Synthesis of Some Novel Fused Pyrimido[4″,5″:5′,6′]-[1,2,4]triazino[3′,4′:3,4] [1,2,4]triazino[5,6-b]indoles with Expected Anticancer Activity



Abstract
:1. Introduction
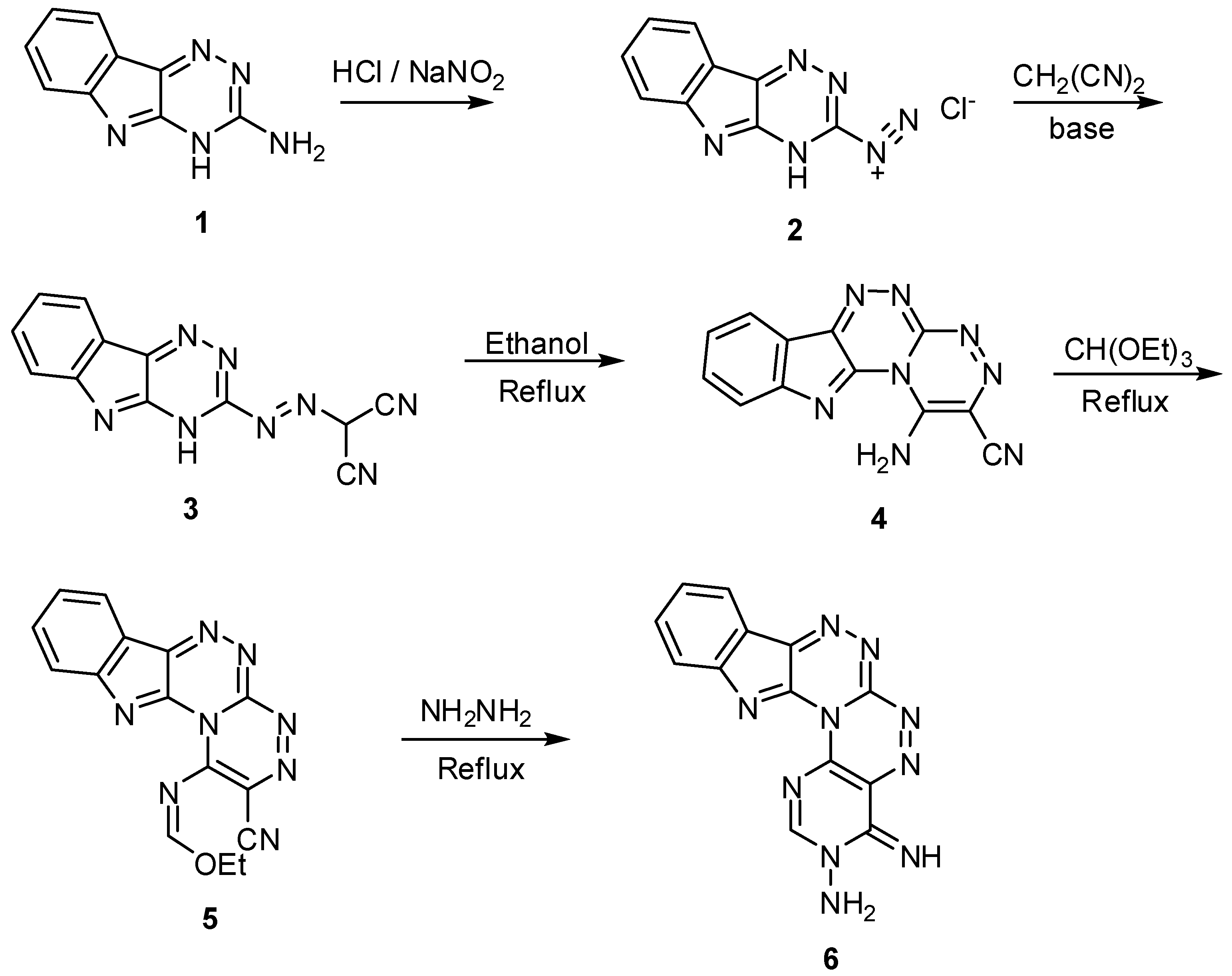
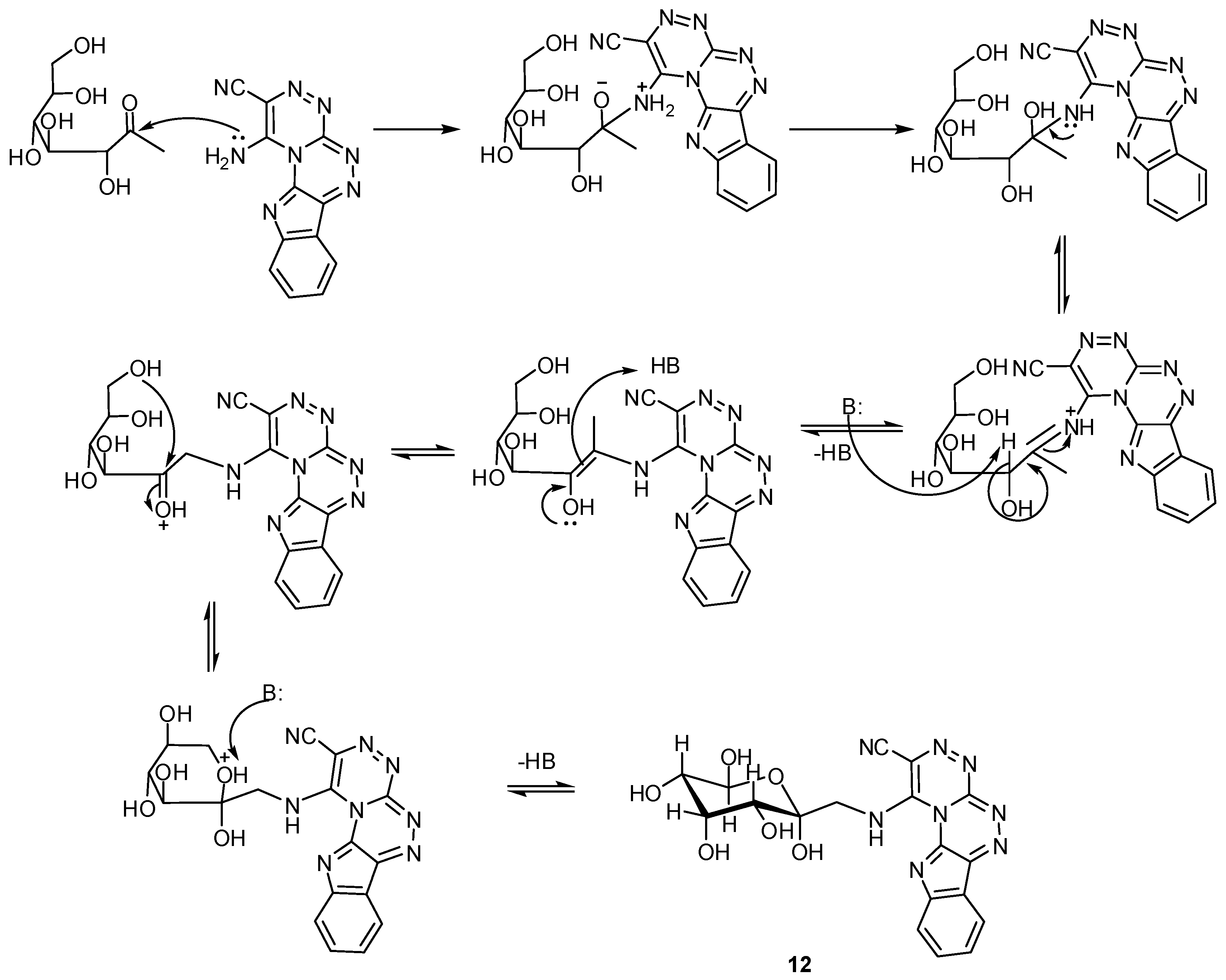
2. Results and Discussion
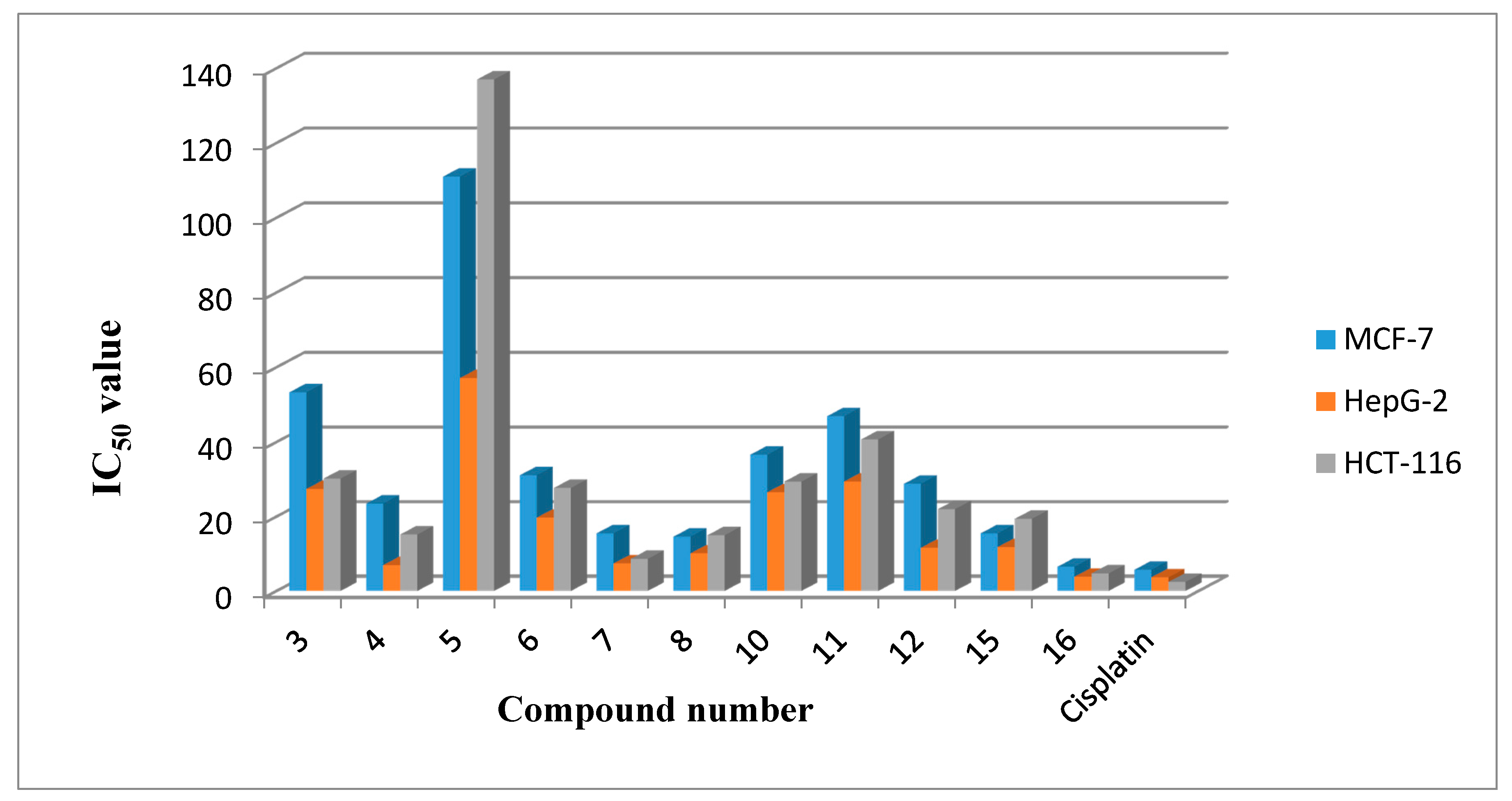
3. Cytotoxic Activity
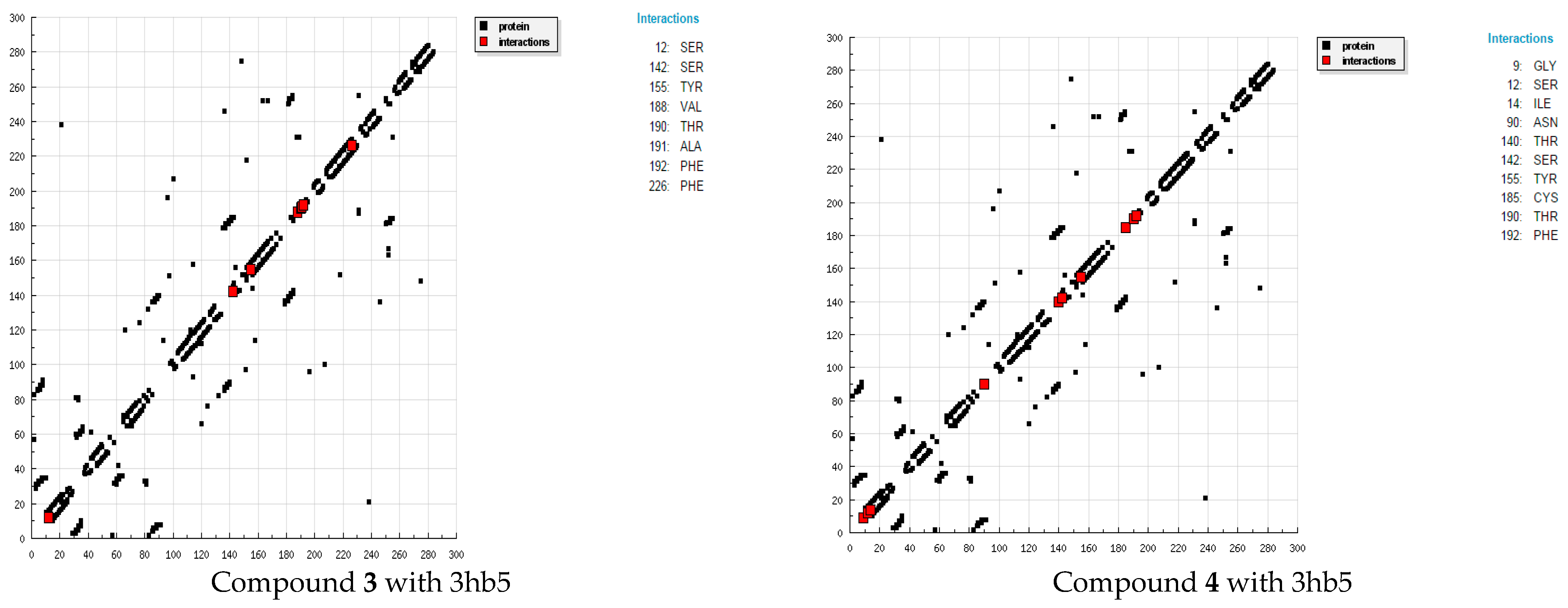
4. Molecular Docking Studies
5. Materials and Methods
5.1. General Information
5.2. Antitumor Activity Assay
5.3. Theoretical Molecular Docking Techniques
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Saad, H.A.; Youssef, M.M.; Mosselhi, M.A. Microwave Assisted Synthesis of Some New Fused 1,2,4-Triazines Bearing Thiophene Moieties with Expected Pharmacological Activity. Molecules 2011, 16, 4937–4957. [Google Scholar] [CrossRef] [PubMed]
- Saad, H.A.; Moustafa, A.H. Synthesis and Anticancer Activity of Some New S-Glycosyl and S-Alkyl 1,2,4-Triazinone Derivatives. Molecules 2011, 16, 5682–5700. [Google Scholar] [CrossRef] [PubMed]
- Saad, H.A. Synthesis of novel fused heterocyclic compounds from 7-amino-[1,2,4]triazino[3,4-b][1,3,4]thiadiazine-8-carbonitrile. Curr. Org. Synth. 2012, 9, 573–582. [Google Scholar] [CrossRef]
- Aly, M.R.E.; Saad, H.A.; Hafez, S.H.A. Three-Component Process for the Synthesis of Pyrimido[2,1-c][1,2,4]triazine Derivatives via Knoevenagel Condensation under Thermal Aqueous Conditions. Curr. Org. Synth. 2015, 12, 208–219. [Google Scholar]
- Aly, M.R.E.; Gobouri, A.A.; Hafez, S.H.A.; Saad, H.A. Synthesis, Reactions and Biological Activity of some Triazine Derivatives Containing Sulfa Drug Moieties. Russ. J. Bioorg. Chem. 2015, 4, 491–504. [Google Scholar]
- Amin, M.A.; Saad, H.A. Synthesis and Biological Activity of Fused Heteropolycyclic Systems Containing an Indole Moiety. Curr. Org. Synth. 2016, 13, 116–125. [Google Scholar] [CrossRef]
