
Radical-Mediated Reactions of α-Bromo Aluminium Thioacetals, α-Bromothioesters, and Xanthates for Thiolactone Synthesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
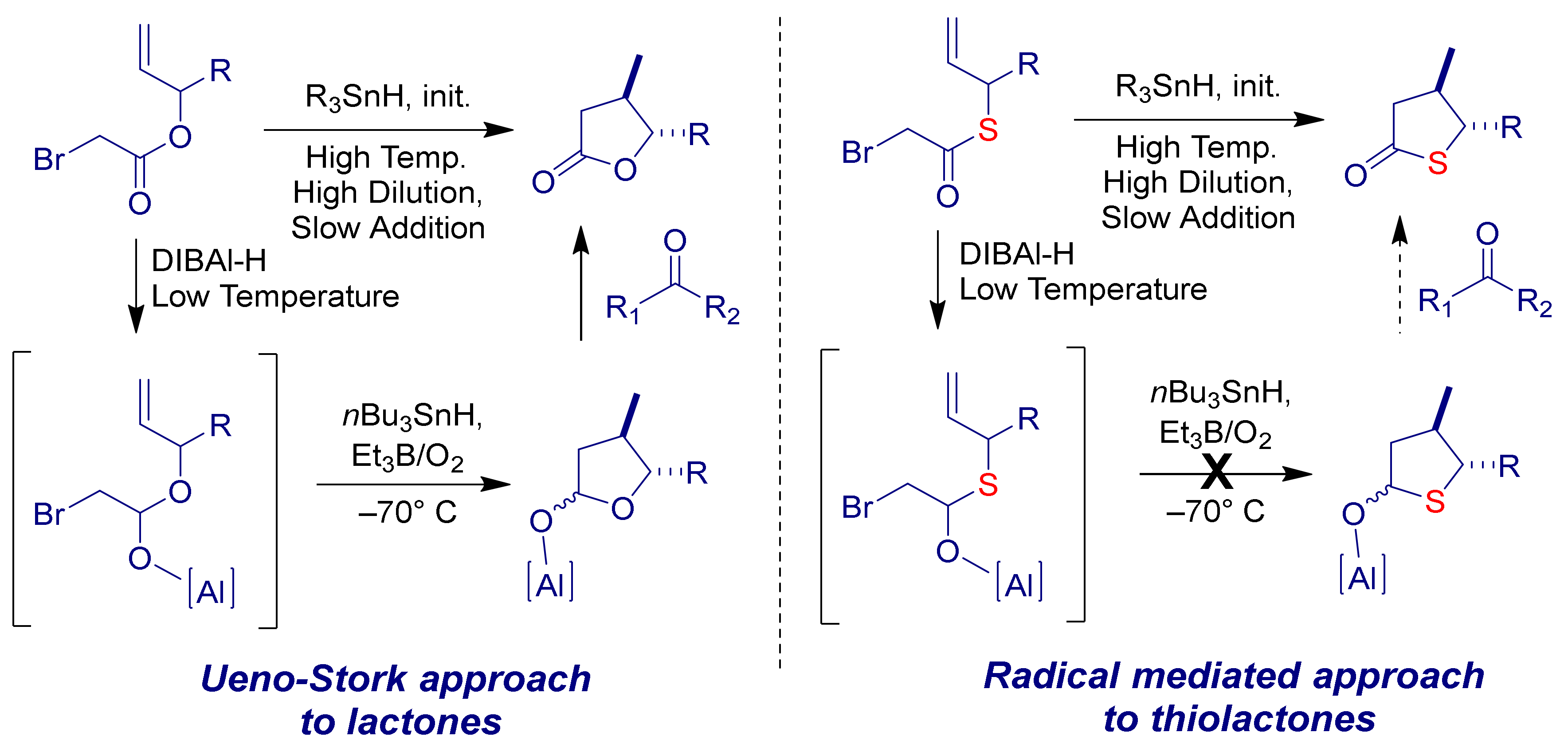
:1. Introduction
2. Results & Discussion
3. Materials and Methods
Instrumental and General Considerations
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ueno, Y.; Chino, K.; Watanabe, M.; Moriya, O.; Okawara, M. Homolytic carbocyclization by use of a heterogeneous supported organotin catalyst. A new synthetic route to 2-alkoxytetrahydrofurans and gamma-butyrolactones. J. Am. Chem. Soc. 1982, 104, 5564–5566. [Google Scholar] [CrossRef]
- Stork, G.; Mook, R.; Biller, S.A.; Rychnovsky, S.D. Free-radical cyclization of bromo acetals. Use in the construction of bicyclic acetals and lactones. J. Am. Chem. Soc. 1983, 105, 3741–3742. [Google Scholar] [CrossRef]
- Salom-Roig, X.J.; Dénès, F.; Renaud, P. Radical Cyclization of Haloacetals: The Ueno-Stork Reaction. Synthesis 2004, 1903–1928. [Google Scholar] [CrossRef]
- Zhang, J.-J.; Yan, C.-S.; Peng, Y.; Luo, Z.-B.; Xu, X.-B.; Wang, Y.-W. Total synthesis of (+/−)-sacidumlignans D and A through Ueno-Stork radical cyclization reaction. Org. Biomol. Chem. 2013, 11, 2498–2513. [Google Scholar] [CrossRef] [PubMed]
- Grieco, P.A.; Oguri, T.; Yokoyama, Y. One-step conversion of protected lactols into lactones. Tetrahedron Lett. 1978, 19, 419–420. [Google Scholar] [CrossRef]
- Gu, X.; Lu, P.; Fan, W.; Li, P.; Yao, Y. Visible light photoredox atom transfer Ueno-Stork reaction. Org. Biomol. Chem. 2013, 11, 7088–7091. [Google Scholar] [CrossRef] [PubMed]
- Boussonnière, A.; Dénès, F.; Lebreton, J. Radical Cyclization of α-Bromo Aluminum Acetals: An Easy Approach to γ-Lactols. Angew. Chem. Int. Ed. 2009, 48, 9549–9552. [Google Scholar] [CrossRef] [PubMed]
- Boussonnière, A.; Bénéteau, R.; Lebreton, J.; Dénès, F. Aluminum Acetals in Organic Synthesis. Eur. J. Org. Chem. 2013, 2013, 7853–7866. [Google Scholar] [CrossRef]
- Maruoka, K.; Yamamoto, H. Tetrahedron report number 239. Tetrahedron 1988, 44, 5001–5032. [Google Scholar] [CrossRef]
- Boussonnière, A.; Bénéteau, R.; Zimmermann, N.; Lebreton, J.; Dénès, F. Radical Cyclization of α-Bromo Aluminum Acetals onto Alkenes and Alkynes (Radic[Al] Process): A Simple Access to γ-Lactols and 4-Methylene-γ-Lactols. Chem. Eur. J. 2011, 17, 5613–5627. [Google Scholar] [CrossRef] [PubMed]
