
Clinically Applicable Inhibitors Impacting Genome Stability




Abstract
:
1. Introduction
1.1. Macro and Micro Targeting of Genome Stability Processes
2. Small Molecule Inhibitors Targeted at the Chromosomal Level
2.1. Inhibitors Targeting Proteins Involved in Centrosome Duplication
2.2. Centrosome Amplification Inhibitors
2.3. Centrosome Clustering Inhibitors
3. Small Molecule Inhibitors Targeting Chromatin
3.1. Inhibitors Targeting DNA Damage Signalling and Processing
3.1.1. PARP Inhibitors
3.1.2. ATM and ATR Kinase Inhibitors
3.1.3. DNA Helicase Inhibitors
3.1.4. Topoisomerase Inhibitors
3.1.5. Mre11 Inhibitors
3.1.6. ERCC1–XPF Inhibitors
3.2. NHEJ Inhibitors
3.2.1. DNA-PK Inhibitors
3.2.2. Ligase IV Inhibitors
3.3. HR Inhibitors
3.3.1. RAD51 Inhibitors
3.3.2. RAD52 Inhibitors
3.3.3. RAD54 Inhibitors
3.4. Targeting Chromatin Remodelling
3.4.1. Acetylation Inhibitors
3.4.2. HDAC Inhibitors
3.4.3. Histone Acetyltransferase Inhibitors
3.4.4. Methylation Inhibitors
3.4.5. KMT Inhibitors
3.4.6. KDM Inhibitors
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DDR | DNA damage response |
| FDA | Food and Drug Administration (United States) |
| HAT | Histone acetyltransferase |
| HATi | HAT inhibitors |
| HDAC | Histone de-acetyltransferase |
| HDACi | Histone de-acetyltransferase inhibitor |
| HMT | Histone methyltransferases |
| KDM | Lysine demethylases |
| DSB | Double strand break |
| SSB | Single Strand Break |
| SSA | Single Strand Annealing |
| HR | Homologous recombination |
| NHEJ | Non-Homologous End Joining |
| CA | Centrosome Amplification |
| PARPi | PARP inhibitors |
| ATM | Ataxia-telangiectasia mutated |
| ATR | Ataxia and Rad3-related |
| PIKK | Phosphatidylinositol 3-kinase-related kinase |
| IR | Ionizing radiation |
| FA | Fanconi Anemia |
| MMC | Mitomycin C |
| MMEJ | Microhomology-Mediated End Joining |
| PTM | Post translational modifications |
References
- Matchett, K.B.; Lynam-Lennon, N.; Watson, R.W.; Brown, J.A.L. Advances in Precision Medicine: Tailoring Individualized Therapies. Cancers (Basal) 2017, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.L.; Ni Chonghaile, T.; Matchett, K.B.; Lynam-Lennon, N.; Kiely, P.A. Big Data-Led Cancer Research, Application, and Insights. Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Arnold, D.; Casali, P.G.; Cervantes, A.; Douillard, J.-Y.; Eggermont, A.; Eniu, A.; McGregor, K.; Peters, S.; Piccart, M.R.; et al. Delivering precision medicine in oncology today and in future-the promise and challenges of personalised cancer medicine: A position paper by the European Society for Medical Oncology (ESMO). Ann. Oncol. 2014, 25, 1673–1678. [Google Scholar] [CrossRef] [PubMed]
- Girotti, M.R.; Gremel, G.; Lee, R.; Galvani, E.; Rothwell, D.; Viros, A.; Mandal, A.K.; Lim, K.H.J.; Saturno, G.; Furney, S.J.; et al. Application of Sequencing, Liquid Biopsies, and Patient-Derived Xenografts for Personalized Medicine in Melanoma. Cancer Discov. 2016, 6, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; Gómez-González, B. Genome instability: A mechanistic view of its causes and consequences. Nat. Rev. Genet. 2008, 9, 204–217. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (Amst.) 2008, 7, 1765–1771. [Google Scholar] [CrossRef] [PubMed]
- Conduit, P.T.; Wainman, A.; Raff, J.W. Centrosome function and assembly in animal cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Lerit, D.A.; Poulton, J.S. Centrosomes are multifunctional regulators of genome stability. Chromosome Res. 2016, 24, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Bornens, M. The centrosome in cells and organisms. Science 2012, 335, 422–426. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.A.; Holland, A.J. Once and only once: Mechanisms of centriole duplication and their deregulation in disease. Nat. Rev. Mol. Cell Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Serçin, Ö.; Larsimont, J.-C.; Karambelas, A.E.; Marthiens, V.; Moers, V.; Boeckx, B.; Mercier, M.L.; Lambrechts, D.; Basto, R.; Blanpain, C. Transient PLK4 overexpression accelerates tumorigenesis in p53-deficient epidermis. Nat. Cell Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.S.; Bakker, B.; Boeckx, B.; Moyett, J.; Lu, J.; Vitre, B.; Spierings, D.C.; Lansdorp, P.M.; Cleveland, D.W.; Lambrechts, D.; et al. Centrosome Amplification Is Sufficient to Promote Spontaneous Tumorigenesis in Mammals. Dev. Cell 2017, 40, 313–322.e5. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.Y. A clinical overview of centrosome amplification in human cancers. Int. J. Biol. Sci. 2011, 7, 1122–1144. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Scorsone, K.; Ge, G.; Kaffes, C.C.; Dobrolecki, L.E.; Mukherjee, M.; Lewis, M.T.; Berg, S.; Stephan, C.C.; Pati, D. Identification and Characterization of Separase Inhibitors (Sepins) for Cancer Therapy. J. Biomol. Screen. 2014, 19, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Do, H.T.; Zhang, N.; Pati, D.; Gilbertson, S.R. Synthesis and activity of benzimidazole-1,3-dioxide inhibitors of separase. Bioorg. Med. Chem. Lett. 2016, 26, 4446–4450. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, R.E.A.; Ndiaye, M.A.; Liu, X.; Ahmad, N. Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol. Cancer Ther. 2016, 15, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Cholewa, B.D.; Ndiaye, M.A.; Huang, W.; Liu, X.; Ahmad, N. Small molecule inhibition of polo-like kinase 1 by volasertib (BI 6727) causes significant melanoma growth delay and regression in vivo. Cancer Lett. 2017, 385, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kim, J. PLK-1 Targeted Inhibitors and Their Potential against Tumorigenesis. Biomed. Res. Int. 2015, 2015, 705745. [Google Scholar] [CrossRef] [PubMed]
