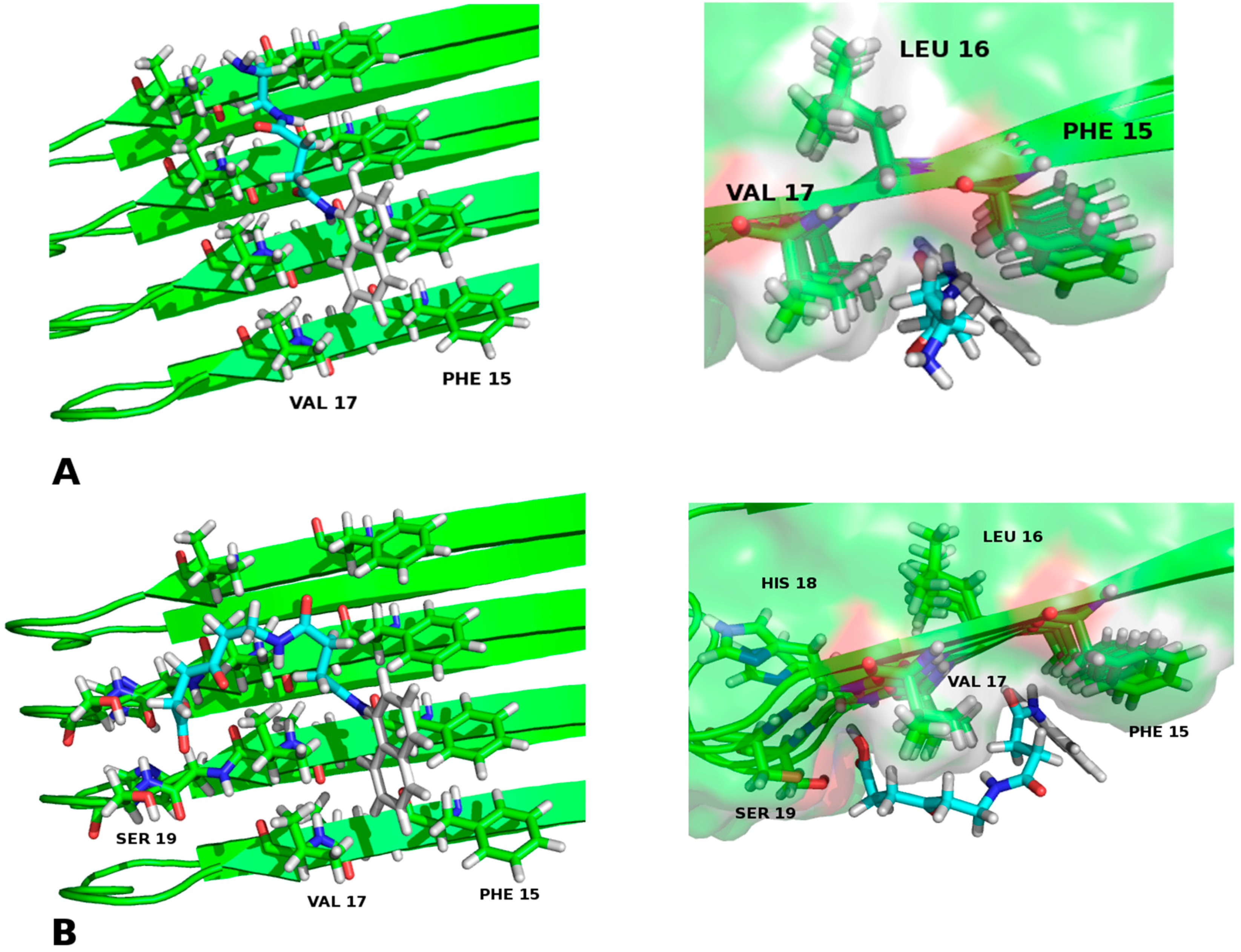
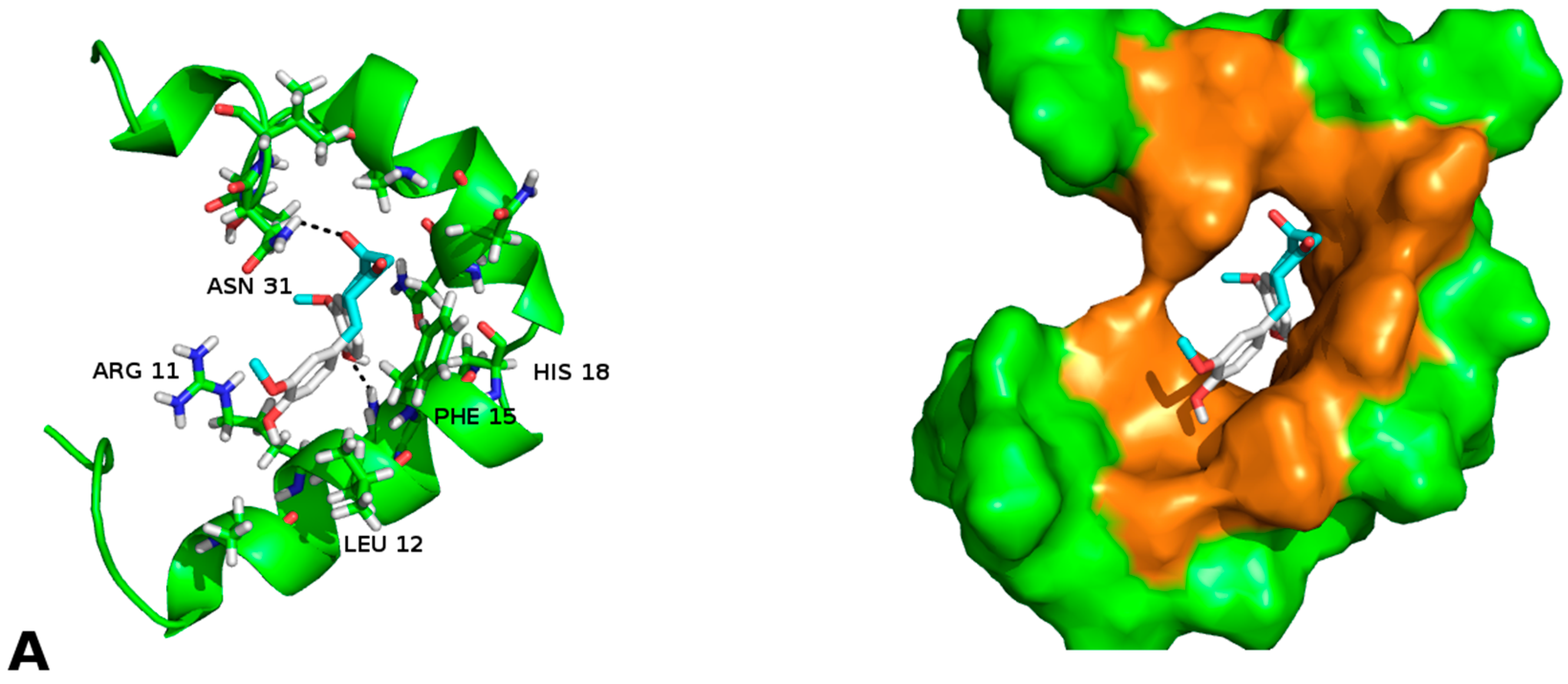
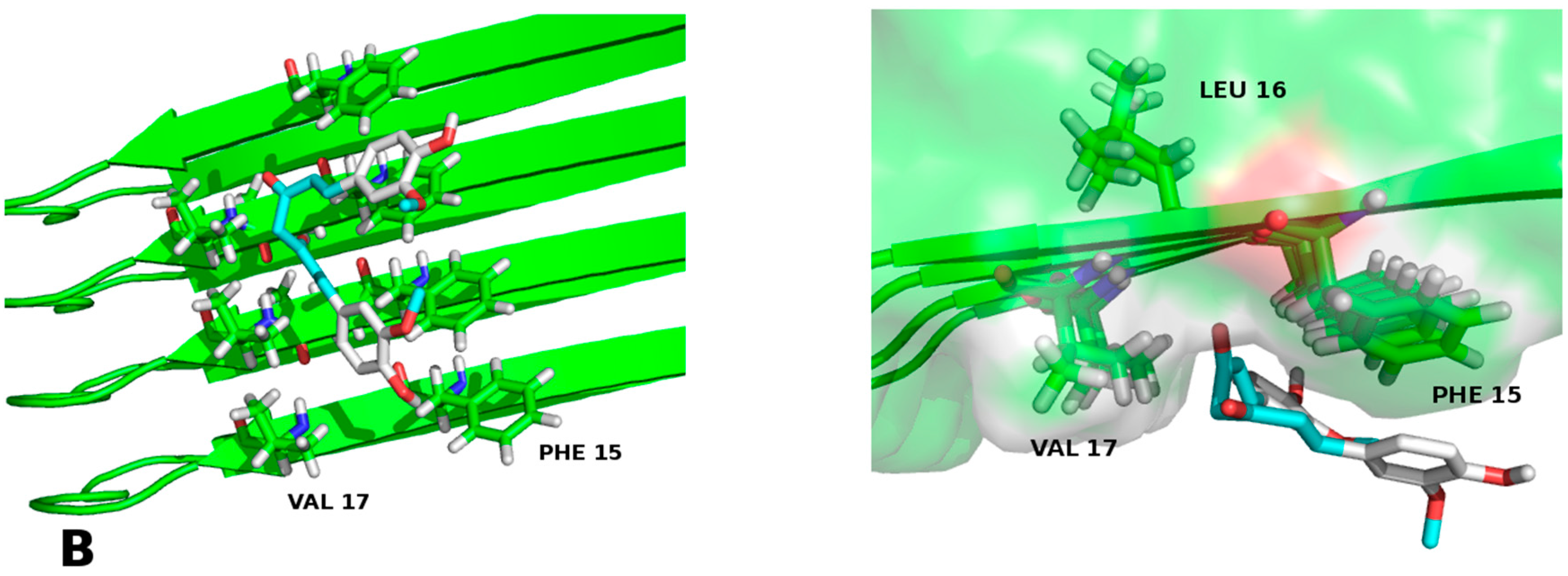
2.2. Molecular Framework of Interaction of Pharmacological Chaperones A and E with the Human Islet Amyloid Polypeptide Fibrillar Structure
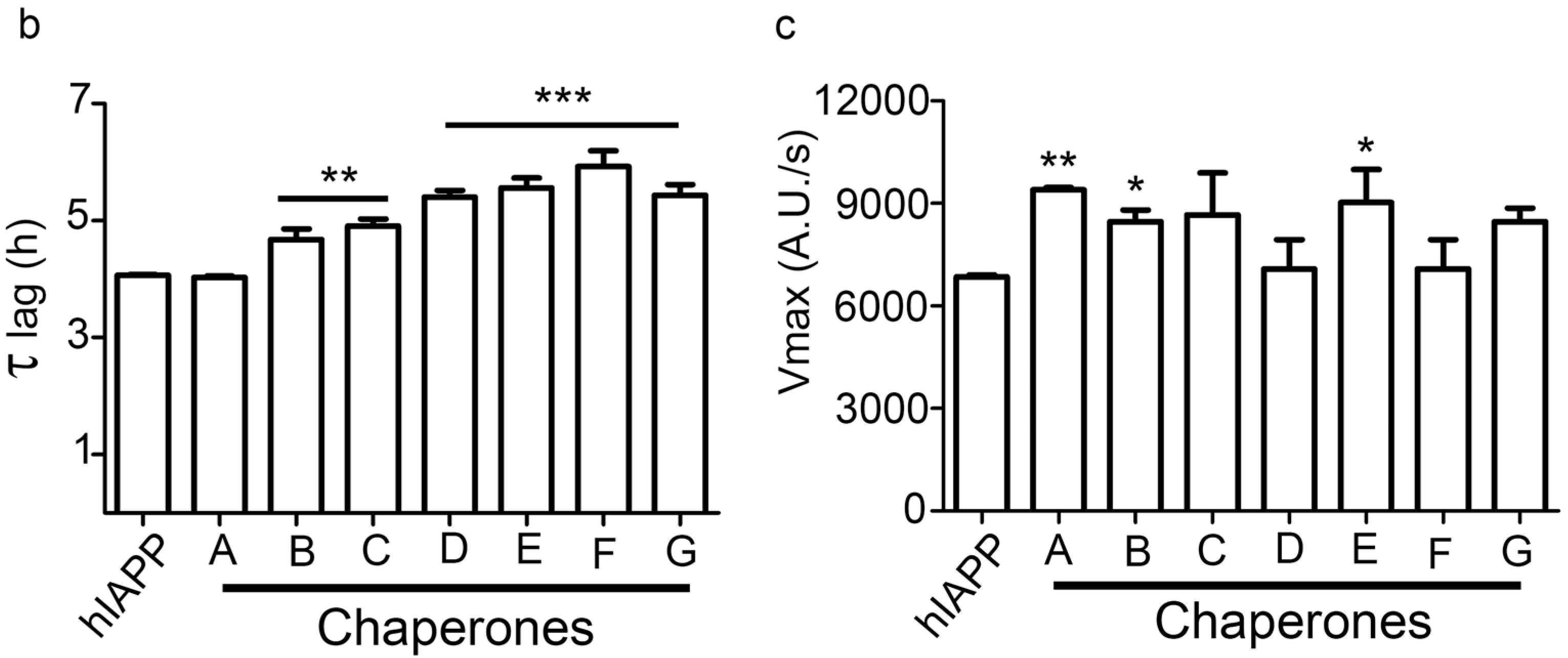
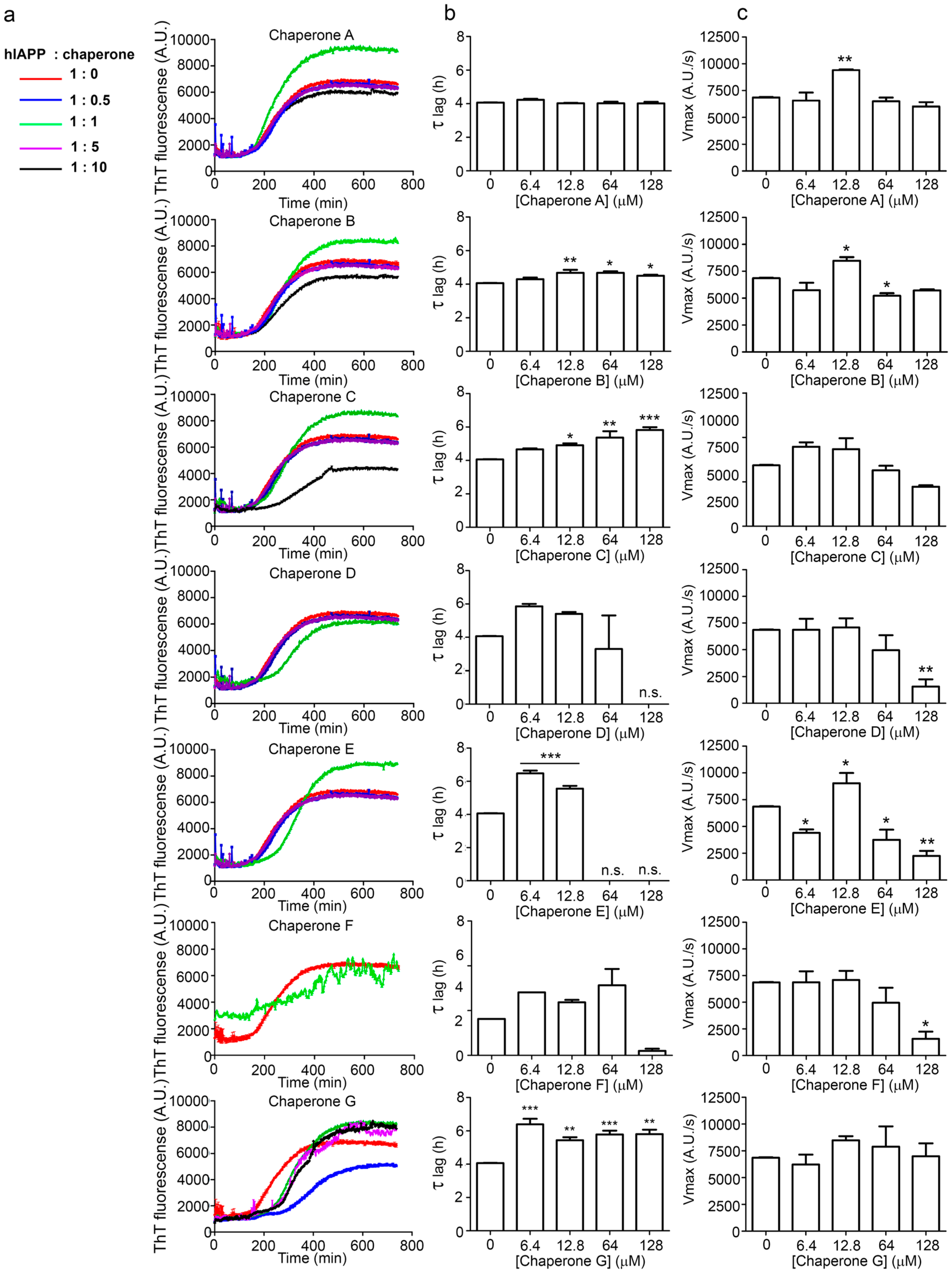
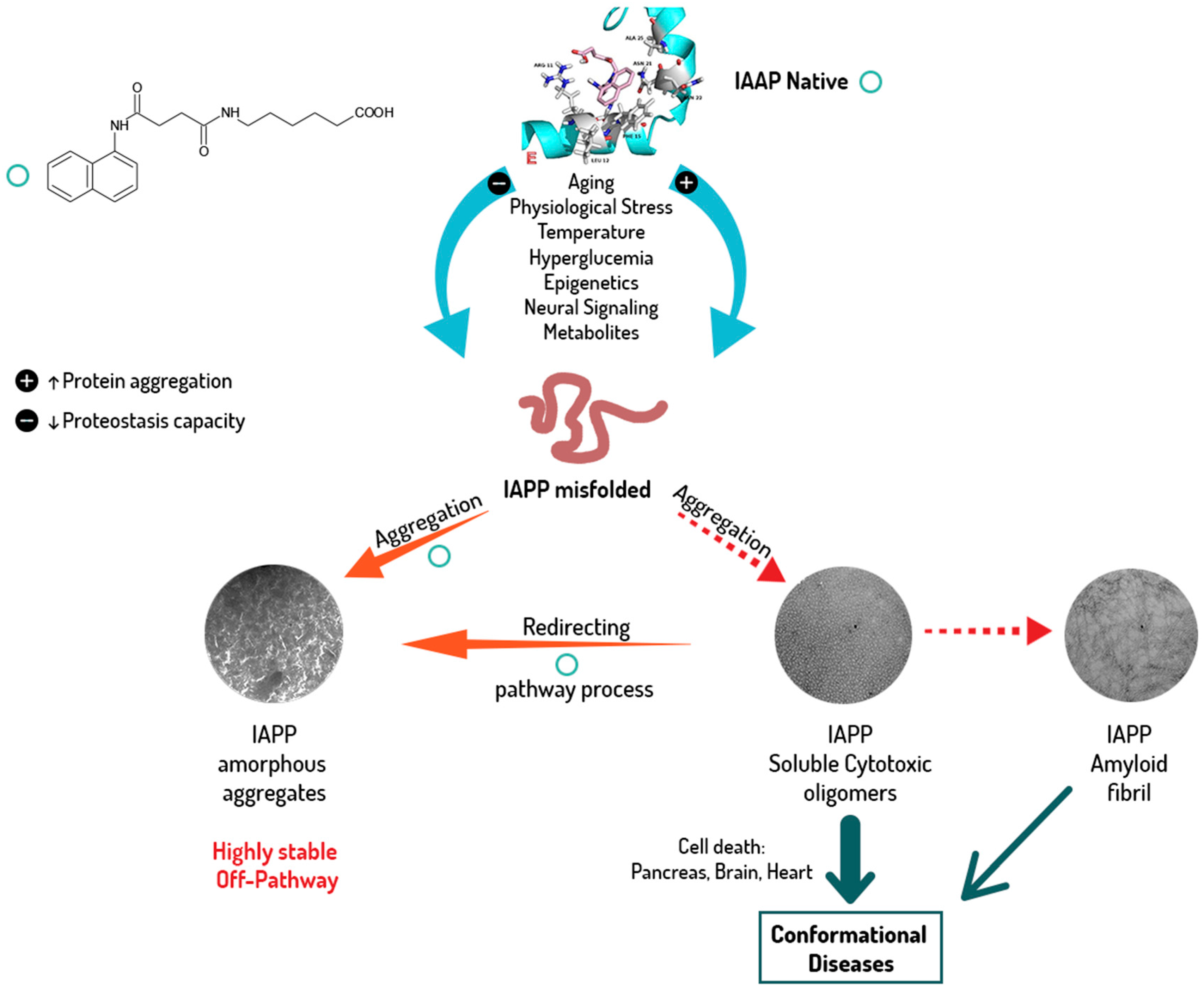
2.3. hIAPP1–37 Misfolding: Aggregation and Amyloid Formation of Toxic Species and Its Modulations by the Family of Pharmaco-Chaperones
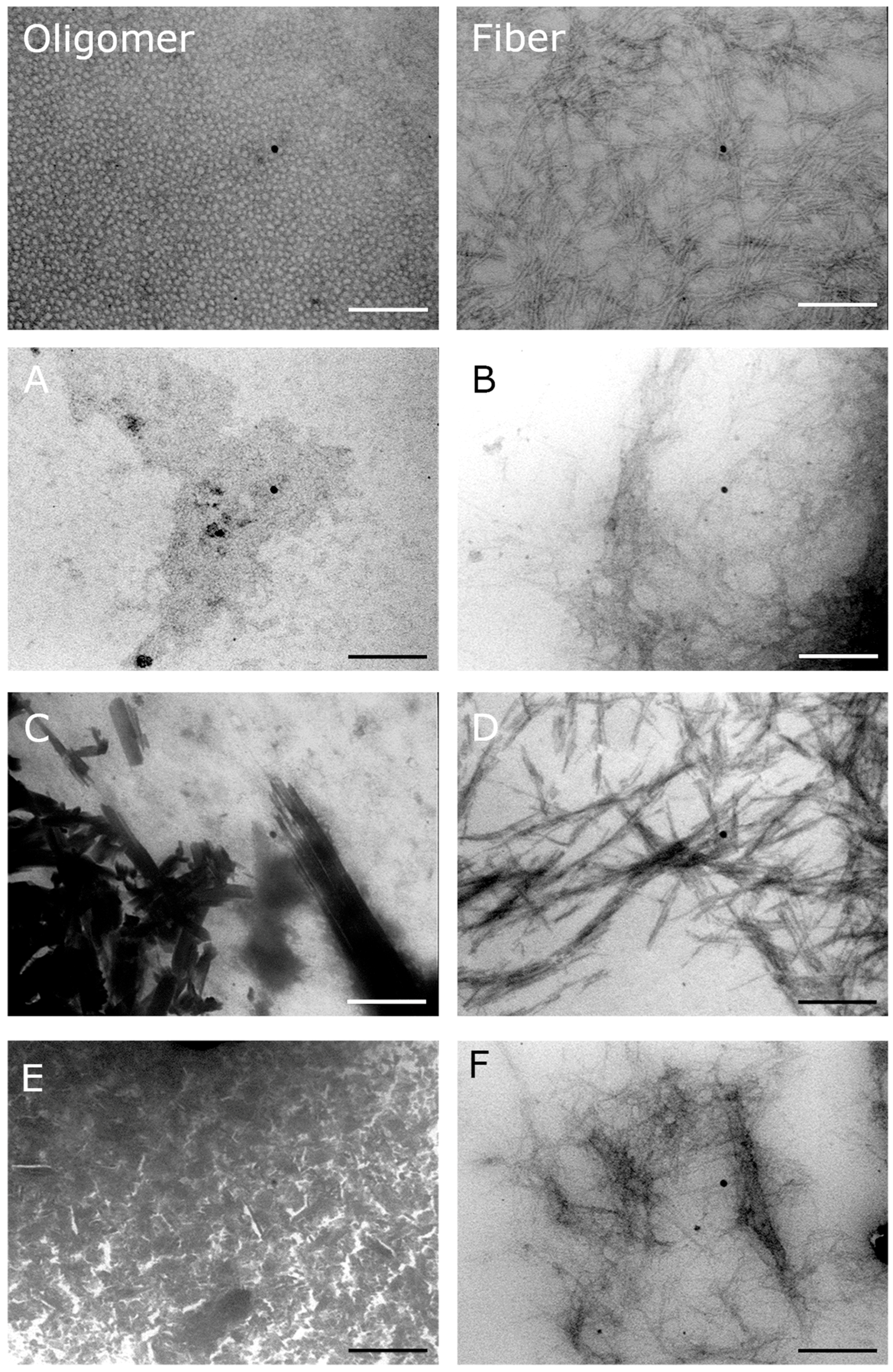
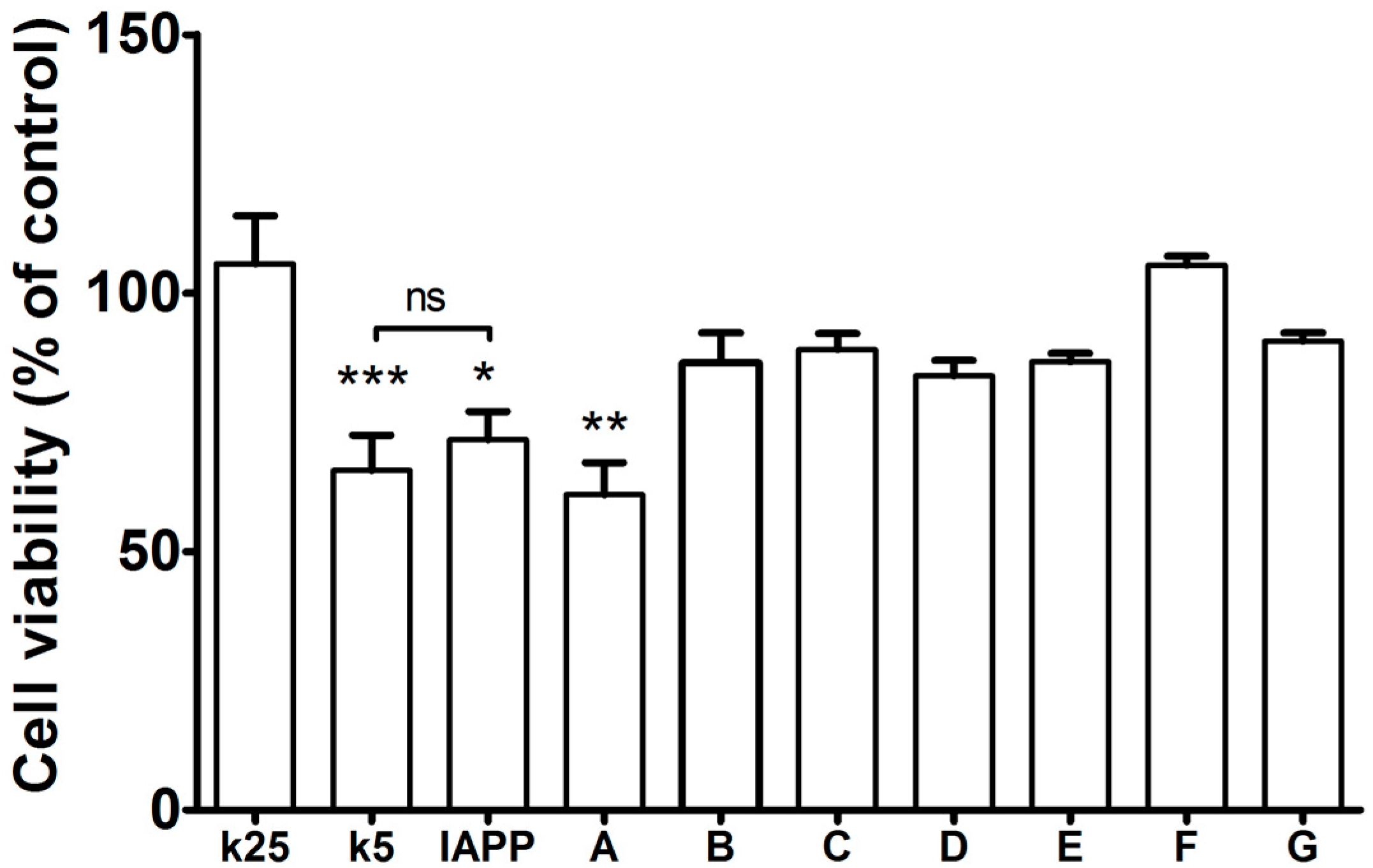
2.4. The Pharmaco-Chaperones Protect Granular Cells of Mouse Cerebellum from Toxicity of the hIAPP1–37 Oligomers
3. Discussion
4. Materials and Methods
4.1. Preparation and Characterization of Chaperones and hIAPP1–37
4.2. Oligomer Preparation
4.3. Molecular Docking
4.4. Thioflavin T (ThT) Fluorescence Assay
4.5. Transmission Electron Microscopy
4.6. Primary Cell Cultures
4.7. Cell Viability
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Bemporad, F.; Ramazzotti, M. From the Evolution of Protein Sequences Able to Resist Self-Assembly to the Prediction of Aggregation Propensity. Int. Rev. Cell Mol. Biol. 2017, 329, 1–47. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Amyloid formation by globular proteins under native conditions. Nat. Chem. Biol. 2009, 5, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Carrell, R.W.; Lomas, D.A. Alpha1-antitrypsin deficiency—A model for conformational diseases. N. Engl. J. Med. 2002, 346, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Carrell, R.W. Cell toxicity and conformational disease. Trends Cell Biol. 2005, 15, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Carrell, R.W.; Lomas, D.A. Conformational disease. Lancet 1997, 350, 134–138. [Google Scholar] [CrossRef]
- Eisenberg, D.; Jucker, M. The amyloid state of proteins in human diseases. Cell 2012, 148, 1188–1203. [Google Scholar] [CrossRef] [PubMed]
- Leyva-García, E.; Lara-Martínez, R.; Morán-Zanabria, L.; Revilla-Monsalve, C.; Jiménez-García, L.F.; Oviedo, N.; Murata, C.; Garrido-Magaña, E.; Altamirano-Bustamante, N.F.; Altamirano-Bustamante, M.M. Novel insight into streptozotocin-induced diabetic rats from the protein misfolding perspective. Sci. Rep. 2017, 7, 11552. [Google Scholar] [CrossRef] [PubMed]
- Avila-Vazquez, M.; Altamirano-Bustamante, N.; Altamirano-Bustamante, M. Amyloid Biomarkers in Conformational Diseases at Face Value: A Systematic Review. Molecules 2017, 23, 79. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. National Diabetes Statistics Report, 2014 Estimates of Diabetes and Its Burden in the Epidemiologic estimation methods. Natl. Diabetes Stat. Rep. 2014, 2009–2012. [Google Scholar]