- AlHarthi, R.R.; Ali, R.S.; Amin, M.A.; Saad, H.A. Synthesis of Some New Fused 1,2,4-Triazines of Expected Antimicrobial Activity. Russ. J. Gen. Chem. 2016, 86, 2906–2913. [Google Scholar]
- El-Nahass, M.M.; Ashour, A.; Atta, A.A.; Saad, H.A.; Hassanien, A.M.; AlBaradi, A.M.; El-Zaidia, E.F.M. Dielectric relaxation and optical properties of 4-amino-3-mercapto-6-(2-(2thienyl)vinyl)-1,2,4-triazin-5(4H)-one donor. Pramana J. Physics 2017, 88, 6. [Google Scholar] [CrossRef]
- Shehab, W.S.; Saad, H.A.; Mouneir, S.M. Synthesis and Antitumor/Antiviral Evaluation of 6-Thienyl-5-cyano-2-thiouracil Derivatives and Their Thiogalactosides Analogs. Curr. Org. Synth. 2017, 14, 291–298. [Google Scholar] [CrossRef]
- Al Osaimi, A.G.; Ali, R.S.; Saad, H.A.; El Sayed Aly, M.R. Synthesis, Reactions and Biological Activity of Some New Derivatives of Fused [1,2,4]triazino[5,6-b]indole. Russ. J. Gen. Chem. 2017, 87, 1246–1255. [Google Scholar] [CrossRef]
- Anderson, E.A. Cascade polycyclisations in natural product synthesis. Org. Biomol. Chem. 2011, 9, 3997–4006. [Google Scholar] [CrossRef] [PubMed]
- Dancik, V.; Seiler, K.P.; Young, D.W.; Schreiber, S.L.; Clemons, P.A. Distinct Biological Network Properties between the Targets of Natural Products and Disease Genes. J. Am. Chem. Soc. 2010, 132, 9259–9261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Khedkar, V.; Baskar, B.; Schürmann, M.; Kumar, K. Branching Cascades: A Concise Synthetic Strategy Targeting Diverse and Complex Molecular Frameworks. Angew. Chem. Int. Ed. 2011, 50, 6900–6905. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.S.; Sakr, S.I.; El-Senousy, W.M.; Madkour, H.M.F. Synthesis, antibacterial, and antiviral evaluation of new heterocycles containing the pyridine moiety. Arch. Pharm. (Weinheim) 2013, 346, 766–773. [Google Scholar] [CrossRef] [PubMed]
- El-salam, N.M.A.; Mostafa, M.S.; Ahmed, G.A.; Alothman, O.Y. Synthesis and Antimicrobial Activities of Some New Heterocyclic Compounds Based on 6-Chloropyridazine-3 (2H)-thione. J. Chem. 2013, 2013, 1–8. [Google Scholar] [CrossRef]