- Chohan, T.A.; Qian, H.; Pan, Y.; Chen, J.-Z. Cyclin-dependent kinase-2 as a target for cancer therapy: Progress in the development of CDK2 inhibitors as anti-cancer agents. Curr. Med. Chem. 2015, 22, 237–263. [Google Scholar] [CrossRef] [PubMed]
- Lane, M.E.; Yu, B.; Rice, A.; Lipson, K.E.; Liang, C.; Sun, L.; Tang, C.; McMahon, G.; Pestell, R.G.; Wadler, S. A novel cdk2-selective inhibitor, SU9516, induces apoptosis in colon carcinoma cells. Cancer Res. 2001, 61, 6170–6177. [Google Scholar] [PubMed]
- Matsumoto, Y.; Hayashi, K.; Nishida, E. Cyclin-dependent kinase 2 (Cdk2) is required for centrosome duplication in mammalian cells. Curr. Biol. CB 1999, 9, 429–432. [Google Scholar] [CrossRef]
- Yamamoto, H.; Monden, T.; Miyoshi, H.; Izawa, H.; Ikeda, K.; Tsujie, M.; Ohnishi, T.; Sekimoto, M.; Tomita, N.; Monden, M. Cdk2/cdc2 expression in colon carcinogenesis and effects of cdk2/cdc2 inhibitor in colon cancer cells. Int. J. Oncol. 1998, 13, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Mariaule, G.; Belmont, P. Cyclin-dependent kinase inhibitors as marketed anticancer drugs: Where are we now? A short survey. Molecules 2014, 19, 14366–14382. [Google Scholar] [CrossRef] [PubMed]
- DePinto, W.; Chu, X.-J.; Yin, X.; Smith, M.; Packman, K.; Goelzer, P.; Lovey, A.; Chen, Y.; Qian, H.; Hamid, R.; et al. In vitro and in vivo activity of R547: A potent and selective cyclin-dependent kinase inhibitor currently in phase I clinical trials. Mol. Cancer Ther. 2006, 5, 2644–2658. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, S.R.; Walton, M.I.; Garrett, M.D.; Workman, P. The Cyclin-dependent kinase inhibitor CYC202 (R-roscovitine) inhibits retinoblastoma protein phosphorylation, causes loss of Cyclin D1, and activates the mitogen-activated protein kinase pathway. Cancer Res. 2004, 64, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Raje, N. Seliciclib (CYC202 or R-roscovitine), a small-molecule cyclin-dependent kinase inhibitor, mediates activity via down-regulation of Mcl-1 in multiple myeloma. Blood 2005, 106, 1042–1047. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Wierda, W.G.; Chubb, S.; Hawtin, R.E.; Fox, J.A.; Keating, M.J.; Gandhi, V.; Plunkett, W. Mechanism of action of SNS-032, a novel cyclin-dependent kinase inhibitor, in chronic lymphocytic leukemia. Blood 2009, 113, 4637–4645. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.M.; Lin, D.C.-C.; Wei, X.; Che, Y.; Yao, Y.; Kiarash, R.; Cescon, D.W.; Fletcher, G.C.; Awrey, D.E.; Bray, M.R.; et al. Functional characterization of CFI-400945, a Polo-like kinase 4 inhibitor, as a potential anticancer agent. Cancer Cell 2014, 26, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Bavetsias, V.; Linardopoulos, S. Aurora Kinase Inhibitors: Current Status and Outlook. Front. Oncol. 2015, 5, 278. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, G.C.; Brokx, R.D.; Denny, T.A.; Hembrough, T.A.; Plum, S.M.; Fogler, W.E.; Sidor, C.F.; Bray, M.R. ENMD-2076 is an orally active kinase inhibitor with antiangiogenic and antiproliferative mechanisms of action. Mol. Cancer Ther. 2011, 10, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Woolery, J.E.; Mahadevan, D. Update on Aurora Kinase Targeted Therapeutics in Oncology. Expert Opin. Drug Discov. 2011, 6, 291–307. [Google Scholar] [CrossRef] [PubMed]
- Shiotsu, Y.; Kiyoi, H.; Ishikawa, Y.; Tanizaki, R.; Shimizu, M.; Umehara, H.; Ishii, K.; Mori, Y.; Ozeki, K.; Minami, Y.; et al. KW-2449, a novel multikinase inhibitor, suppresses the growth of leukemia cells with FLT3 mutations or T315I-mutated BCR/ABL translocation. Blood 2009, 114, 1607–1617. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Blagg, J.; Linardopoulos, S.; Pearson, A.D.J. Aurora kinase inhibitors: Novel small molecules with promising activity in acute myeloid and Philadelphia-positive leukemias. Leukemia 2010, 24, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Hoar, K.; Chakravarty, A.; Rabino, C.; Wysong, D.; Bowman, D.; Roy, N.; Ecsedy, J.A. MLN8054, a small-molecule inhibitor of Aurora A, causes spindle pole and chromosome congression defects leading to aneuploidy. Mol. Cell. Biol. 2007, 27, 4513–4525. [Google Scholar] [CrossRef] [PubMed]
- Sessa, F.; Villa, F. Structure of Aurora B-INCENP in complex with barasertib reveals a potential transinhibitory mechanism. Acta Crystallogr. F Struct. Biol. Commun. 2014, 70, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Jani, J.P.; Arcari, J.; Bernardo, V.; Bhattacharya, S.K.; Briere, D.; Cohen, B.D.; Coleman, K.; Christensen, J.G.; Emerson, E.O.; Jakowski, A.; et al. PF-03814735, an orally bioavailable small molecule aurora kinase inhibitor for cancer therapy. Mol. Cancer Ther. 2010, 9, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Henise, J.C.; Taunton, J. Irreversible Nek2 kinase inhibitors with cellular activity. J. Med. Chem. 2011, 54, 4133–4146. [Google Scholar] [CrossRef] [PubMed]