- Cameron, F.J.; Wherrett, D.K. Care of diabetes in children and adolescents: controversies, changes, and consensus. Lancet (Lond. Engl.) 2015, 385, 2096–2106. [Google Scholar] [CrossRef]
- Clark, A.; Cooper, G.J.; Lewis, C.E.; Morris, J.F.; Willis, A.C.; Reid, K.B.; Turner, R.C. Islet amyloid formed from diabetes-associated peptide may be pathogenic in type-2 diabetes. Lancet (Lond. Engl.) 1987, 2, 231–234. [Google Scholar] [CrossRef]
- Engel, M.F.M. Membrane permeabilization by Islet Amyloid Polypeptide. Chem. Phys. Lipids 2009, 160, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zraika, S.; Hull, R.L.; Verchere, C.B.; Clark, A.; Potter, K.J.; Fraser, P.E.; Raleigh, D.P.; Kahn, S.E. Toxic oligomers and islet beta cell death: Guilty by association or convicted by circumstantial evidence? Diabetologia 2010, 53, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Ankarcrona, M.; Winblad, B.; Monteiro, C.; Fearns, C.; Powers, E.T.; Johansson, J.; Westermark, G.T.; Presto, J.; Ericzon, B.-G.; Kelly, J.W. Current and future treatment of amyloid diseases. J. Intern. Med. 2016, 280, 177–202. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-J.; Haataja, L.; Gurlo, T.; Butler, A.E.; Wu, X.; Soeller, W.C.; Butler, P.C. Induction of endoplasmic reticulum stress-induced beta-cell apoptosis and accumulation of polyubiquitinated proteins by human islet amyloid polypeptide. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1656–E1662. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Engstrom, U.; Johnson, K.H.; Westermark, G.T.; Betsholtz, C. Islet amyloid polypeptide: Pinpointing amino acid residues linked to amyloid fibril formation. Proc. Natl. Acad. Sci. USA 1990, 87, 5036–5040. [Google Scholar] [CrossRef] [PubMed]
- Herczenik, E.; Gebbink, M.F. Molecular and cellular aspects of protein misfolding and disease. FASEB J. 2008, 22, 2115–2133. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, B.; Winkelmann, J.; Tiribilli, B.; Chiti, F. Searching for conditions to form stable protein oligomers with amyloid-like characteristics: The unexplored basic pH. Biochim. Biophys. Acta 2010, 1804, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Zanuy, D.; Ma, B.; Nussinov, R. Short peptide amyloid organization: Stabilities and conformations of the islet amyloid peptide NFGAIL. Biophys. J. 2003, 84, 1884–1894. [Google Scholar] [CrossRef]