- Azab, M.E.; Youssef, M.M.; El-Bordany, E.A. Synthesis and antibacterial evaluation of novel heterocyclic compounds containing a sulfonamido moiety. Molecules 2013, 18, 832–844. [Google Scholar] [CrossRef] [PubMed]
- El-Sawy, E.R.; Ebaid, M.S.; Abo-Salem, H.M.; Al-Sehemi, A.G.; Mandour, A.H. Synthesis, anti-inflammatory, analgesic and anticonvulsant activities of some new 4,6-dimethoxy-5(heterocycles)benzofuran starting from naturally occurring visnagin. Arab. J. Chem. 2013, 7, 914–923. [Google Scholar] [CrossRef]
- Cao, X.; Sun, Z.; Cao, Y.; Wang, R.; Cai, T.; Chu, W.; Hu, W.; Yang, Y. Design, Synthesis, and Structure—Activity Relationship Studies of Novel Fused Heterocycles-Linked Triazoles with Good Activity and Water Solubility. J. Med. Chem. 2014, 57, 3687–3706. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yu, K.; Tan, N.Y.; Qiu, R.H.; Liu, W.; Luo, N.L.; Tong, L.; Au, C.T.; Luo, Z.Q.; Yin, S.F. Synthesis, characterization and anti-proliferative activity of heterocyclic hypervalent organoantimony compounds. Eur. J. Med. Chem. 2014, 79, 391–398. [Google Scholar] [CrossRef] [PubMed]
- El-Sawy, E.R.; Mandour, A.H.; El-Hallouty, S.M.; Shaker, K.H.; Abo-Salem, H.M. Synthesis, antimicrobial and anticancer activities of some new N-methylsulphonyl and N-benzenesulphonyl-3-indolyl heterocycles. 1st Cancer Update. Arab. J. Chem. 2013, 6, 67–78. [Google Scholar] [CrossRef]
- Mabkhot, Y.N.; Barakat, A.; Al-Majid, A.M.; Alshahrani, S.; Yousuf, S.; Choudhary, M.I. Synthesis, reactions and biological activity of some new bis-heterocyclic ring compounds containing sulphur atom. Chem. Cent. J. 2013, 7, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Bergman, J.; Koch, E.; Pelcman, B. 2,2′-Biindolyl revisited. Synthesis and reactions. Tetrahedron 1995, 51, 5631–5642. [Google Scholar] [CrossRef]
