
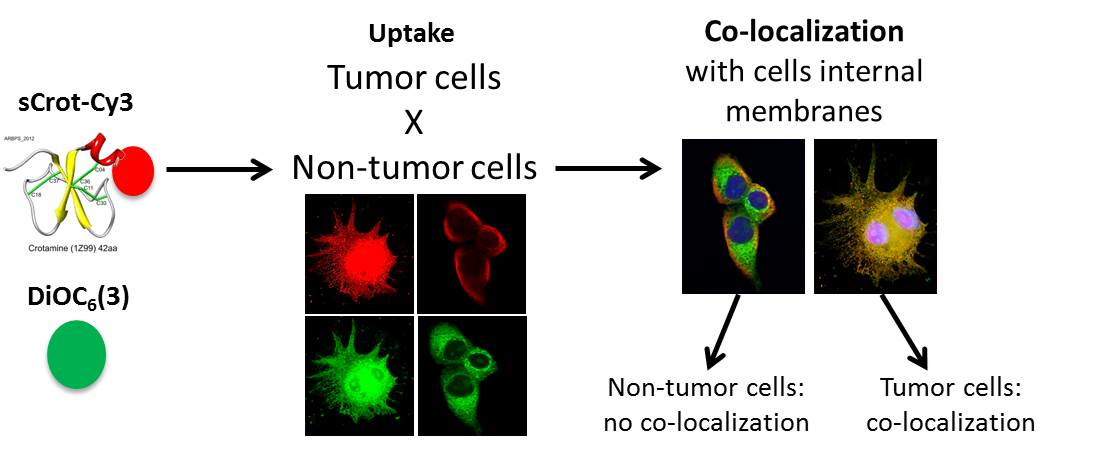
Co-Localization of Crotamine with Internal Membranes and Accentuated Accumulation in Tumor Cells

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Comparison of sCrot and nCrot
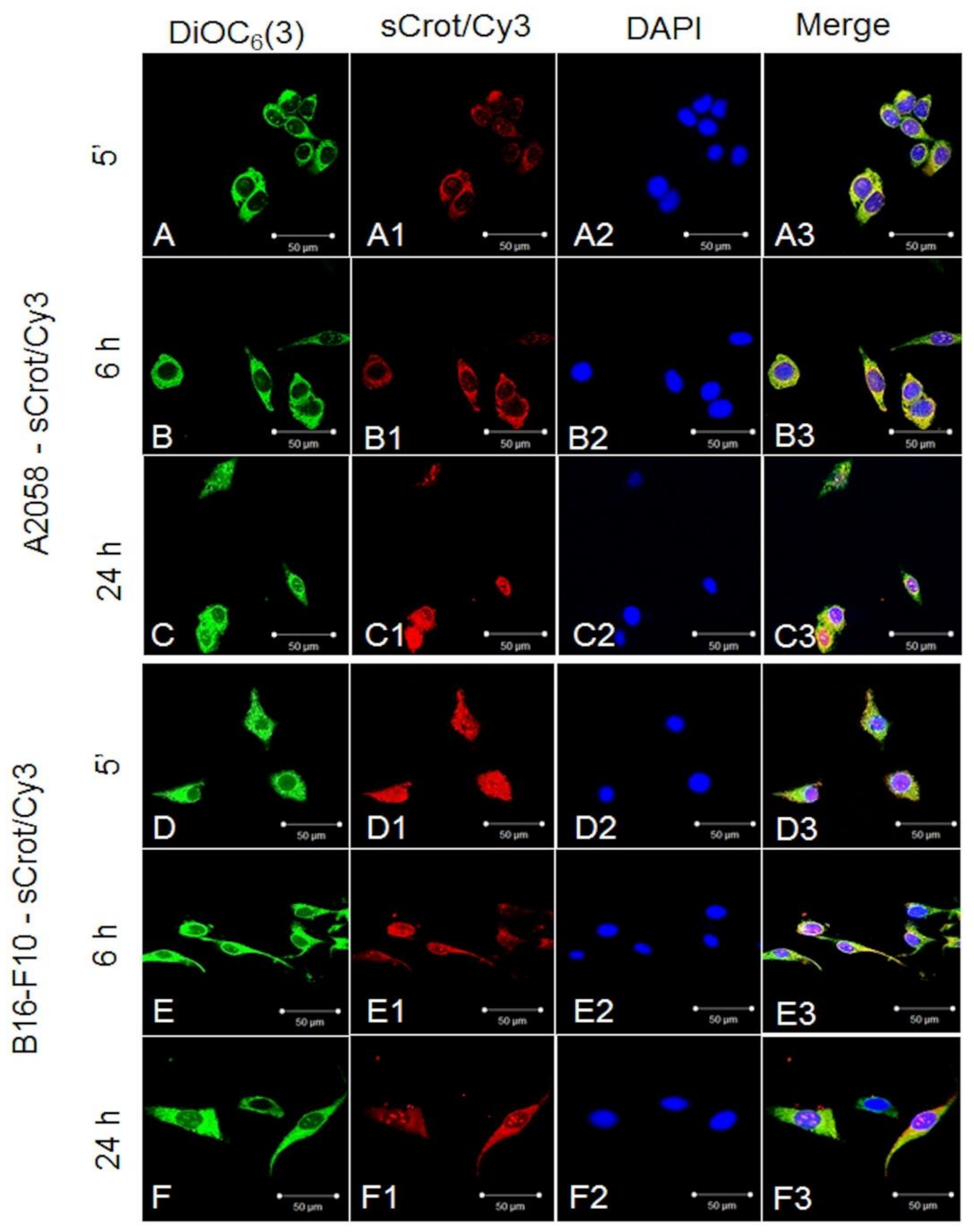
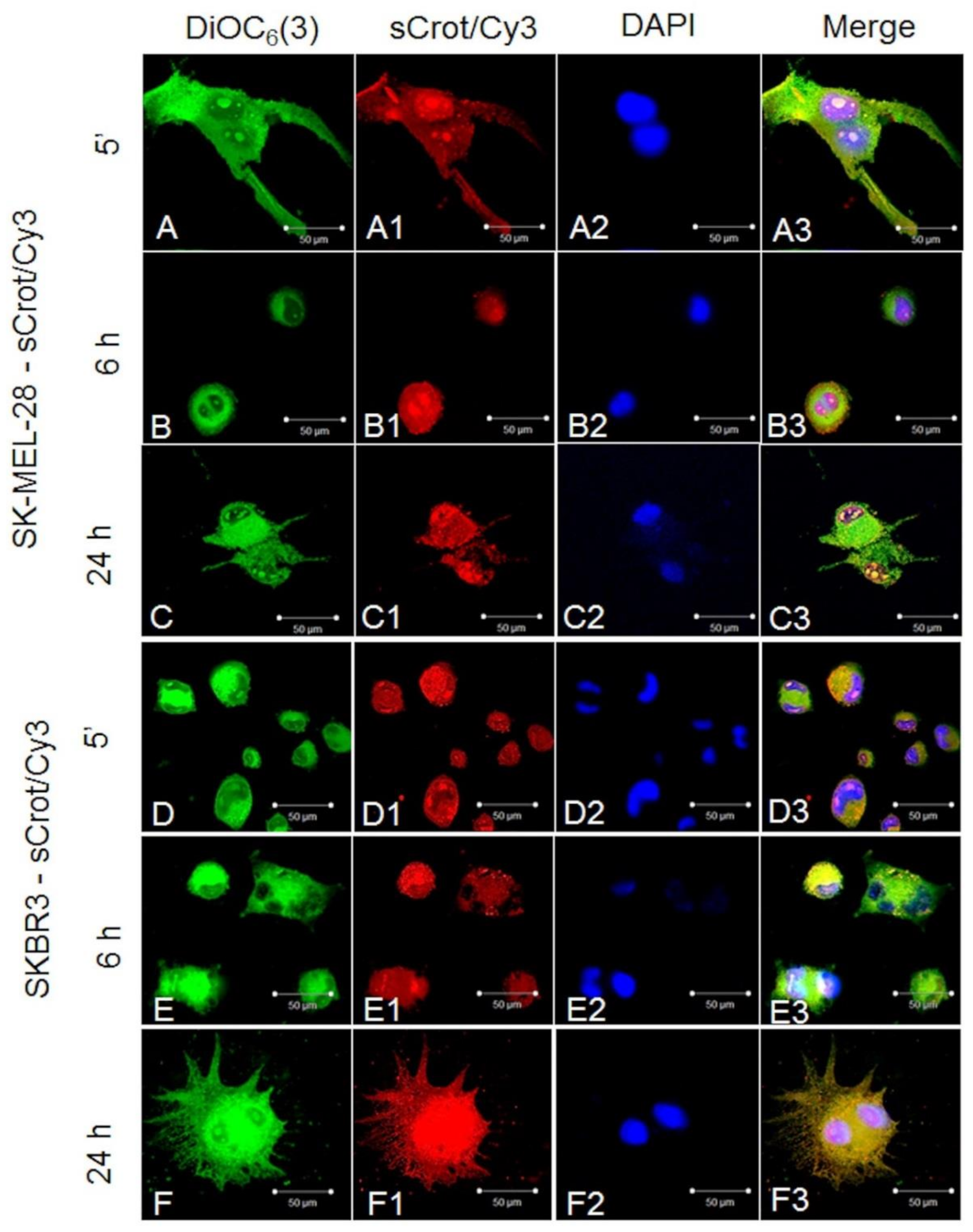
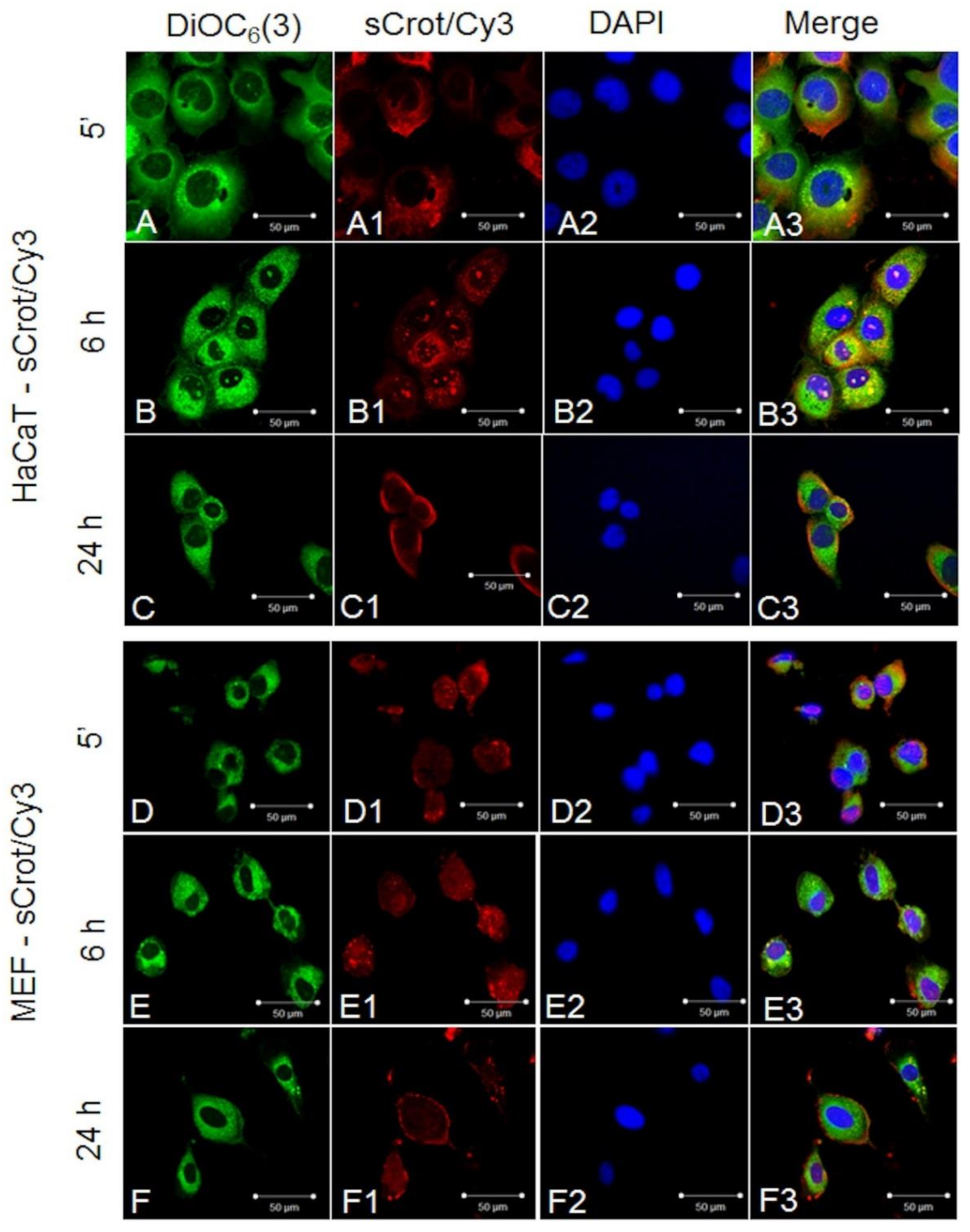
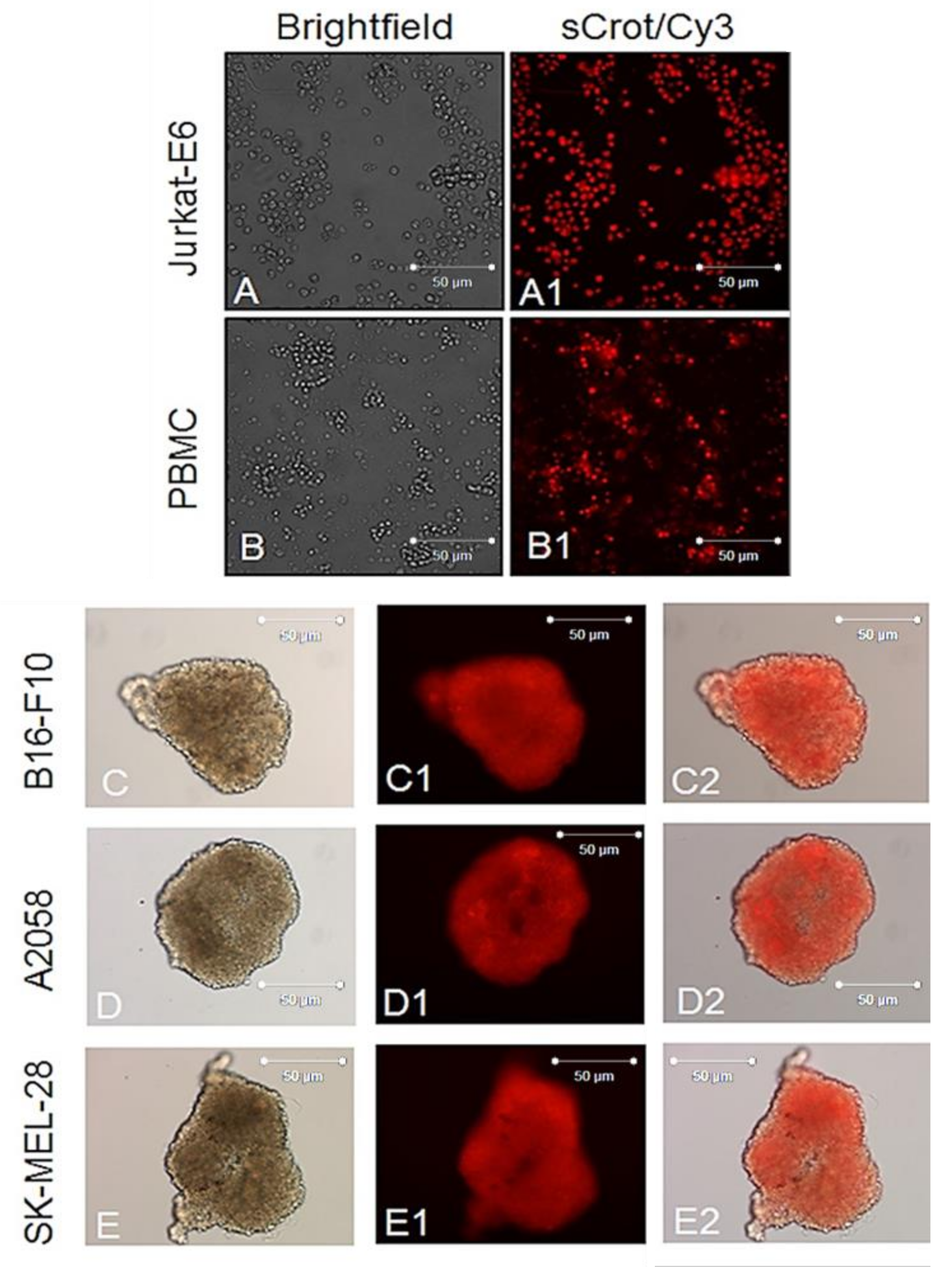
2.2. sCrot-Cy3 Uptake
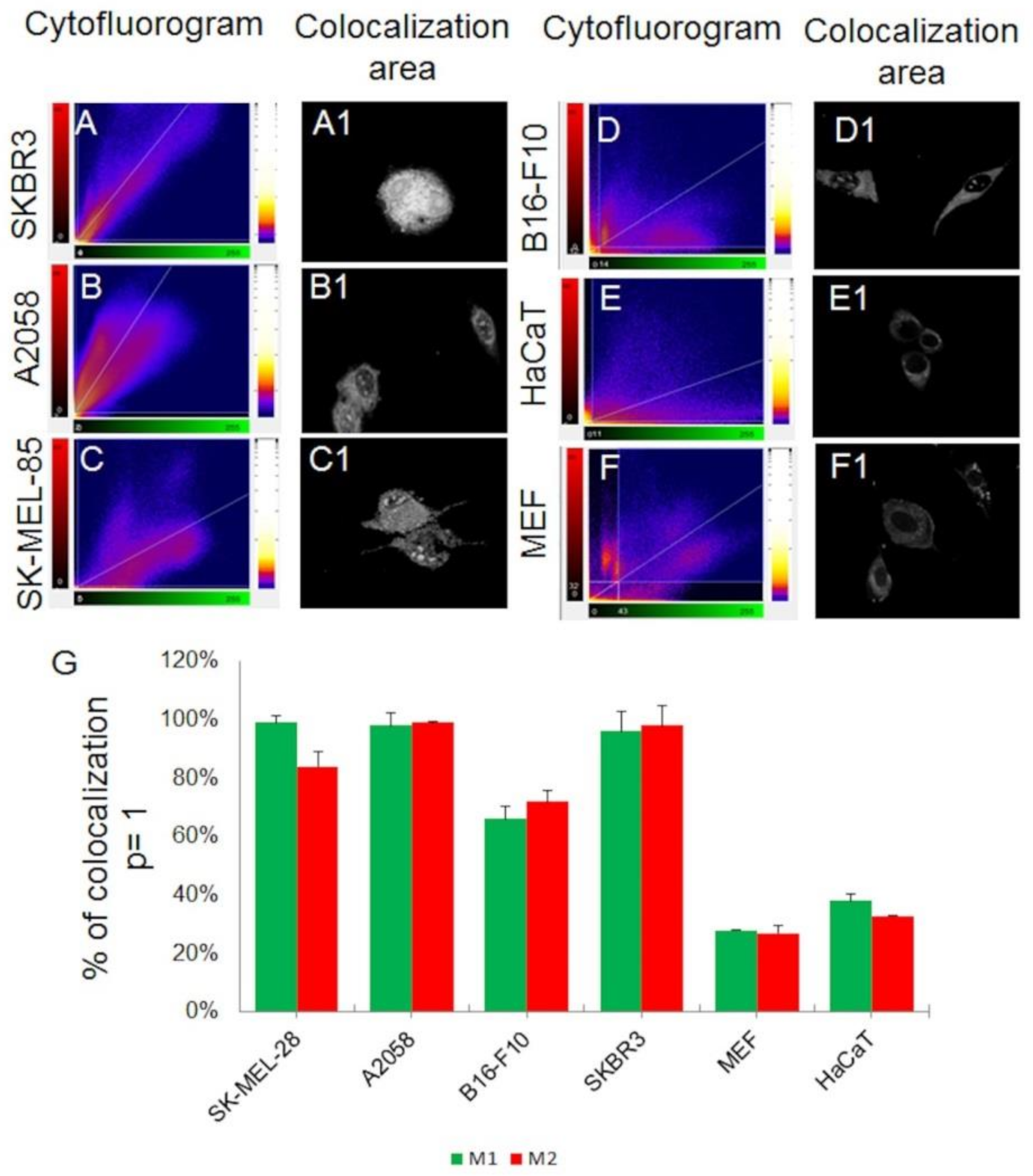
2.3. Uptake of sCrot-Cy3 in 3-D Models
2.4. Live-Cell Imaging by Time-Lapse Microscopy
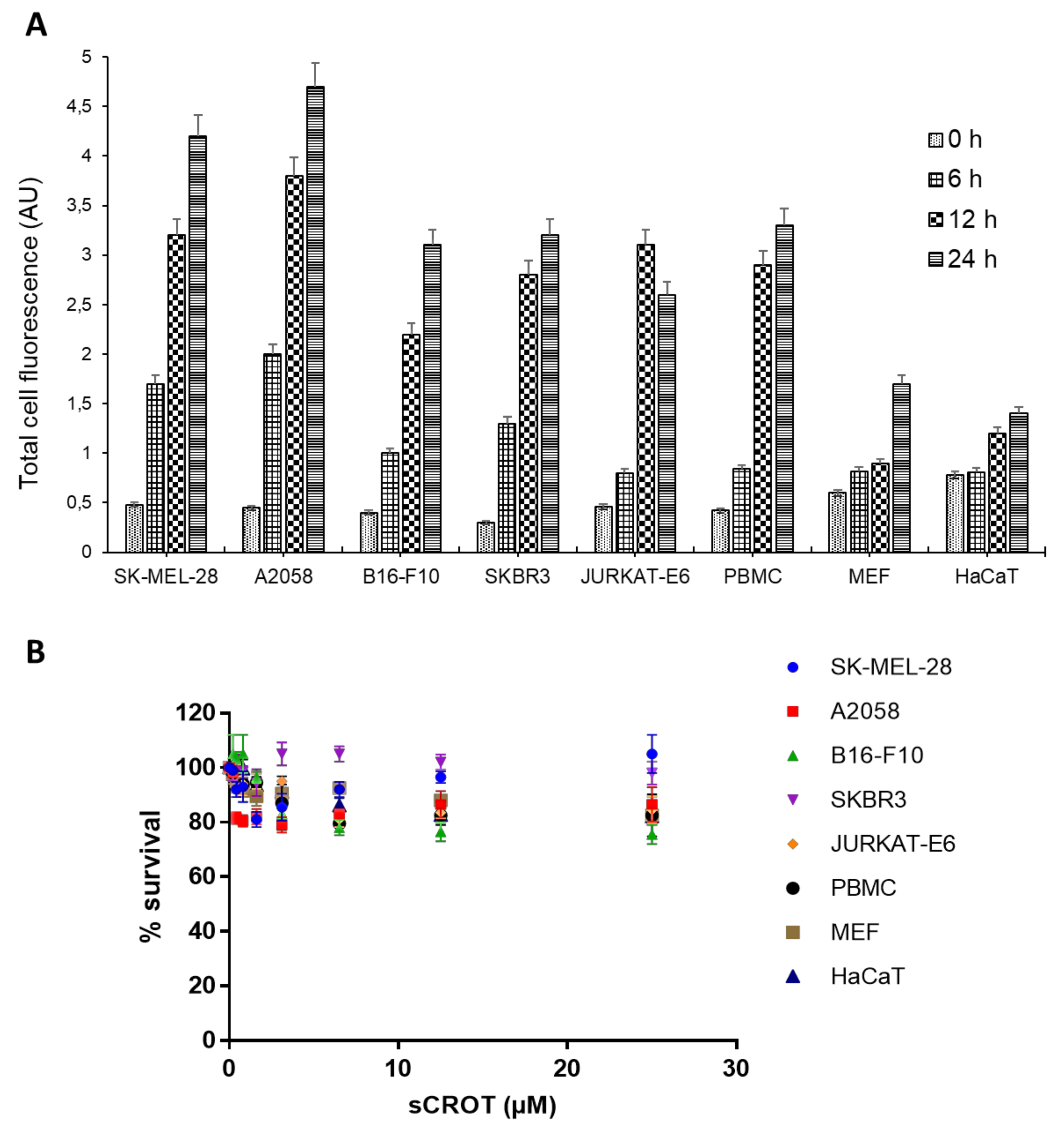
2.5. sCrot-Cy3 Fluorescence Intensity and Cell Viability
3. Discussion
4. Materials and Methods
4.1. Crotamine
4.2. Cell Lines and Culture Condition
4.3. Generation of Melanoma Spheroids and sCrot-Cy3 Uptake
4.4. Uptake Experiments
4.4.1. Confocal Laser Scanning Microscopy
4.4.2. High-Content Imaging: Time Lapse
4.5. Cell Viability Assay
4.6. Statistical Analyses
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kerkis, A.; Kerkis, I.; Radis-Baptista, G.; Oliveira, E.B.; Vianna-Morgante, A.M.; Pereira, L.V.; Yamane, T. Crotamine is a novel cell-penetrating protein from the venom of rattlesnake Crotalus durissus terrificus. FASEB J. 2004, 18, 1407–1409. [Google Scholar] [CrossRef] [PubMed]
- Kerkis, A.; Hayashi, M.A.F.; Yamane, T.; Kerkis, I. Properties of cell penetrating peptides (CPPs). IUBMB Life 2006, 58, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Kerkis, I.; Hayashi, M.A.F.; Prieto da Silva, A.R.B.; Pereira, A.; De Sa Junior, P.L.; Zaharenko, A.J.; Radis-Baptista, G.; Kerkis, A.; Yamane, T. State of the art in the studies on crotamine, a cell penetrating peptide from South American rattlesnake. Biomed. Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nascimento, F.D.; Hayashi, M.A.F.; Kerkis, A.; Oliveira, V.; Oliveira, E.B.; Radis-Baptista, G.; Nader, H.B.; Yamane, T.; Tersariol, I.L.D.; Kerkis, I. Crotamine mediates gene delivery into cells through the binding to heparan sulfate proteoglycans. J. Biol. Chem. 2007, 282, 21349–21360. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, A.; Futaki, S.; Harashima, H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: Ways to overcome endosomal entrapment. AAPS J. 2009, 11, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Raucher, D.; Ryu, J.S. Cell-penetrating peptides: Strategies for anticancer treatment. Trends Mol. Med. 2015, 21, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Bechara, C.; Sagan, S. Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013, 587, 1693–1702. [Google Scholar] [CrossRef] [PubMed]
- Gupta, B.; Levchenko, T.S.; Torchilin, V.P. Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides. Adv. Drug Deliv. Rev. 2005, 57, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Tsui, T.Y.; Ma, W.X. Intracellular delivery of molecular cargo using cell-penetrating peptides and the combination strategies. Int. J. Mol. Sci. 2015, 16, 19518–19536. [Google Scholar] [CrossRef] [PubMed]
- Munyendo, W.L.; Lv, H.; Benza-Ingoula, H.; Baraza, L.D.; Zhou, J. Cell penetrating peptides in the delivery of biopharmaceuticals. Biomolecules 2012, 2, 187–202. [Google Scholar] [CrossRef] [PubMed]