- Shigihara, N.; Fukunaka, A.; Hara, A.; Komiya, K.; Honda, A.; Uchida, T.; Abe, H.; Toyofuku, Y.; Tamaki, M.; Ogihara, T.; et al. Human IAPP-induced pancreatic β cell toxicity and its regulation by autophagy. J. Clin. Investig. 2014, 124, 3634–3644. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Functional Amyloid, and Human Disease. Annu. Rev. Biochem. 2009, 756, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Papp, E.; Csermely, P. Chemical chaperones: Mechanisms of action and potential use. Handb. Exp. Pharmacol. 2006, 172, 405–416. [Google Scholar]
- Scheibel, T.; Buchner, J. Protein aggregation as a cause for disease. Handb. Exp. Pharmacol. 2006, 172, 199–219. [Google Scholar]
- Scheidt, H.A.; Morgado, I.; Huster, D. Solid-state NMR reveals a close structural relationship between amyloid-β protofibrils and oligomers. J. Biol. Chem. 2012, 287, 22822–22826. [Google Scholar] [CrossRef] [PubMed]
- Mogk, A.; Tomoyasu, T.; Goloubinoff, P.; Rudiger, S.; Roder, D.; Langen, H.; Bukau, B. Identification of thermolabile Escherichia coli proteins: Prevention and reversion of aggregation by DnaK and ClpB. EMBO J. 1999, 18, 6934–6949. [Google Scholar] [CrossRef] [PubMed]
- Seong, I.S.; Oh, J.Y.; Lee, J.W.; Tanaka, K.; Chung, C.H. The HslU ATPase acts as a molecular chaperone in prevention of aggregation of SulA, an inhibitor of cell division in Escherichia coli. FEBS Lett. 2000, 477, 224–229. [Google Scholar] [CrossRef]
- Zahn, R.; Perrett, S.; Stenberg, G.; Fersht, A.R. Catalysis of amide proton exchange by the molecular chaperones GroEL and SecB. Science 1996, 271, 642–645. [Google Scholar] [CrossRef] [PubMed]
- Sablón-Carrazana, M.; Fernández, I.; Bencomo, A.; Lara-Martínez, R.; Rivera-Marrero, S.; Domínguez, G.; Pérez-Perera, R.; Jiménez-García, L.F.; Altamirano-Bustamante, N.F.; Diaz-Delgado, M.; et al. Drug Development in Conformational Diseases: A Novel Family of Chemical Chaperones that Bind and Stabilise Several Polymorphic Amyloid Structures. PLoS ONE 2015, 10, e0135292. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yu, X.; Li, L.; Zheng, J. Inhibition of amyloid-β aggregation in Alzheimer’s disease. Curr. Pharm. Des. 2014, 20, 1223–1243. [Google Scholar] [CrossRef] [PubMed]
- Doig, A.J.; Derreumaux, P. Inhibition of protein aggregation and amyloid formation by small molecules. Curr. Opin. Struct. Biol. 2015, 30, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.-P.; Yu, S.; McGeer, P.L. Simple in vitro assays to identify amyloid-beta aggregation blockers for Alzheimer’s disease therapy. J. Alzheimers Dis. 2010, 19, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Bu, X.-L.; Rao, P.P.N.; Wang, Y.-J. Anti-amyloid Aggregation Activity of Natural Compounds: Implications for Alzheimer’s Drug Discovery. Mol. Neurobiol. 2016, 53, 3565–3575. [Google Scholar] [CrossRef] [PubMed]