- 40. Carbain, B.; Bayliss, R.; Boxall, K.; Coxon, C.; Lebraud, H.; Matheson, C.; Turner, D.; Zhen-Wang, L.; Griffin, R.J. 188 2-arylamino-6-ethynylpurines as Potent Irreversible Inhibitors of the Mitotic Kinase Nek2. Eur. J. Cancer 2012, 48, 37. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Tovar, C.; Chen, S.; Knezevic, D.; Zhao, X.; Sun, H.; Heimbrook, D.C.; Chen, L. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc. Natl. Acad. Sci. USA 2006, 103, 10660–10665. [Google Scholar] [CrossRef] [PubMed]
- Steere, N.; Wagner, M.; Beishir, S.; Smith, E.; Breslin, L.; Morrison, C.G.; Hochegger, H.; Kuriyama, R. Centrosome amplification in CHO and DT40 cells by inactivation of cyclin-dependent kinases. Cytoskeleton (Hoboken) 2011, 68, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Furet, P.; Zimmermann, J.; Capraro, H.G.; Meyer, T.; Imbach, P. Structure-based design of potent CDK1 inhibitors derived from olomoucine. J. Comput. Aided Mol. Des. 2000, 14, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Dent, P.; Grant, S. Induction of apoptosis in human leukemia cells by the CDK1 inhibitor CGP74514A. Cell Cycle 2002, 1, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.M.; Collins, I. Recent findings and future directions for interpolar mitotic kinesin inhibitors in cancer therapy. Future Med. Chem. 2016, 8, 463–489. [Google Scholar] [CrossRef] [PubMed]
- Sarli, V.; Giannis, A. Targeting the kinesin spindle protein: Basic principles and clinical implications. Clin. Cancer Res. 2008, 14, 7583–7587. [Google Scholar] [CrossRef] [PubMed]
- Rath, O.; Kozielski, F. Kinesins and cancer. Nat Rev Cancer. 2012, 12, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Woessner, R.; Tunquist, B.; Lemieux, C.; Chlipala, E.; Jackinsky, S.; Dewolf, W., Jr.; Voegtli1, W.; Cox1, A.; Rana, S.; Lee, P.; Walker, D. ARRY-520, a novel KSP inhibitor with potent activity in hematological and taxane-resistant tumor models. Anticancer Res. 2009, 29, 4373–4380. [Google Scholar] [PubMed]
- Mardin, B.R.; Schiebel, E. Breaking the ties that bind: New advances in centrosome biology. J. Cell Biol. 2012, 197, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Agircan, F.G.; Schiebel, E.; Mardin, B.R. Separate to operate: Control of centrosome positioning and separation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130461. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.; Fofanov, V.; Panigrahi, A.; Merchant, F.; Zhang, N.; Pati, D. Overexpression and mislocalization of the chromosomal segregation protein separase in multiple human cancers. Clin. Cancer Res. 2009, 15, 2703–2710. [Google Scholar] [CrossRef] [PubMed]
- Kishi, K.; van Vugt, M.A.T.M.; Okamoto, K.-I.; Hayashi, Y.; Yaffe, M.B. Functional dynamics of Polo-like kinase 1 at the centrosome. Mol. Cell. Biol. 2009, 29, 3134–3150. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, K.; Rhee, K. PLK1 regulation of PCNT cleavage ensures fidelity of centriole separation during mitotic exit. Nat. Commun. 2015, 6, 10076. [Google Scholar] [CrossRef] [PubMed]
- Tsou, M.-F.B.; Wang, W.-J.; George, K.A.; Uryu, K.; Stearns, T.; Jallepalli, P.V. Polo kinase and separase regulate the mitotic licensing of centriole duplication in human cells. Dev. Cell 2009, 17, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Lončarek, J.; Hergert, P.; Khodjakov, A. Centriole Reduplication during Prolonged Interphase Requires Procentriole Maturation Governed by Plk1. Curr. Biol. 2010, 20, 1277–1282. [Google Scholar] [CrossRef] [PubMed]
- Zyss, D.; Gergely, F. Centrosome function in cancer: Guilty or innocent? Trends Cell Biol. 2009, 19, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Yim, H. Current clinical trials with polo-like kinase 1 inhibitors in solid tumors. Anticancer Drugs 2013, 24, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hong, M.J.; Chow, J.P.H.; Man, W.Y.; Mak, J.P.Y.; Ma, H.T.; Poon, R.Y.C. Co-inhibition of polo-like kinase 1 and Aurora kinases promotes mitotic catastrophe. Oncotarget 2015, 6, 9327–9340. [Google Scholar] [CrossRef] [PubMed]
- Godinho, S.A.; Pellman, D. Causes and consequences of centrosome abnormalities in cancer. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 1650. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Indjeian, V.B.; McManus, M.; Wang, L.; Dynlacht, B.D. CP110, a cell cycle-dependent CDK substrate, regulates centrosome duplication in human cells. Dev. Cell 2002, 3, 339–350. [Google Scholar] [CrossRef]
- Bourke, E.; Brown, J.A.L.; Takeda, S.; Hochegger, H.; Morrison, C.G. DNA damage induces Chk1-dependent threonine-160 phosphorylation and activation of Cdk2. Oncogene 2010, 29, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Korzeniewski, N.; Hohenfellner, M.; Duensing, S. The centrosome as potential target for cancer therapy and prevention. Expert Opin. Ther. Targets 2013, 17, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Duensing, A.; Ghanem, L.; Steinman, R.A.; Liu, Y.; Duensing, S. p21(Waf1/Cip1) deficiency stimulates centriole overduplication. Cell Cycle 2006, 5, 2899–2902. [Google Scholar] [CrossRef] [PubMed]
- Tetsu, O.; McCormick, F. Proliferation of cancer cells despite CDK2 inhibition. Cancer Cell 2003, 3, 233–245. [Google Scholar] [CrossRef]