- Rathnayake, P.V.; Gunathunge, B.G.; Wimalasiri, P.N.; Karunaratne, D.N.; Ranatunga, R.J. Trends in the binding of cell penetrating peptides to siRNA: A molecular docking study. J. Biophys. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.C.; Doherty, T.; Waring, A.J.; Puchala, P.; Hong, M. Roles of arginine and lysine residues in the translocation of a cell-penetrating peptide from c-13, p-31, and f-19 solid-state NMR. Biochemistry 2009, 48, 4587–4595. [Google Scholar] [CrossRef] [PubMed]
- Zorko, M.; Langel, U. Cell-penetrating peptides: Mechanism and kinetics of cargo delivery. Adv. Drug Deliv. Rev. 2005, 57, 529–545. [Google Scholar] [CrossRef] [PubMed]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Esteve, E.; Mabrouk, K.; Dupuis, A.; Smida-Rezgui, S.; Altafaj, X.; Grunwald, D.; Platel, J.C.; Andreotti, N.; Marty, I.; Sabatier, J.M.; et al. Transduction of the scorpion toxin maurocalcine into cells—Evidence that the toxin crosses the plasma membrane. J. Biol. Chem. 2005, 280, 12833–12839. [Google Scholar] [CrossRef] [PubMed]
- Boisseau, S.; Mabrouk, K.; Ram, N.; Garmy, N.; Collin, V.; Tadmouri, A.; Mikati, M.; Sabatier, J.M.; Ronjat, M.; Fantini, J.; et al. Cell penetration properties of maurocalcine, a natural venom peptide active on the intracellular ryanodine receptor. Biochim. Biophys. Acta-Biomembr. 2006, 1758, 308–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahbazzadeh, D.; Srairi-Abid, N.; Feng, W.; Ram, N.; Borchani, L.; Ronjat, M.; Akbari, A.; Pessah, I.N.; De Waard, M.; El Ayeb, M. Hemicalcin, a new toxin from the iranian scorpion Hemiscorpius lepturus which is active on ryanodine-sensitive Ca2+ channels. Biochem. J. 2007, 404, 89–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurrola, G.B.; Capes, E.M.; Zamudio, F.Z.; Possani, L.D.; Valdivia, H.H. Imperatoxin a, a cell-penetrating peptide from scorpion venom, as a probe of Ca2+-release channels/ryanodine receptors. Pharmaceuticals (Basel) 2010, 3, 1093–1107. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, F.D.; Sancey, L.; Pereira, A.; Rome, C.; Oliveira, V.; Oliveira, E.B.; Nader, H.B.; Yamane, T.; Kerkis, I.; Tersariol, I.L.S.; et al. The natural cell-penetrating peptide crotamine targets tumor tissue in vivo and triggers a lethal calcium-dependent pathway in cultured cells. Mol. Pharm. 2012, 9, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.J.; Sung, B.H.; Shin, J.R.; Lee, Y.W.; Kim, D.J.; Yang, K.S.; Kim, S.C. A cancer specific cell-penetrating peptide, BR2, for the efficient delivery of an scFv into cancer cells. PLoS ONE 2013, 8, e66084. [Google Scholar] [CrossRef]
- Ponnappan, N.; Chugh, A. Cell-penetrating and cargo-delivery ability of a spider toxin-derived peptide in mammalian cells. Eur. J. Pharm. Biopharm. 2017, 114, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Kerkis, I.; de Brandão Prieto da Silva, A.R.; Pompeia, C.; Tytgat, J.; de Sá Junior, P.L. Toxin bioportides: Exploring toxin biological activity and multifunctionality. Cell Mol. Life Sci. 2017, 74, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Valdivia, H.H.; Kirby, M.S.; Lederer, W.J.; Coronado, R. Scorpion toxins targeted against the sarcoplasmic-reticulum Ca2+-release channel of skeletal and cardiac-muscle. Proc. Natl. Acad. Sci. USA 1992, 89, 12185–12189. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.A.F.; Nascimento, F.D.; Kerkis, A.; Oliveira, V.; Oliveira, E.B.; Pereira, A.; Radis-Baptista, G.; Nader, H.B.; Yamane, T.; Kerkis, I.; et al. Cytotoxic effects of crotamine are mediated through lysosomal membrane permeabilization. Toxicon 2008, 52, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, E.F.; Capes, E.M.; Diego-Garcia, E.; Zamudio, F.Z.; Fuentes, O.; Possani, L.D.; Valdivia, H.H. Characterization of hadrucalcin, a peptide from Hadrurus gertschi scorpion venom with pharmacological activity on ryanodine receptors. Br. J. Pharmacol. 2009, 157, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Radis-Baptista, G.; de la Torre, B.G.; Andreu, D. A novel cell-penetrating peptide sequence derived by structural minimization of a snake toxin exhibits preferential nucleolar localization. J. Med. Chem. 2008, 51, 7041–7044. [Google Scholar] [CrossRef] [PubMed]
- Radis-Baptista, G.; de la Torre, B.G.; Andreu, D. Insights into the uptake mechanism of NrTP, a cell-penetrating peptide preferentially targeting the nucleolus of tumour cells. Chem. Biol. Drug Des. 2012, 79, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Jha, D.; Mishra, R.; Gottschalk, S.; Wiesmuller, K.H.; Ugurbil, K.; Maier, M.E.; Engelmann, J. Cylop-1: A novel cysteine-rich cell-penetrating peptide for cytosolic delivery of cargoes. Bioconjug. Chem. 2011, 22, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Ponnappan, N.; Budagavi, D.P.; Chugh, A. Cylop-1: Membrane-active peptide with cell-penetrating and antimicrobial properties. Biochim. Biophys. Acta-Biomembr. 2017, 1859, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, M.; Furukawa, K.; Tu, A.T.; Ohizumi, Y. Calsequestrin is a major binding protein of myotoxin alpha and an endogenous Ca2+ releaser in sarcoplasmic reticulum. Eur. J. Pharmacol. 1994, 268, R1–R2. [Google Scholar] [CrossRef]