- Breitner, J.C.; Welsh, K.A.; Gau, B.A.; McDonald, W.M.; Steffens, D.C.; Saunders, A.M.; Magruder, K.M.; Helms, M.J.; Plassman, B.L.; Folstein, M.F. Alzheimer’s disease in the National Academy of Sciences-National Research Council Registry of Aging Twin Veterans. III. Detection of cases, longitudinal results, and observations on twin concordance. Arch. Neurol. 1995, 52, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Agdeppa, E.D.; Kepe, V.; Petri, A.; Satyamurthy, N.; Liu, J.; Huang, S.-C.C.; Small, G.W.; Cole, G.M.; Barrio, J.R. In vitro detection of (S)-naproxen and ibuprofen binding to plaques in the Alzheimer’s brain using the positron emission tomography molecular imaging probe 2-(1-{6-[(2-[18F] fluoroethyl)(methyl) amino]-2-naphthyl} ethylidene) malononitrile. Neurosciences 2003, 117, 723–730. [Google Scholar] [CrossRef]
- Fortin, J.S.; Benoit-Biancamano, M.-O. Inhibition of islet amyloid polypeptide aggregation and associated cytotoxicity by nonsteroidal anti-inflammatory drugs. Can. J. Physiol. Pharmacol. 2016, 94, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Wiltzius, J.J.W.; Sievers, S.A.; Sawaya, M.R.; Eisenberg, D. Atomic structures of IAPP (amylin) fusions suggest a mechanism for fibrillation and the role of insulin in the process. Protein Sci. 2009, 18, 1521–1530. [Google Scholar] [CrossRef] [PubMed]
- Soriaga, A.B.; Sangwan, S.; Macdonald, R.; Sawaya, M.R.; Eisenberg, D. Crystal Structures of IAPP Amyloidogenic Segments Reveal a Novel Packing Motif of Out-of-Register Beta Sheets. J. Phys. Chem. B 2016, 120, 5810–5816. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.M.; Xu, S.; Sheftic, S.R.; Alexandrescu, A.T. Dynamic α-helix structure of micelle-bound human amylin. J. Biol. Chem. 2009, 284, 11982–11991. [Google Scholar] [CrossRef] [PubMed]
- Nanga, R.P.R.; Brender, J.R.; Vivekanandan, S.; Ramamoorthy, A. Structure and membrane orientation of IAPP in its natively amidated form at physiological pH in a membrane environment. Biochim. Biophys. Acta Biomembr. 2011, 1808, 2337–2342. [Google Scholar] [CrossRef] [PubMed]
- Smaoui, M.R.; Poitevin, F.; Delarue, M.; Koehl, P.; Orland, H.; Waldispühl, J. Computational assembly of polymorphic amyloid fibrils reveals stable aggregates. Biophys. J. 2013, 104, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Smaoui, M.R.; Waldispühl, J. Computational re-engineering of Amylin sequence with reduced amyloidogenic potential. BMC Struct. Biol. 2015, 15, 7. [Google Scholar] [CrossRef] [PubMed]
- Smaoui, M.R.; Orland, H.; Waldispühl, J. Probing the binding affinity of amyloids to reduce toxicity of oligomers in diabetes. Bioinformatics 2015, 31, 2294–2302. [Google Scholar] [CrossRef] [PubMed]