- Lee, M.; Seo, M.Y.; Chang, J.; Hwang, D.S.; Rhee, K. PLK4 phosphorylation of CP110 is required for efficient centriole assembly. Cell Cycle 2017, 16, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.J.; Cleveland, D.W. Polo-like kinase 4 inhibition: A strategy for cancer therapy? Cancer Cell 2014, 26, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Kuriyama, R.; Bettencourt-Dias, M.; Hoffmann, I.; Arnold, M.; Sandvig, L. Gamma-tubulin-containing abnormal centrioles are induced by insufficient Plk4 in human HCT116 colorectal cancer cells. J. Cell Sci. 2009, 122, 2014–2023. [Google Scholar] [CrossRef] [PubMed]
- Sillibourne, J.E.; Bornens, M. Polo-like kinase 4: The odd one out of the family. Cell Div. 2010, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Meraldi, P.; Honda, R.; Nigg, E.A. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53-/- cells. EMBO J. 2002, 21, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Abbruzzese, J.L.; Izzo, J.; Hittelman, W.N.; Li, D. AURKA amplification, chromosome instability, and centrosome abnormality in human pancreatic carcinoma cells. Cancer Genet. Cytogenet. 2005, 159, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Conroy, P.C.; Saladino, C.; Dantas, T.J.; Lalor, P.; Dockery, P.; Morrison, C.G. C-NAP1 and rootletin restrain DNA damage-induced centriole splitting and facilitate ciliogenesis. Cell Cycle 2012, 11, 3769–3778. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Zhang, X. Targeting NEK2 as a promising therapeutic approach for cancer treatment. Cell Cycle 2016, 15, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Velimezi, G.; Liontos, M.; Vougas, K.; Roumeliotis, T.; Bartkova, J.; Sideridou, M.; Dereli-Oz, A.; Kocylowski, M.; Pateras, I.S.; Evangelou, K.; et al. Functional interplay between the DNA-damage-response kinase ATM and ARF tumour suppressor protein in human cancer. Nat. Cell Biol. 2013. [Google Scholar] [CrossRef]
- Manchado, E.; Guillamot, M.; Cárcer, G.; Eguren, M.; Trickey, M.; García-Higuera, I.; Moreno, S.; Yamano, H.; Cañamero, M.; Malumbres, M. Targeting mitotic exit leads to tumor regression in vivo: Modulation by Cdk1, Mastl, and the PP2A/B55α,δ phosphatase. Cancer Cell 2010, 18, 641–654. [Google Scholar] [CrossRef] [PubMed]
- McCloy, R.A.; Rogers, S.; Caldon, C.E.; Lorca, T.; Castro, A.; Burgess, A. Partial inhibition of Cdk1 in G2 phase overrides the SAC and decouples mitotic events. Cell Cycle 2014, 13, 1400–1412. [Google Scholar] [CrossRef] [PubMed]
- Chavali, P.L.; Chandrasekaran, G.; Barr, A.R.; Tátrai, P.; Taylor, C.; Papachristou, E.K.; Woods, C.G.; Chavali, S.; Gergely, F. A CEP215-HSET complex links centrosomes with spindle poles and drives centrosome clustering in cancer. Nat. Commun. 2016, 7, 11005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarli, V.; Huemmer, S.; Sunder-Plassmann, N.; Mayer, T.U.; Giannis, A. Synthesis and Biological Evaluation of Novel Eg5 Inhibitors. ChemBioChem 2005, 6, 2005–2013. [Google Scholar] [CrossRef] [PubMed]
- Bourke, E.; Dodson, H.; Merdes, A.; Cuffe, L.; Zachos, G.; Walker, M.; Gillespie, D.; Morrison, C.G. DNA damage induces Chk1-dependent centrosome amplification. EMBO Rep. 2007, 8, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Saladino, C.; Bourke, E.; Morrison, C.G. The Centrosome; Schatten, H., Ed.; Humana Press: New York, NY, USA, 2012; pp. 223–241. [Google Scholar] [CrossRef]
- Brown, J.A.L.; Bourke, E.; Liptrot, C.; Dockery, P.; Morrison, C.G. MCPH1/BRIT1 limits ionizing radiation-induced centrosome amplification. Oncogene 2010. [Google Scholar] [CrossRef] [PubMed]
- Saladino, C.; Bourke, E.; Conroy, P.C.; Morrison, C.G. Centriole separation in DNA damage-induced centrosome amplification. Environ. Mol. Mutagen. 2009, 50, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Dodson, H.; Bourke, E.; Jeffers, L.J.; Vagnarelli, P.; Sonoda, E.; Takeda, S.; Earnshaw, W.C.; Merdes, A.; Morrison, C. Centrosome amplification induced by DNA damage occurs during a prolonged G2 phase and involves ATM. EMBO J. 2004, 23, 3864–3873. [Google Scholar] [CrossRef] [PubMed]
- Hollander, M.C.; Fornace, A.J. Genomic instability, centrosome amplification, cell cycle checkpoints and Gadd45a. Oncogene 2002, 21, 6228–6233. [Google Scholar] [CrossRef] [PubMed]
- Bode, A.M.; Dong, Z. The enigmatic effects of caffeine in cell cycle and cancer. Cancer Lett. 2007, 247, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Gatei, M.; Sloper, K.; Sörensen, C.; Syljuäsen, R.; Falck, J.; Hobson, K.; Savage, K.; Lukas, J.; Zhou, B.-B.; Bartek, J.; et al. Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of Chk1 on Ser-317 in response to ionizing radiation. J. Biol. Chem. 2003, 278, 14806–14811. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Kumagai, A.; Wang, S.X.; Dunphy, W.G. Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev. 2000, 14, 2745–2756. [Google Scholar] [CrossRef] [PubMed]
- Kanai, M.; Tong, W.-M.; Sugihara, E.; Wang, Z.-Q.; Fukasawa, K.; Miwa, M. Involvement of poly(ADP-Ribose) polymerase 1 and poly(ADP-Ribosyl)ation in regulation of centrosome function. Mol. Cell. Biol. 2003, 23, 2451–2462. [Google Scholar] [CrossRef] [PubMed]
- Tentori, L.; Leonetti, C.; Scarsella, M.; d’Amati, G.; Portarena, I.; Zupi, G.; Bonmassar, E.; Graziani, G. Combined treatment with temozolomide and poly(ADP-ribose) polymerase inhibitor enhances survival of mice bearing hematologic malignancy at the central nervous system site. Blood 2002, 99, 2241–2244. [Google Scholar] [CrossRef] [PubMed]