- Gribble, G.W.; Pelcman, B. Total syntheses of the marine sponge pigments fascaplysin and homofascaplysin B and C. J. Org. Chem. 1992, 57, 3636–3642. [Google Scholar] [CrossRef]
- Carter, D.S.; Vranken, D.L.V. Synthesis of Homofascaplysin C and Indolo[2,3-a]carbazole from Ditryptophans. J. Org. Chem. 1999, 64, 8537–8545. [Google Scholar] [CrossRef]
- Segraves, N.L.; Robinson, S.J.; Garcia, D.; Said, S.A.; Fu, X.; Schmitz, F.J.; Pietraszkiewicz, H.; Valeriote, F.A.; Crews, P. Comparison of Fascaplysin and Related Alkaloids: A Study of Structures, Cytotoxicities, and Sources. J. Nat. Prod. 2004, 67, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Dubovitskii, S.V. Method for synthesis of 12H-pyrido[1,2-a:3,4-b′]diindoles. Total synthesis of homofascaplysin C. Tetrahedron Lett. 1996, 37, 5207–5208. [Google Scholar] [CrossRef]
- Lawrie, A.M.; Noble, M.E.M.; Tunnah, P.; Brown, N.R.; Johnson, L.N.; Endicott, J.A. Protein kinase inhibition by staurosporine revealed in details of the molecular interaction with CDK2. Nat. Struct. Mol. Biol. 1997, 4, 796–801. [Google Scholar] [CrossRef]
- Sasaki, T.; Ohtani, I.I.; Tanaka, J.; Higa, T. Iheyamines, new cytotoxic bisindole pigments from a colonial ascidian, Polycitorellasp. Tetrahedron Lett. 1999, 40, 303–306. [Google Scholar] [CrossRef]
- Bush, J.A.; Long, B.H.; Catino, J.J.; Bradner, W.T.; Tomita, K. Production and biological activity of rebeccamycin, a novel antitumor agent. J. Antibiot. 1987, 40, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Tomchin, A.B.; Uryupoy, O.Y.; Smirnov, A.V. Thiourea and thiosemicarbazide derivatives: Structure, transformations, and pharmacological activity. Part II. Antihypoxic activity of 1,2,4-triazino[5,6-b]indole derivatives. Pharm. Chem. J. 1997, 31, 125. [Google Scholar] [CrossRef]
- Bikadi, Z.; Hazai, E. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J. Cheminf. 2009, 1, 15. [Google Scholar] [CrossRef] [PubMed]