- Nedumpully-Govindan, P.; Kakinen, A.; Pilkington, E.H.; Davis, T.P.; Chun Ke, P.; Ding, F. Stabilizing Off-pathway Oligomers by Polyphenol Nanoassemblies for IAPP Aggregation Inhibition. Sci. Rep. 2016, 6, 19463. [Google Scholar] [CrossRef] [PubMed]
- Lolicato, F.; Raudino, A.; Milardi, D.; La Rosa, C. Resveratrol interferes with the aggregation of membrane-bound human-IAPP: A molecular dynamics study. Eur. J. Med. Chem. 2015, 92, 876–881. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Misra, A.; Sri Vaishnavi, T.; Thota, C.; Gupta, M.; Ramakumar, S.; Chauhan, V.S. Conformationally restricted short peptides inhibit human islet amyloid polypeptide (hIAPP) fibrillization. Chem. Commun. 2013, 49, 2688. [Google Scholar] [CrossRef] [PubMed]
- Landau, M.; Sawaya, M.R.; Faull, K.F.; Laganowsky, A.; Jiang, L.; Sievers, S.A.; Liu, J.; Barrio, J.R.; Eisenberg, D. Towards a pharmacophore for amyloid. PLoS Biol. 2011, 9, e1001080. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.; Sawaya, M.R.; Balbirnie, M.; Madsen, A.Ø.; Riekel, C.; Grothe, R.; Eisenberg, D. Structure of the cross-beta spine of amyloid-like fibrils. Nature 2005, 435, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Riek, R.; Eisenberg, D.S. The activities of amyloids from a structural perspective. Nature 2016, 539, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Wiltzius, J.J.; Sievers, S.A.; Sawaya, M.R.; Cascio, D.; Popov, D.; Riekel, C.; Eisenberg, D. Atomic structure of the cross-β spine of islet amyloid polypeptide (amylin). Insulin 2008, 17, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.S.; Sawaya, M.R. Structural Studies of Amyloid Proteins at the Molecular Level. Annu. Rev. Biochem. 2017, 86, 69–95. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar] [CrossRef] [PubMed]
- Reinke, A.A.; Gestwicki, J.E. Structure-activity relationships of amyloid beta-aggregation inhibitors based on curcumin: Influence of linker length and flexibility. Chem. Biol. Drug Des. 2007, 70, 206–215. [Google Scholar] [CrossRef] [PubMed]
- De Matos, A.M.; de Macedo, M.P.; Rauter, A.P. Bridging Type 2 Diabetes and Alzheimer’s Disease: Assembling the Puzzle Pieces in the Quest for the Molecules with Therapeutic and Preventive Potential. Med. Res. Rev. 2017, 38, 261–324. [Google Scholar] [CrossRef] [PubMed]