- Tu, A.T.; Morita, M. Attachment of rattlesnake venom myotoxin a to sarcoplasmic reticulum: Peroxidase conjugated method. Br. J. Exp. Pathol. 1983, 64, 633–637. [Google Scholar] [PubMed]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-penetrating peptides: From basic research to clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef] [PubMed]
- Vanregenmortel, M.H.V. Synthetic peptides versus natural antigens in immunoassays. Ann. Biol. Clin. 1993, 51, 39–41. [Google Scholar]
- Kolomin, T.; Shadrina, M.; Slominsky, P.; Limborska, S.; Myasoedov, N. A new generation of drugs: Synthetic peptides based on natural regulatory peptides. Neurosci. Med. 2013, 4, 223–252. [Google Scholar] [CrossRef]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L.J. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Sztiller-Sikorska, M.; Koprowska, K.; Jakubowska, J.; Zalesna, I.; Stasiak, M.; Duechler, M.; Czyz, M.E. Sphere formation and self-renewal capacity of melanoma cells is affected by the microenvironment. Melanoma Res. 2012, 22, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Pearson, K. On certain points connected with scale order in the case of the correlation of two characters which for some arrangement give a linear regression line. Biometrika 1906, 5, 176–178. [Google Scholar] [CrossRef]
- Manders, E.M.M.; Verbeek, F.J.; Aten, J.A. Measurement of colocalization of objects in dual-color confocal images. J. Microsc.-Oxf. 1993, 169, 375–382. [Google Scholar] [CrossRef]
- Costes, S.V.; Daelemans, D.; Cho, E.H.; Dobbin, Z.; Pavlakis, G.; Lockett, S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys. J. 2004, 86, 3993–4003. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lau, A.; Morris, T.J.; Guo, L.; Fordyce, C.B.; Stanley, E.F. A syntaxin 1, Galpha(o), and N-type calcium channel complex at a presynaptic nerve terminal: Analysis by quantitative immunocolocalization. J. Neurosci. 2004, 24, 4070–4081. [Google Scholar] [CrossRef] [PubMed]