- Lesueur, P.; Chevalier, F.; Austry, J.-B.; Waissi, W.; Burckel, H.; Noël, G.; Habrand, J.-L.; Saintigny, Y.; Joly, F. Poly-(ADP-ribose)-polymerase inhibitors as radiosensitizers: A systematic review of pre-clinical and clinical human studies. Oncotarget 2017, 8, 69105–69124. [Google Scholar] [CrossRef] [PubMed]
- De Soto, J.A.; Wang, X.; Tominaga, Y.; Wang, R.-H.; Cao, L.; Qiao, W.; Li, C.; Xu, X.; Skoumbourdis, A.P.; Prindiville, S.A.; et al. The inhibition and treatment of breast cancer with poly (ADP-ribose) polymerase (PARP-1) inhibitors. Int. J. Biol. Sci. 2006, 2, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M.; Tanaka, M.; Mushiake, M.; Takahashi, J.; Tanaka, K.; Watase, J.; Fujisawa, J.-I.; Miwa, M. Novel pathway of centrosome amplification that does not require DNA lesions. Cancer Sci. 2012, 103, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Bower, M.J.; McDevitt, P.J.; Zhao, H.; Davis, S.T.; Johanson, K.O.; Green, S.M.; Concha, N.O.; Zhou, B.-B.S. Structural basis for Chk1 inhibition by UCN-01. J. Biol. Chem. 2002, 277, 46609–46615. [Google Scholar] [CrossRef] [PubMed]
- Bridges, K.A.; Chen, X.; Liu, H.; Rock, C.; Buchholz, T.A.; Shumway, S.D.; Skinner, H.D.; Meyn, R.E. MK-8776, a novel chk1 kinase inhibitor, radiosensitizes p53-defective human tumor cells. Oncotarget 2016, 7, 71660–71672. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef] [PubMed]
- Sarker, D.; Ang, J.E.; Baird, R.; Kristeleit, R.; Shah, K.; Moreno, V.; Clarke, P.A.; Raynaud, F.I.; Levy, G.; Ware, J.A.; et al. First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2015, 21, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Berenjeno, I.M.; Piñeiro, R.; Castillo, S.D.; Pearce, W.; McGranahan, N.; Dewhurst, S.M.; Meniel, V.; Birkbak, N.J.; Lau, E.; Sansregret, L.; et al. Oncogenic PIK3CA induces centrosome amplification and tolerance to genome doubling. Nat. Commun. 2017, 8, 1773. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, S.; Flanagan, J.U.; Kolekar, S.; Buchanan, C.; Kendall, J.D.; Lee, W.-J.; Rewcastle, G.W.; Denny, W.A.; Singh, R.; Dickson, J.; et al. A drug targeting only p110α can block phosphoinositide 3-kinase signalling and tumour growth in certain cell types. Biochem. J. 2011, 438, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Mehta, P.P.; Yin, M.-J.; Sun, S.; Zou, A.; Chen, J.; Rafidi, K.; Feng, Z.; Nickel, J.; Engebretsen, J.; et al. PF-04691502, a potent and selective oral inhibitor of PI3K and mTOR kinases with antitumor activity. Mol. Cancer Ther. 2011, 10, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Surma, M.; Shi, S.; Lambert-Cheatham, N.; Shi, J. Novel Insights into the Roles of Rho Kinase in Cancer. Arch. Immunol. Ther. Exp. (Warsz.) 2016, 64, 259–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, H.-J.; Chae, S.; Jang, S.-H.; Cho, H.; Lee, J.-H. The PI3K-Akt mediates oncogenic Met-induced centrosome amplification and chromosome instability. Carcinogenesis 2010, 31, 1531–1540. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.W. Initiating breast cancer by PIK3CA mutation. Breast Cancer Res. BCR 2012, 14, 301. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.A. Centrosome aberrations: Cause or consequence of cancer progression? Nat. Rev. Cancer 2002, 2, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Coelho, P.A.; Bury, L.; Shahbazi, M.N.; Liakath-Ali, K.; Tate, P.H.; Wormald, S.; Hindley, C.J.; Huch, M.; Archer, J.; Skarnes, W.C.; et al. Over-expression of Plk4 induces centrosome amplification, loss of primary cilia and associated tissue hyperplasia in the mouse. Open Biol. 2015, 5, 150209. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Ferreira, I.; Bento, I.; Pimenta-Marques, A.; Jana, S.C.; Lince-Faria, M.; Duarte, P.; Borrego-Pinto, J.; Gilberto, S.; Amado, T.; Brito, D.; et al. Regulation of Autophosphorylation Controls PLK4 Self-Destruction and Centriole Number. Curr. Biol. CB 2013, 23, 2245–2254. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues-Martins, A.; Riparbelli, M.; Callaini, G.; Glover, D.M.; Bettencourt-Dias, M. Revisiting the role of the mother centriole in centriole biogenesis. Science 2007, 316, 1046–1050. [Google Scholar] [CrossRef] [PubMed]
- Rogers, G.C.; Rusan, N.M.; Roberts, D.M.; Peifer, M.; Rogers, S.L. The SCF Slimb ubiquitin ligase regulates Plk4/Sak levels to block centriole reduplication. J. Cell Biol. 2009, 184, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Bettencourt-Dias, M.; Hildebrandt, F.; Pellman, D.; Woods, G.; Godinho, S.A. Centrosomes and cilia in human disease. Trends Genet. 2011, 27, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Lohse, I.; Mason, J.; Mary, P.C.; Pintilie, M.; Bray, M.; Hedley, D.W. Activity of the novel polo-like kinase 4 inhibitor CFI-400945 in pancreatic cancer patient-derived xenografts. Oncotarget 2017, 8, 3064–3071. [Google Scholar] [CrossRef] [PubMed]
- Sredni, S.T.; Bailey, A.W.; Suri, A.; Hashizume, R.; He, X.; Louis, N.; Gokirmak, T.; Piper, D.R.; Watterson, D.M.; Tomita1, T. Inhibition of polo-like kinase 4 (PLK4): A new therapeutic option for rhabdoid tumors and pediatric medulloblastoma. Oncotarget 2017, 8, 111190–111212. [Google Scholar] [CrossRef] [PubMed]