- Beteringhe, A.; Racuciu, C.; Balan, C.; Stoican, E.; Patron, L. Molecular Docking Studies Involving Transitional Metal Complexes (Zn(II), Co(II), Cu(II), Fe(II), Ni(II) with Cholic Acid (AC) as Ligand against Aurora A Kinase. Adv. Mater. Res. 2013, 787, 236–240. [Google Scholar] [CrossRef]
- Bikadi, Z.; Demko, L.; Hazai, E. Functional and structural characterization of a protein based on analysis of its hydrogen bonding network by hydrogen bonding plot. Arch. Biochem. Biophys. 2007, 461, 225–234. [Google Scholar] [CrossRef] [PubMed]
- McDonald, I.K.; Thornton, J.M. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 1994, 238, 777–793. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Ghorab, M.M.; Alsaid, M.S.; El-Gaby, M.S.A.; Safwat, N.A.; Elaasser, M.M.; Soliman, A.M. Biological evaluation of some new N-(2,6-dimethoxypyrimidinyl)thioureidobenzenesulfonamide derivatives as potential antimicrobial and anticancer agents. Eur. J. Med. Chem. 2016, 124, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Solis, F.J.; Wets, R.J.B. Minimization by Random Search Techniques. Math. Operat. Res. 1981, 6, 19–30. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Number | MCF-7 | HepG-2 | HCT-116 |
|---|---|---|---|
| 3 | 53.3 ± 1.9 | 27.4 ± 0.8 | 30.2 ± 0.7 |
| 4 | 23.5 ± 0.8 | 6.93 ± 0.3 | 15.2 ± 0.4 |
| 5 | 111 ± 2.9 | 57.1 ± 1.4 | 137 ± 6.4 |
| 6 | 31.1 ± 0.7 | 19.7 ± 0.9 | 27.7 ± 0.9 |
| 7 | 15.5 ± 0.8 | 7.4 ± 0.5 | 8.64 ± 0.8 |
| 8 | 14.6 ± 1.2 | 10.1 ± 0.9 | 15 ± 0.6 |
| 10 | 36.6 ± 1.8 | 26.5 ± 0.4 | 29.4 ± 0.7 |
| 11 | 46.9 ± 3.4 | 29.4 ± 0.8 | 40.7 ± 2.3 |
| 12 | 28.8 ± 0.8 | 11.6 ± 0.8 | 22 ± 0.7 |
| 15 | 15.5 ± 0.7 | 11.8 ± 0.6 | 19.4 ± 1.8 |
| 16 | 6.52 ± 0.6 | 3.82 ± 0.3 | 4.73 ± 0.7 |
| Cisplatin | 5.71 ± 0.4 | 3.67 ± 0.2 | 2.43 ± 0.2 |
| Compd. | Est. Free Energy of Binding kcal/mol | Est. Inhibition Constant, Ki uM | vdW + Hbond + desolv Energy kcal/mol | Electrostatic Energy kcal/mol | Total Intermolec. Energy kcal/mol | Interact Surface |
|---|---|---|---|---|---|---|