- Bénéteau, R.; Lebreton, J.; Dénès, F. A Convenient Access to γ-Lactones from O-Allyl-α-Bromoesters using a One-Pot Ionic–Radical–Ionic Sequence. Chem. Asian J. 2012, 7, 1516–1520. [Google Scholar] [CrossRef] [PubMed]
- Bénéteau, R.; Despiau, C.F.; Rouaud, J.-C.; Boussonnière, A.; Silvestre, V.; Lebreton, J.; Dénès, F. Synthesis of Polysubstituted γ-Butenolides via a Radical Pathway: Cyclization of α-Bromo Aluminium Acetals and Comparison with the Cyclization of α-Bromoesters at High Temperature. Chem. Eur. J. 2015, 21, 11378–11386. [Google Scholar] [CrossRef] [PubMed]
- Espeel, P.; Du Prez, F.E. One-pot multi-step reactions based on thiolactone chemistry: A powerful synthetic tool in polymer science. Eur. Polym. J. 2015, 62, 247–272. [Google Scholar] [CrossRef]
- Espeel, P.; Goethals, F.; Du Prez, F.E. One-Pot Multistep Reactions Based on Thiolactones: Extending the Realm of Thiol−Ene Chemistry in Polymer Synthesis. J. Am. Chem. Soc. 2011, 133, 1678–1681. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, D.V.; Majer, P.; Ni, C.; Slusher, C.E.; Rais, R.; Wu, Y.; Wozniak, K.M.; Alt, J.; Rojas, C.; Slusher, B.S.; et al. δ-Thiolactones as Prodrugs of Thiol-Based Glutamate Carboxypeptidase II (GCPII) Inhibitors. J. Med. Chem. 2014, 57, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Bénéteau, R.; Boussonnière, A.; Rouaud, J.-C.; Lebreton, J.; Graton, J.; Jacquemin, D.; Sebban, M.; Oulyadi, H.; Hamdoun, G.; Hancock, A.N.; et al. Radical Cyclisation of α-Halo Aluminium Acetals: A Mechanistic Study. Chem. Eur. J. 2016, 22, 4809–4824. [Google Scholar] [CrossRef] [PubMed]
- Bastug, G.; Dierick, S.; Lebreux, F.; Markó, I.E. Highly Chemoselective Reduction of Carbonyl Groups in the Presence of Aldehydes. Org. Lett. 2012, 14, 1306–1309. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C.; Ferreri, C.; Melchiorre, M.; Sansone, A.; Torreggiani, A. Lipid Geometrical Isomerism: From Chemistry to Biology and Diagnostics. Chem. Rev. 2014, 114, 255–284. [Google Scholar] [CrossRef] [PubMed]
- Dénès, F.; Pichowicz, M.; Povie, G.; Renaud, P. Thiyl Radicals in Organic Synthesis. Chem. Rev. 2014, 114, 2587–2693. [Google Scholar] [CrossRef] [PubMed]
- Ram, R.N.; Gupta, D.K.; Soni, V.K. Copper(I)-Promoted Synthesis of Highly Substituted and Functionalized Tetrahydrothiophenes. Eur. J. Org. Chem. 2016, 2016, 3434–3440. [Google Scholar] [CrossRef]
- Frizon, T.E.; Rafique, J.; Saba, S.; Bechtold, I.H.; Gallardo, H.; Braga, A.L. Synthesis of Functionalized Organoselenium Materials: Selenides and Diselenides Containing Cholesterol. Eur. J. Org. Chem. 2015, 2015, 3470–3476. [Google Scholar] [CrossRef]