- Metge, B.; Ofori-Acquah, S.; Stevens, T.; Balczon, R. Stat3 activity is required for centrosome duplication in Chinese hamster ovary cells. J. Biol. Chem. 2004, 279, 41801–41806. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.J.; Dedhar, S. Stat3 in mitosis: A new role in clustering excess centrosomes. Cell Cycle 2017, 16, 1557–1559. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.J.; Kawamura, E.; Gillespie, J.A.; Balgi, A.; Kannan, N.; Muller, W.J.; Roberge, M.; Dedhar, S. Stat3 regulates centrosome clustering in cancer cells via Stathmin/PLK1. Nat. Commun. 2017, 8, 15289. [Google Scholar] [CrossRef] [PubMed]
- Avalle, L.; Pensa, S.; Regis, G.; Novelli, F.; Poli, V. STAT1 and STAT3 in tumorigenesis: A matter of balance. JAKSTAT 2012, 1, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Kushner, E.J.; Ferro, L.S.; Liu, J.Y.; Durrant, J.R.; Rogers, S.L.; Dudley, A.C.; Bautch, V.L. Excess centrosomes disrupt endothelial cell migration via centrosome scattering. J. Cell Biol. 2014, 206, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; Zhang, L.; Gamper, A.M.; Fujita, T.; Wan, Y. APC/C-Cdh1: From cell cycle to cellular differentiation and genomic integrity. Cell Cycle 2010, 9, 3904–3912. [Google Scholar] [CrossRef] [PubMed]
- Hatano, T.; Sluder, G. The interrelationship between APC/C and Plk1 activities in centriole disengagement. Biol. Open 2012, 1, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; He, M.; Shah, A.A.; Wan, Y. Insights into APC/C: From cellular function to diseases and therapeutics. Cell Div. 2016, 11, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drosopoulos, K.; Tang, C.; Chao, W.C.H.; Linardopoulos, S. APC/C is an essential regulator of centrosome clustering. Nat. Commun. 2014, 5, 3686. [Google Scholar] [CrossRef] [PubMed]
- Sackton, K.L.; Dimova, N.; Zeng, X.; Tian, W.; Zhang, M.; Sackton, T.B.; Meaders, J.; Pfaff, K.L.; Sigoillot, F.; Yu, H.; et al. Synergistic blockade of mitotic exit by two chemical inhibitors of the APC/C. Nature 2014, 514, 646–649. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, J.; Wan, L.; Zhou, X.; Wang, Z.; Wei, W. Targeting Cdc20 as a novel cancer therapeutic strategy. Pharmacol. Ther. 2015, 151, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Huang, E.Y.-H.; Huang, S.-C.; Chung, B.-C. DNA-PK/Chk2 induces centrosome amplification during prolonged replication stress. Oncogene 2015, 34, 1263–1269. [Google Scholar] [CrossRef] [PubMed]
- Watts, C.A.; Watts, C.A.; Richards, F.M.; Bender, A.; Bond, P.J.; Korb, O.; Kern, O.; Riddick, M.; Owen, P.; Myers, R.M.; Raff, J.; et al. Design, synthesis, and biological evaluation of an allosteric inhibitor of HSET that targets cancer cells with supernumerary centrosomes. Chem. Biol. 2013, 20, 1399–1410. [Google Scholar] [CrossRef] [PubMed]
- Castiel, A.; Visochek, L.; Mittelman, L.; Dantzer, F.; Izraeli, S.; Cohen-Armon, M. A phenanthrene derived PARP inhibitor is an extra-centrosomes de-clustering agent exclusively eradicating human cancer cells. BMC Cancer 2011, 11, 412. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.-X.; Yang, W.-X. KIFC1: A promising chemotherapy target for cancer treatment? Oncotarget 2016, 7, 48656–48670. [Google Scholar] [CrossRef] [PubMed]
- Rønnest, M.H.; Rebacz, B.; Markworth, L.; Terp, A.; Larsen, T.; Krämer, A.; Clausen, M. Synthesis and structure-activity relationship of griseofulvin analogues as inhibitors of centrosomal clustering in cancer cells. J. Med. Chem. 2009, 52, 3342–3347. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Alpmann, P.; Blaum-Feder, S.; Krämer, S.; Endo, T.; Lu, D.; Carson, D.; Schmidt-Wolf, I.G.H. In vivo efficacy of griseofulvin against multiple myeloma. Leuk. Res. 2011, 35, 1070–1073. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.S.; Breitkreutz, I.; Anderhub, S.; Rønnest, M.H.; Leber, B.; Larsen, T.O.; Weiz, L.; Konotop, G.; Hayden, P.J.; Podar, K.; et al. GF-15, a novel inhibitor of centrosomal clustering, suppresses tumor cell growth in vitro and in vivo. Cancer Res. 2012, 72, 5374–5385. [Google Scholar] [CrossRef] [PubMed]
- Fielding, A.B.; Lim, S.; Montgomery, K.; Dobreva, I.; Dedhar, S. A critical role of integrin-linked kinase, ch-TOG and TACC3 in centrosome clustering in cancer cells. Oncogene 2011, 30, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Johannes, J.W.; Almeida, L.; Daly, K.; Ferguson, A.D.; Grosskurth, S.E.; Guan, H.; Howard, T.; Ioannidis, S.; Kazmirski, S.; Lamb, M.L.; et al. Discovery of AZ0108, an orally bioavailable phthalazinone PARP inhibitor that blocks centrosome clustering. Bioorg. Med. Chem. Lett. 2015, 25, 5743–5747. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.T.-J.; Gronthos, S.; Shi, S. Mesenchymal stem cells derived from dental tissues vs. those from other sources: Their biology and role in regenerative medicine. J. Dent. Res. 2009, 88, 792–806. [Google Scholar] [CrossRef] [PubMed]
- Visochek, L.; Castiel, A.; Mittelman, L.; Elkin, M.; Atias, D.; Golan, T.; Izraeli, S.; Peretz, T.; Cohen-Armon, M. Exclusive destruction of mitotic spindles in human cancer cells. Oncotarget 2017, 8, 20813–20824. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Zhang, Y.; Morris, J.; Ji, J.; Takeda, S.; Doroshow, J.H.; Pommier, Y. Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J. Pharmacol. Exp. Ther. 2014, 349, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shi, Y.; Maag, D.X.; Palma, J.P.; Patterson, M.J.; Ellis, P.A.; Surber, B.W.; Ready, D.B.; Soni, N.B.; Ladror, U.S.; et al. Iniparib nonselectively modifies cysteine-containing proteins in tumor cells and is not a bona fide PARP inhibitor. Clin. Cancer Res. 2012, 18, 510–523. [Google Scholar] [CrossRef] [PubMed]