| 3 | −7.04 | 6.90 | −8.22 | +0.04 | −8.17 | 509.119 |
| 4 | −7.51 | 3.12 | −8.09 | −0.02 | −8.11 | 465.35 |
| 5 | −8.31 | 8.14 | −9.44 | −0.04 | −9.48 | 523.761 |
| 6 | −7.98 | 1.41 | −8.03 | −0.25 | −8.28 | 508.448 |
| 7 | −8.13 | 1.09 | −9.18 | −0.03 | −9.21 | 519.896 |
| 8 | −6.35 | 22.23 | −6.41 | +0.06 | −6.35 | 522.705 |
| 12 | −0.96 | 1.97 | −1.87 | +0.61 | −1.26 | 583.589 |
| 13 | −0.26 | 6.47 | −7.64 | +0.01 | −7.63 | 576.654 |
| 16 | −7.95 | 1.49 | −8.16 | −0.40 | −8.56 | 540.988 |
| Compd. | Est. Free Energy of Binding kcal/mol | Est. Inhibition Constant, Ki uM | vdW + Hbond + desolv Energy kcal/mol | Electrostatic Energy kcal/mol | Total Intermolec. Energy kcal/mol | Interact. Surface |
|---|---|---|---|---|---|---|
| 3 | −7.55 | 2.90 | −8.28 | −0.18 | −8.47 | 700.877 |
| 4 | −7.66 | 2.43 | −8.03 | −0.06 | −8.09 | 639.72 |
| 5 | −7.68 | 2.34 | −8.94 | +0.03 | −8.91 | 740.419 |
| 6 | −7.93 | 1.53 | −8.22 | −0.01 | −8.23 | 719.146 |
| 9 | −9.31 | 14.94 | −11.12 | −0.85 | −11.97 | 1288.56 |
| 12 | −9.15 | 19.78 | −9.76 | +0.31 | −9.44 | 851.928 |
| 13 | −8.01 | 1.35 | −10.31 | −0.08 | −10.39 | 911.407 |
| 14 | −8.87 | 31.24 | −9.49 | −0.59 | −10.08 | 908.153 |
| 15 | −8.74 | 39.33 | −9.08 | +0.34 | −8.74 | 846.707 |
| Compd. | Hydrogen Bonds | Polar | cation-pi | Hydrophobic | Other |
|---|---|---|---|---|---|
| 3 | LEU704 (−0.9588) ASN705 (−0.2861) THR877 (0.0967) LEU873 (0.2094) | PHE764 (−0.9244) LEU707 (−0.5332) PHE876 (−0.4595) MET780 (−0.3859) MET749 (−0.3551) LEU701 (−0.1735) LEU880 (0.031) | GLN711 (−0.3009) ARG752 (−0.2282) PHE891 (−0.1823) | ||
| 4 | ASN705 (−0.6684) THR877 (−0.4461) | PHE764 (−1.321) LEU873 (−0.6886) LEU707 (−0.4798) LEU701 (−0.2013) | LEU704 (−1.2982) PHE876 (−0.5103) MET742 (−0.4068) PHE891 (−0.3538) LEU880 (0.2352) | ||
| 5 | PHE764 (−1.0213) GLN711 (−0.5111) | ARG752 (−0.2593) | LEU704 (−1.3263) LEU873 (−0.8151) LEU707 (−0.6963) MET745 (−0.6501) MET742 (−0.5522) PHE876 (−0.4912) VAL746 (−0.4163 TRP741 (−0.3903 LEU701 (−0.2362) MET780 (−0.0943) | MET749 (−0.5969) ASN705 (−0.5916) MET787 (−0.2392) | |