- Manders, E.M.M.; Stap, J.; Brakenhoff, G.J.; Vandriel, R.; Aten, J.A. Dynamics of 3-dimensional replication patterns during the s-phase, analyzed by double labeling of DNA and confocal microscopy. J. Cell Sci. 1992, 103, 857–862. [Google Scholar] [PubMed]
- Hoppe, A.; Christensen, K.; Swanson, J.A. Fluorescence resonance energy transfer-based stoichiometry in living cells. Biophys. J. 2002, 83, 3652–3664. [Google Scholar] [CrossRef]
- Bolte, S.; Cordelieres, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc.-Oxf. 2006, 224, 213–232. [Google Scholar] [CrossRef] [PubMed]
- Schenberg, S. Geographical pattern of crotamine distribution in the same rattlesnake subspecies. Science 1959, 129, 1361–1363. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Tang, Z.Y.; Zhao, Y.; Yao, R.; Li, L.S.; Sun, W. Three-dimensional in vitro cancer models: A short review. Biofabrication 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Bielecka, Z.F.; Maliszewska-Olejniczak, K.; Safir, I.J.; Szczylik, C.; Czarnecka, A.M. Three-dimensional cell culture model utilization in cancer stem cell research. Biol. Rev. Camb. Philos. Soc. 2016, 92, 1505–1520. [Google Scholar] [CrossRef] [PubMed]
- Penfornis, P.; Vallabhaneni, K.C.; Janorkar, A.V.; Pochampally, R.R. Three dimensional tumor models for cancer studies. Front. Biosci. 2017, 9, 162–173. [Google Scholar]