- Scanlan, E.M.; Corce, V.; Malone, A. Synthetic applications of intramolecular thiol-ene ‘click’ reactions. Molecules 2014, 19, 19137–19151. [Google Scholar] [CrossRef] [PubMed]
- McSweeney, L.; Dénès, F.; Scanlan, E.M. Thiyl-Radical Reactions in Carbohydrate Chemistry: From Thiosugars to Glycoconjugate Synthesis. Eur. J. Org. Chem. 2016, 2016, 2080–2095. [Google Scholar] [CrossRef]
- Beckwith, A.L.J.; Pigou, P.E. Formation of lactones via a radical ring closure mechanism. J. Chem. Soc. Chem. Commun. 1986, 85–86. [Google Scholar] [CrossRef]
- Beckwith, A.; Glover, S. Determination of the Rates of Ring-Closure of Oxygen-Containing Analogs of Hex-5-Enyl Radical by Kinetic Electron Spin Resonance Spectroscopy. Aust. J. Chem. 1987, 40, 157–173. [Google Scholar] [CrossRef]
- Clough, J.M.; Pattenden, G.; Wight, P.G. Radical cyclisations of propargyl bromoamides and propargyl bromoesters. New routes to tetramic acids, pyrrolinones, tetronic acids and butenolides. Tetrahedron Lett. 1989, 30, 7469–7472. [Google Scholar] [CrossRef]
- Belletire, J.L.; Mahmoodi, N.O. Direct butyrolactone production using tin hydride. Tetrahedron Lett. 1989, 30, 4363–4366. [Google Scholar] [CrossRef]
- Hanessian, S.; Di Fabio, R.; Marcoux, J.F.; Prud’homme, M. The synthesis of substituted lactones by intramolecular chirality transfer with stereodifferentiating chiral. alpha.-ester radical intermediates. J. Org. Chem. 1990, 55, 3436–3438. [Google Scholar] [CrossRef]
- Deerfield, D.W.; Pedersen, L.G. An ab initio quantum mechanical study of thioesters. J. Mol. Struct. THEOCHEM 1995, 358, 99–106. [Google Scholar] [CrossRef]
- Liebman, J.F.; Greenberg, A. The origin of rotational barriers in amides and esters. Biophys. Chem. 1974, 1, 222–226. [Google Scholar] [CrossRef]
- Wiberg, K.B.; Laidig, K.E. Barriers to rotation adjacent to double bonds. 3. The carbon-oxygen barrier in formic acid, methyl formate, acetic acid, and methyl acetate. The origin of ester and amide resonance. J. Am. Chem. Soc. 1987, 109, 5935–5943. [Google Scholar] [CrossRef]
- Zard, S.Z. On the Trail of Xanthates: Some New Chemistry from an Old Functional Group. Angew. Chem. Int. Ed. 1997, 36, 673–685. [Google Scholar] [CrossRef]
- Forbes, J.E.; Tailhan, C.; Zard, S.Z. Dithiocarbonate group transfer in radical chain reactions. Tetrahedron Lett. 1990, 31, 2565–2568. [Google Scholar] [CrossRef]