| 6 | MET742 (0.5327) ASN705 (1.1422) | THR877 (−12.988) | PHE891 (0.4955) PHE876 (1.6611) | PHE764 (−1.6911) MET749 (−0.4918) LEU707 (−0.1047) LEU704 (0.1685) LEU873 (1.6514) | GLN711 (−0.3671) VAL746 (−0.3662) ARG752 (−0.132) LEU880 (0.7122 LEU701 (1.3032) |
| 7 | GLN711 (−0.2307) MET787 (2.5309) | ARG752 (−0.3853) | LEU873 (−9.3441) PHE764 (−0.6167) MET749 (−0.5857) LEU704 (−0.5848) LEU701 (−0.1264) PHE876 (0.1242) MET780 (0.6784) | MET745 (−0.7229) LEU707 (−0.3795) ASN705 (−0.255) TRP741 (−0.2472) VAL746 (−0.1409 THR877 (0.2176 | |
| 8 | LEU704 (−2.7674) ASN705 (21.4001) | THR877 (−1.5585) | PHE876 (−0.703) | PHE764 (−2.7458) LEU701 (−2.0955) LEU873 (−1.5548) MET780 (−0.6025 MET749 (−0.457 VAL746 (1.5949) MET787 (3.0657) | PHE891 (−1.637) LEU880 (−0.6967) |
| 16 | GLN711 (−0.4793) MET745 (0.1156) | ASN705 (−1.6327) ARG752 (0.3987) | LEU701 (−2.1158) LEU880 (−0.6528) PHE764 (−0.5839) MET749 (−0.3359) PHE876 (1.1185) | LEU873 (−0.4761) LEU707 (−0.4553) MET787 (−0.2292 MET895 (−0.0748) THR877 (1.3407) |
| Compd. | Hydrogen Bonds | Polar | cation-pi | Hydrophobic | Other |
|---|---|---|---|---|---|
| 3 | SER12 (−0.443) THR190 (0.8132) | PHE192 (−1.7843) VAL188 (−1.341) PHE226 (−0.4523) | ALA191 (−0.3955) TYR155 (−0.2826) SER142 (−0.0999) | ||
| 4 | ASN90 (−1.0823) SER12 (−0.2674) GLY9 (−0.009) THR190 (0.5483) | ILE14 (−0.9193) CYS185 (1.4843) | TYR155 (−1.2233) PHE192 (−1.1514) THR140 (−0.9162) SER142 (4.0984) | ||
| 5 | ASN90 (−0.9996) SER12 (−0.3889) GLY9 (−0.145) THR190 (1.0804) | PHE192 (−1.0039) ILE14 (−0.9349) CYS185 (−0.1611) | TYR155 (−0.5008) LYS159 (−0.2892) SER142 (−0.2522) | ||
| 6 | VAL188 (−1.233) GLY186 (−0.7452) TYR155 (−0.6104) SER142 (−0.2699) THR190 (0.5399) | PHE192 (−1.5739) ILE14 (−1.0434) | ASN90 (−0.9337) PHE226 (−0.238) THR140 (−0.2162) PRO187 (1.0923) | ||