- Geisler, I.; Chmielewski, J. Cationic amphiphilic polyproline helices: Side-chain variations and cell-specific internalization. Chem. Biol. Drug Des. 2009, 73, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Martín, I.; Teixidó, M.; Giralt, E. Building cell selectivity into CPP-mediated strategies. Pharmaceuticals (Basel) 2010, 3, 1456–1490. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.; Kerkis, A.; Hayashi, M.A.F.; Pereira, A.S.P.; Silva, F.S.; Oliveira, E.B.; Prieto da Silva, A.R.B.; Yamane, T.; Radis-Baptista, G.; Kerkis, I. Crotamine toxicity and efficacy in mouse models of melanoma. Expert Opin. Investig. Drugs 2011, 20, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Foty, R. A simple hanging drop cell culture protocol for generation of 3D spheroids. J. Vis. Exp. 2011, 51. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mambelli-Lisboa, N.C.; Sciani, J.M.; Brandão Prieto da Silva, A.R.; Kerkis, I. Co-Localization of Crotamine with Internal Membranes and Accentuated Accumulation in Tumor Cells. Molecules 2018, 23, 968. https://doi.org/10.3390/molecules23040968
Mambelli-Lisboa NC, Sciani JM, Brandão Prieto da Silva AR, Kerkis I. Co-Localization of Crotamine with Internal Membranes and Accentuated Accumulation in Tumor Cells. Molecules. 2018; 23(4):968. https://doi.org/10.3390/molecules23040968
Chicago/Turabian StyleMambelli-Lisboa, Nicole Caroline, Juliana Mozer Sciani, Alvaro Rossan Brandão Prieto da Silva, and Irina Kerkis. 2018. "Co-Localization of Crotamine with Internal Membranes and Accentuated Accumulation in Tumor Cells" Molecules 23, no. 4: 968. https://doi.org/10.3390/molecules23040968
APA StyleMambelli-Lisboa, N. C., Sciani, J. M., Brandão Prieto da Silva, A. R., & Kerkis, I. (2018). Co-Localization of Crotamine with Internal Membranes and Accentuated Accumulation in Tumor Cells. Molecules, 23(4), 968. https://doi.org/10.3390/molecules23040968