- Profenno, L.A.; Porsteinsson, A.P.; Faraone, S.V. Meta-analysis of Alzheimer’s disease risk with obesity, diabetes, and related disorders. Biol. Psychiatry 2010, 67, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Despa, S.; Margulies, K.B.; Chen, L.; Knowlton, A.A.; Havel, P.J.; Taegtmeyer, H.; Bers, D.M.; Despa, F. Hyperamylinemia Contributes to Cardiac Dysfunction in Obesity and Diabetes: A Study in Humans and Rats. Circ. Res. 2012, 110, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Srodulski, S.; Sharma, S.; Bachstetter, A.B.; Brelsfoard, J.M.; Pascual, C.; Xie, X.S.; Saatman, K.E.; Van Eldik, L.J.; Despa, F. Neuroinflammation and neurologic deficits in diabetes linked to brain accumulation of amylin. Mol. Neurodegener. 2014, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Benedet, A.L.; Labbe, A.; Lemay, P.; Zimmer, E.R.; Pascoal, T.A.; Leuzy, A.; Mathotaarachchi, S.; Mohades, S.; Shin, M.; Dionne-Laporte, A.; et al. CSF Biomarkers and Incipient Alzheimer Disease in Patients With Mild Cognitive Impairment. Neurobiol. Aging 2017, 6, 643–648. [Google Scholar] [CrossRef]
- Alavez, S.; Pedroza, D.; Morán, J. Mechanisms of cell death by deprivation of depolarizing conditions during cerebellar granule neurons maturation. Neurochem. Int. 2003, 43, 581–590. [Google Scholar] [CrossRef]
- Xifro, X.; Malagelada, C.; Miñano, A.; Rodríguez-Álvarez, J. Brief exposure to NMDA produces long-term protection of cerebellar granule cells from apoptosis. Eur. J. Neurosci. 2005, 21, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Moran, J.; Patel, A.J. Stimulation of the N-methyl-d-aspartate receptor promotes the biochemical differentiation of cerebellar granule neurons and not astrocytes. Brain Res. 1989, 486, 15–25. [Google Scholar] [CrossRef]
- Meier, J.J.; Kayed, R.; Lin, C.-Y.; Gurlo, T.; Haataja, L.; Jayasinghe, S.; Langen, R.; Glabe, C.G.; Butler, P.C. Inhibition of human IAPP fibril formation does not prevent β-cell death: Evidence for distinct actions of oligomers and fibrils of human IAPP. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E1317–E1324. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Knowles, T.P.J.; Linse, S. On the lag phase in amyloid fibril formation. Phys. Chem. Chem. Phys. 2015, 17, 7606–7618. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R. Common Structure of Soluble Amyloid Oligomers Implies Common Mechanism of Pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, U.; Nilson, A.N.; Kayed, R. The Role of Amyloid-β Oligomers in Toxicity, Propagation, and Immunotherapy. EBioMedicine 2016, 6, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Selkoe, D.J. A beta oligomers—A decade of discovery. J. Neurochem. 2007, 101, 1172–1184. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Liu, C.; Leibly, D.; Landau, M.; Zhao, M.; Hughes, M.P.; Eisenberg, D.S. Structure-based discovery of fiber-binding compounds that reduce the cytotoxicity of amyloid beta. Elife 2013, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Sablón, C.M.; Rodriguez-Tanty, C.; Perera, P.A.; Rivera, M.S.; Perez, P.R.; López, B.R.M.; Prats, C.A.; Vélez, C.H.; Pérez, M.C.S.; Valdés, S.P. Method for Obtaining Novel Derivatives of Naphthalene for the In Vivo Diagnosis of Alzheimer´s Disease. U.S. Patent 9,764,047 B2, 19 September 2017. [Google Scholar]