| 8 | VAL188 (−1.1105) ASN90 (−0.4933) THR190 (0.7801) | TYR155 (−0.4568) | PHE192 (−1.5267) ILE14 (−1.0426) | THR140 (−0.3635) LYS159 (−0.2711) SER142 (−0.0517) | |
| 9 | GLY94 (−0.9894) GLY92 (−0.811) SER11 (−0.6928) SER12 (9.0552) | LYS195 (−1.1394) TYR155 (−1.0113) | PHE192 (5.8101) | LEU93 (−1.8663) ILE14 (−1.7045) VAL196 (−1.0286) ALA106 (−0.3699) LEU95 (1.3807) | ASN90 (−2.3374) ALA91 (−1.5627) ARG37 (−0.5212) SER142 (−0.3955) CYS185 (−0.3436) |
| 14 | ASN90 (−0.8782) GLY92 (−0.822) SER12 (−0.5469) | ARG37 (−1.3972) SER11 (−0.4518) LYS159 (−0.3494) | PHE192 (−1.2886) ALA91 (−0.6477) VAL113 (−0.4438) VAL66 (−0.273) | LEU93 (−0.5816) THR140 (−0.3649) TYR155 (−0.3056) | |
| 15 | GLY92 (−0.5283) GLY9 (−0.4326) | ARG37 (−1.8678) SER12 (−0.5819) THR190 (−0.2891) | PHE192 (−1.547) | VAL113 (−0.8616) ALA91 (−0.383) VAL66 (−0.2579) | LEU93 (−0.6114) ARG67 (−0.4835) ALA191 (−0.1625) ASP65 (−0.0007) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, R.S.; Saad, H.A. Synthesis of Some Novel Fused Pyrimido[4″,5″:5′,6′]-[1,2,4]triazino[3′,4′:3,4] [1,2,4]triazino[5,6-b]indoles with Expected Anticancer Activity. Molecules 2018, 23, 693. https://doi.org/10.3390/molecules23030693
Ali RS, Saad HA. Synthesis of Some Novel Fused Pyrimido[4″,5″:5′,6′]-[1,2,4]triazino[3′,4′:3,4] [1,2,4]triazino[5,6-b]indoles with Expected Anticancer Activity. Molecules. 2018; 23(3):693. https://doi.org/10.3390/molecules23030693
Chicago/Turabian StyleAli, Rania S., and Hosam A. Saad. 2018. "Synthesis of Some Novel Fused Pyrimido[4″,5″:5′,6′]-[1,2,4]triazino[3′,4′:3,4] [1,2,4]triazino[5,6-b]indoles with Expected Anticancer Activity" Molecules 23, no. 3: 693. https://doi.org/10.3390/molecules23030693
APA StyleAli, R. S., & Saad, H. A. (2018). Synthesis of Some Novel Fused Pyrimido[4″,5″:5′,6′]-[1,2,4]triazino[3′,4′:3,4] [1,2,4]triazino[5,6-b]indoles with Expected Anticancer Activity. Molecules, 23(3), 693. https://doi.org/10.3390/molecules23030693