- Axon, J.; Boiteau, L.; Boivin, J.; Forbes, J.E.; Zard, S.Z. A new radical based synthesis of lactams and indolones from dithiocarbonates (xanthates). Tetrahedron Lett. 1994, 35, 1719–1722. [Google Scholar] [CrossRef]
- Simonet-Davin, R.; Zard, S.Z. A Direct Xanthate-Based Route to γ-Thiolactones. Synlett 2018, 29, 815–819. [Google Scholar]
- Curran, D.P.; Chang, C.T. Atom transfer cyclization reactions of .alpha.-iodo esters, ketones, and malonates: Examples of selective 5-exo, 6-endo, 6-exo, and 7-endo ring closures. J. Org. Chem. 1989, 54, 3140–3157. [Google Scholar] [CrossRef]
- Quiclet-Sire, B.; Zard, S.Z. An Unusual Route to Deoxysugars by Hydrogen Atom Transfer from Cyclohexane. Possible Manifestation of Polar Effects in a Radical Process. J. Am. Chem. Soc. 1996, 118, 9190–9191. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- Pennell, M.N.; Turner, P.G.; Sheppard, T.D. Gold- and Silver-Catalyzed Reactions of Propargylic Alcohols in the Presence of Protic Additives. Chem. Eur. J. 2012, 18, 4748–4758. [Google Scholar] [CrossRef] [PubMed]
- Hojo, M.; Sakuragi, R.; Okabe, S.; Hosomi, A. Allyl- and propargylchromium reagents generated by a chromium(iii) ate-type reagent as a reductant and their reactions with electrophiles. Chem. Commun. 2001, 357–358. [Google Scholar] [CrossRef]
- Gao, N.; Zhao, X. Synthesis of Chiral Allylic Thioesters: Enantio- and Regioselective Iridium-Catalyzed Allylations of KSAc. Eur. J. Org. Chem. 2013, 2013, 2708–2714. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available. |




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCourt, R.O.; Dénès, F.; Scanlan, E.M. Radical-Mediated Reactions of α-Bromo Aluminium Thioacetals, α-Bromothioesters, and Xanthates for Thiolactone Synthesis. Molecules 2018, 23, 897. https://doi.org/10.3390/molecules23040897
McCourt RO, Dénès F, Scanlan EM. Radical-Mediated Reactions of α-Bromo Aluminium Thioacetals, α-Bromothioesters, and Xanthates for Thiolactone Synthesis. Molecules. 2018; 23(4):897. https://doi.org/10.3390/molecules23040897
Chicago/Turabian StyleMcCourt, Ruairí O., Fabrice Dénès, and Eoin M. Scanlan. 2018. "Radical-Mediated Reactions of α-Bromo Aluminium Thioacetals, α-Bromothioesters, and Xanthates for Thiolactone Synthesis" Molecules 23, no. 4: 897. https://doi.org/10.3390/molecules23040897
APA StyleMcCourt, R. O., Dénès, F., & Scanlan, E. M. (2018). Radical-Mediated Reactions of α-Bromo Aluminium Thioacetals, α-Bromothioesters, and Xanthates for Thiolactone Synthesis. Molecules, 23(4), 897. https://doi.org/10.3390/molecules23040897