- Kayed, R.; Glabe, C.G. Conformation-dependent anti-amyloid oligomer antibodies. Methods Enzymol. 2006, 413, 326–344. [Google Scholar] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Peinado, J.R.; Sami, F.; Rajpurohit, N.; Lindberg, I. Blockade of islet amyloid polypeptide fibrillation and cytotoxicity by the secretory chaperones 7B2 and proSAAS. FEBS Lett. 2013, 587, 3406–3411. [Google Scholar] [CrossRef] [PubMed]
- Holm, N.K.; Jespersen, S.K.; Thomassen, L.V.; Wolff, T.Y.; Sehgal, P.; Thomsen, L.A.; Christiansen, G.; Andersen, C.B.; Knudsen, A.D.; Otzen, D.E. Aggregation and fibrillation of bovine serum albumin. Biochim. Biophys. Acta 2007, 1774, 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Moran, J.; Patel, A.J. Effect of potassium depolarization on phosphate-activated glutaminase activity in primary cultures of cerebellar granule neurons and astroglial cells during development. Brain Res. Dev. Brain Res. 1989, 46, 97–105. [Google Scholar] [CrossRef]
- Balázs, R.; Hack, N.; Jørgensen, O.S. Selective stimulation of excitatory amino acid receptor subtypes and the survival of cerebellar granule cells in culture: Effect of kainic acid. Neuroscience 1990, 37, 251–258. [Google Scholar] [CrossRef]
Sample Availability: Not available. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Dose (µM) | Vmax | Sd | ν max (%) | τ lag | Sd | τ lag (%) |
|---|---|---|---|---|---|---|---|
| IAPP1–37 | 12.8 | 6944 | 772 | 100 | 3.989 | 0.37388 | 100 |
Chaperone A | 6.4 | 6701 | 775 | 97 | 4.172 | 0.42174 | 105 |
| 12.8 | 9509 | 1032 | 137 | 3.973 | 0.35464 | 100 | |
| 64 | 6582 | 730 | 95 | 3.967 | 0.372 | 99 | |
Chaperone B | 6.4 | 5783 | 707 | 83 | 4.250 | 0.46283 | 107 |
| 12.8 | 8562 | 1100 | 123 | 4.602 | 0.55049 | 115 | |
| 64 | 5328 | 672 | 77 | 4.639 | 0.51981 | 116 | |
Chaperone C | 6.4 | 9128 | 1217 | 131 | 4.650 | 0.60413 | 117 |
| 12.8 | 8791 | 1175 | 127 | 4.797 | 0.6116 | 120 | |
| 64 | 6348 | 885 | 91 | 4.840 | 0.65495 | 121 | |
Chaperone D | 6.4 | 6946 | 1126 | 100 | 5.787 | 1.14215 | 145 |
| 12.8 | 6326 | 944 | 91 | 5.212 | 0.80756 | 131 | |
| 64 | 5255 | 289 | 76 | 2.071 | 0.24877 | 52 | |
Chaperone E | 6.4 | 4341 | 783 | 63 | 6.320 | 1.48342 | 158 |
| 12.8 | 9055 | 1488 | 130 | 5.559 | 1.0796 | 139 | |