- Ekblad, T.; Camaioni, E.; Schüler, H.; Macchiarulo, A. PARP inhibitors: Polypharmacology versus selective inhibition. FEBS J. 2013, 280, 3563–3575. [Google Scholar] [CrossRef] [PubMed]
- Kalra, J.; Warburton, C.; Fang, K.; Edwards, L.; Daynard, T.; Waterhouse, D.; Dragowska, W.; Sutherland, B.W.; Dedhar, S.; Gelmon, K.; et al. QLT0267, a small molecule inhibitor targeting integrin-linked kinase (ILK), and docetaxel can combine to produce synergistic interactions linked to enhanced cytotoxicity, reductions in P-AKT levels, altered F-actin architecture and improved treatment outcomes in an orthotopic breast cancer model. Breast Cancer Res. BCR 2009, 11, R25. [Google Scholar] [PubMed]
- Sampson, J.; O’Regan, L.; Dyer, M.J.S.; Bayliss, R.; Fry, A.M. Hsp72 and Nek6 Cooperate to Cluster Amplified Centrosomes in Cancer Cells. Cancer Res. 2017, 77, 4785–4796. [Google Scholar] [CrossRef] [PubMed]
- Massey, A.J.; Williamson, D.S.; Browne, H.; Murray, J.B.; Dokurno, P.; Shaw, T.; Macias, A.T.; Daniels, Z.; Geoffroy, S.; Dopson, M.; et al. A novel, small molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis in HCT116 colon carcinoma cells. Cancer Chemother. Pharmacol. 2010, 66, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, E.; Fielding, A.B.; Kannan, N.; Balgi, A.; Eaves, C.J.; Roberge, M.; Dedhar, S. Identification of novel small molecule inhibitors of centrosome clustering in cancer cells. Oncotarget 2013, 4, 1763–1776. [Google Scholar] [CrossRef] [PubMed]
- Karna, P.; Rida, P.C.; Pannu, V.; Gupta, K.K.; Dalton, W.B.; Joshi, H.; Yang, V.W.; Zhou, J.; Aneja, R. A novel microtubule-modulating noscapinoid triggers apoptosis by inducing spindle multipolarity via centrosome amplification and declustering. Cell Death Differ. 2011, 18, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Pannu, V.; Karna, P.; Sajja, H.K.; Shukla, D.; Aneja, R. Synergistic antimicrotubule therapy for prostate cancer. Biochem. Pharmacol. 2011, 81, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Konotop, G.; Bausch, E.; Nagai, T.; Turchinovich, A.; Becker, N.; Benner, A.; Boutros, M.; Mizuno, K.; Krämer, A.; Raab, M.S. Pharmacological Inhibition of Centrosome Clustering by Slingshot-Mediated Cofilin Activation and Actin Cortex Destabilization. Cancer Res. 2016, 76, 6690–6700. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Chen, M.; Liu, X.; Lu, Z.; Ding, Y.; Li, D. CP-673451, a platelet-derived growth-factor receptor inhibitor, suppresses lung cancer cell proliferation and migration. OTT 2014, 7, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- Rhys, A.D.; Monteiro, P.; Smith, C.; Vaghela, M.; Arnandis, T.; Kato, T.; Leitinger, B.; Sahai, E.; McAinsh, A.; Charras, G.; et al. Loss of E-cadherin provides tolerance to centrosome amplification in epithelial cancer cells. J. Cell Biol. 2018, 217, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Mountain, V.; Simerly, C.; Howard, L.; Ando, A.; Schatten, G.; Compton, D.A. The kinesin-related protein, HSET, opposes the activity of Eg5 and cross-links microtubules in the mammalian mitotic spindle. J. Cell Biol. 1999, 147, 351–366. [Google Scholar] [CrossRef] [PubMed]
- She, Z.-Y.; Yang, W.-X. Molecular mechanisms of kinesin-14 motors in spindle assembly and chromosome segregation. J. Cell Sci. 2017, 130, 2097–2110. [Google Scholar] [CrossRef] [PubMed]
- Krämer, A.; Maier, B.; Bartek, J. Centrosome clustering and chromosomal (in)stability: A matter of life and death. Mol. Oncol. 2011, 5, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Rebacz, B.; Larsen, T.O.; Clausen, M.H.; Rønnest, M.H.; Löffler, H.; Ho, A.D.; Krämer, A. Identification of griseofulvin as an inhibitor of centrosomal clustering in a phenotype-based screen. Cancer Res. 2007, 67, 6342–6350. [Google Scholar] [CrossRef] [PubMed]
- Pannu, V.; Rida, P.C.; Celik, B.; Turaga, R.C.; Ogden, A.; Cantuaria, G.; Gopalakrishnan, J.; Aneja, R. Centrosome-declustering drugs mediate a two-pronged attack on interphase and mitosis in supercentrosomal cancer cells. Cell Death Dis. 2014, 5, e1538. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-H.; Xiong, M.; Chen, X.P.; Xiao, Z.Y.; Zhao, Y.F.; Huang, Z.Y. PJ34, an inhibitor of PARP-1, suppresses cell growth and enhances the suppressive effects of cisplatin in liver cancer cells. Oncol. Rep. 2008, 20, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Donawho, C.K.; Luo, Y.; Luo, Y.-P.; Penning, T.D.; Bauch, J.L.; Bouska, J.J.; Bontcheva-Diaz, V.D.; Cox, B.F.; DeWeese, T.L.; Dillehay, L.E.; et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin. Cancer Res. 2007, 13, 2728–2737. [Google Scholar] [CrossRef] [PubMed]