| 64 | 4465 | 835 | 64 | 6.569 | 1.76324 | 165 | |
Chaperone G | 6.4 | 5182 | 867 | 75 | 6.322 | 1.36815 | 158 |
| 12.8 | 8455 | 1281 | 122 | 5.294 | 0.86324 | 133 | |
| 64 | 6252 | 847 | 90 | 5.659 | 0.81244 | 142 |
| Contact % with aa of hIAPP1–37 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cys7 | Arg11 | Leu 12 | Asn 14 | Phe 15 | His 18 | Asn 21 | Asn 22 | Ala 25 | Ser 28 | Asn 31 | |
| Chaperone A | - | 90 | 92 | 91 | 89 | 88 | 85 | 90 | 90 | 91 | - |
| Chaperone B | - | 91 | 89 | 91 | 90 | 90 | 80 | - | - | 89 | - |
| Chaperone C | - | 89 | 90 | 91 | 89 | 90 | 82 | 91 | 92 | 90 | - |
| Chaperone D | - | 87 | 91 | 91 | 87 | 88 | 85 | 88 | 88 | 91 | 30 |
| Chaperone E | - | 96 | 98 | 96 | 97 | 95 | 98 | 95 | 98 | 98 | 92 |
| Chaperone F | - | 90 | 92 | 88 | 84 | 80 | 86 | 87 | 89 | 90 | - |
| Chaperone G | 50 | 90 | 92 | 87 | 91 | 90 | 87 | 90 | 90 | 89 | 40 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Gómez, I.; Sablón-Carrazana, M.; Bencomo-Martínez, A.; Domínguez, G.; Lara-Martínez, R.; Altamirano-Bustamante, N.F.; Jiménez-García, L.F.; Pasten-Hidalgo, K.; Castillo-Rodríguez, R.A.; Altamirano, P.; et al. Diabetes Drug Discovery: hIAPP1–37 Polymorphic Amyloid Structures as Novel Therapeutic Targets. Molecules 2018, 23, 686. https://doi.org/10.3390/molecules23030686
Fernández-Gómez I, Sablón-Carrazana M, Bencomo-Martínez A, Domínguez G, Lara-Martínez R, Altamirano-Bustamante NF, Jiménez-García LF, Pasten-Hidalgo K, Castillo-Rodríguez RA, Altamirano P, et al. Diabetes Drug Discovery: hIAPP1–37 Polymorphic Amyloid Structures as Novel Therapeutic Targets. Molecules. 2018; 23(3):686. https://doi.org/10.3390/molecules23030686
Chicago/Turabian StyleFernández-Gómez, Isaac, Marquiza Sablón-Carrazana, Alberto Bencomo-Martínez, Guadalupe Domínguez, Reyna Lara-Martínez, Nelly F. Altamirano-Bustamante, Luis Felipe Jiménez-García, Karina Pasten-Hidalgo, Rosa Angélica Castillo-Rodríguez, Perla Altamirano, and et al. 2018. "Diabetes Drug Discovery: hIAPP1–37 Polymorphic Amyloid Structures as Novel Therapeutic Targets" Molecules 23, no. 3: 686. https://doi.org/10.3390/molecules23030686
APA StyleFernández-Gómez, I., Sablón-Carrazana, M., Bencomo-Martínez, A., Domínguez, G., Lara-Martínez, R., Altamirano-Bustamante, N. F., Jiménez-García, L. F., Pasten-Hidalgo, K., Castillo-Rodríguez, R. A., Altamirano, P., Marrero, S. R., Revilla-Monsalve, C., Valdés-Sosa, P., Salamanca-Gómez, F., Garrido-Magaña, E., Rodríguez-Tanty, C., & Altamirano-Bustamante, M. M. (2018). Diabetes Drug Discovery: hIAPP1–37 Polymorphic Amyloid Structures as Novel Therapeutic Targets. Molecules, 23(3), 686. https://doi.org/10.3390/molecules23030686