- O’Regan, L.; Sampson, J.; Richards, M.W.; Knebel, A.; Roth, D.; Hood, F.E.; Straube, A.; Royle, S.J.; Bayliss, R.; Fry, A.M. Hsp72 is targeted to the mitotic spindle by Nek6 to promote K-fiber assembly and mitotic progression. J. Cell Biol. 2015, 209, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Lopus, M.; Naik, P.K. Taking aim at a dynamic target: Noscapinoids as microtubule-targeted cancer therapeutics. Pharmacol. Rep. 2015, 67, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Hojjat-Farsangi, M. Small-molecule inhibitors of the receptor tyrosine kinases: Promising tools for targeted cancer therapies. Int. J. Mol. Sci. 2014, 15, 13768–13801. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Hengel, S.R.; Spies, M.A.; Spies, M. Small-Molecule Inhibitors Targeting DNA Repair and DNA Repair Deficiency in Research and Cancer Therapy. Cell Chem. Biol. 2017, 24, 1101–1119. [Google Scholar] [CrossRef] [PubMed]
- Jeggo, P.A.; Pearl, L.H.; Carr, A.M. DNA repair, genome stability and cancer: A historical perspective. Nat. Rev. Cancer 2016, 16, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.L.; Ren, E.C. Functional Aspects of PARP1 in DNA Repair and Transcription. Biomolecules 2012, 2, 524–548. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.H. Recognition, signaling, and repair of DNA double-strand breaks produced by ionizing radiation in mammalian cells: The molecular choreography. Mutat. Res. 2012, 751, 158–246. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Pauli, C.; Hopkins, B.D.; Prandi, D.; Shaw, R.; Fedrizzi, T.; Sboner, A.; Sailer, V.; Augello, M.; Puca, L.; Rosati, R.; et al. Personalized In Vitro and In Vivo Cancer Models to Guide Precision Medicine. Cancer Discov. 2017, 7, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of Single-Strand Annealing and its Role in Genome Maintenance. Trends Genet. 2016, 32, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Frit, P.; Barboule, N.; Yuan, Y.; Gomez, D.; Calsou, P. Alternative end-joining pathway(s): Bricolage at DNA breaks. DNA Repair (Amst.) 2014, 17, 81–97. [Google Scholar] [CrossRef] [PubMed]
- McVey, M.; Lee, S.E. MMEJ repair of double-strand breaks (director’s cut): Deleted sequences and alternative endings. Trends Genet. 2008, 24, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Howard, S.M.; Yanez, D.A.; Stark, J.M. DNA damage response factors from diverse pathways, including DNA crosslink repair, mediate alternative end joining. PLoS Genet. 2015, 11, e1004943. [Google Scholar] [CrossRef] [PubMed]
- Chernikova, S.B.; Game, J.C.; Brown, J.M. Inhibiting homologous recombination for cancer therapy. Cancer Biol. Ther. 2012, 13, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Truong, L.N.; Li, Y.; Shi, L.Z.; Hwang, P.Y.; He, J.; Wang, H.; Razavian, N.; Berns, M.W.; Wu, X. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 7720–7725. [Google Scholar] [CrossRef] [PubMed]
- Sousa, F.G.; Matuo, R.; Soares, D.G.; Escargueil, A.E.; Henriques, J.A.; Larsen, A.K.; Saffi, J. PARPs and the DNA damage response. Carcinogenesis 2012, 33, 1433–1440. [Google Scholar] [CrossRef] [PubMed]
- Krietsch, J.; Caron, M.-C.; Gagné, J.-P.; Ethier, C.; Vignard, J.; Vincent, M.; Rouleau, M.; Hendzel, M.J.; Poirier, G.G.; Masson, J.-Y. PARP activation regulates the RNA-binding protein NONO in the DNA damage response to DNA double-strand breaks. Nucleic Acids Res. 2012, 40, 10287–10301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickson, I.; Zhao, Y.; Richardson, C.J.; Green, S.J.; Martin, N.M.; Orr, A.I.; Reaper, P.M.; Jackson, S.P.; Curtin, N.J.; Smith, G.C. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004, 64, 9152–9159. [Google Scholar] [CrossRef] [PubMed]
- Haince, J.-F.; Kozlov, S.; Dawson, V.L.; Dawson, T.M.; Hendzel, M.J.; Lavin, M.F.; Poirier, G.G. Ataxia telangiectasia mutated (ATM) signaling network is modulated by a novel poly(ADP-ribose)-dependent pathway in the early response to DNA-damaging agents. J. Biol. Chem. 2007, 282, 16441–16453. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Murata, S.; Zhang, C.; Finch, N.; Zhang, K.; Campo, L.; Breuer, E.K. Predictors and Modulators of Synthetic Lethality: An Update on PARP Inhibitors and Personalized Medicine. Biomed. Res. Int. 2016, 2016, 2346585. [Google Scholar] [CrossRef] [PubMed]
- Sonnenblick, A.; de Azambuja, E.; Azim, H.A.; Piccart, M. An update on PARP inhibitors—Moving to the adjuvant setting. Nat. Rev. Clin. Oncol. 2015, 12, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps17. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Plummer, R.; Stephens, P.; Aissat-Daudigny, L.; Cambois, A.; Moachon, G.; Brown, P.D.; Campone, M. Phase 1 dose-escalation study of the PARP inhibitor CEP-9722 as monotherapy or in combination with temozolomide in patients with solid tumors. Cancer Chemother. Pharmacol. 2014, 74, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Incorvaia, L.; Passiglia, F.; Rizzo, S.; Galvano, A.; Listì, A.; Barraco, N.; Maragliano, R.; Calò, V.; Natoli, C.; Ciaccio, M.; et al. ‘Back to a false normality’: New intriguing mechanisms of resistance to PARP inhibitors. Oncotarget 2017, 8, 23891–23904. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Burness, M.L.; Martin-Trevino, R.; Guy, J.; Bai, S.; Harouaka, R.; Brooks, M.D.; Shang, L.; Fox, A.; Luther, T.K.; et al. RAD51 Mediates Resistance of Cancer Stem Cells to PARP Inhibition in Triple-Negative Breast Cancer. Clin. Cancer Res. 2017, 23, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [PubMed]
- Domchek, S.M. Reversion Mutations with Clinical Use of PARP Inhibitors: Many Genes, Many Versions. Cancer Discov. 2017, 7, 937–939. [Google Scholar] [CrossRef] [PubMed]
- Golding, S.E.; Rosenberg, E.; Valerie, N.; Hussaini, I.; Frigerio, M.; Cockcroft, X.F.; Chong, W.Y.; Hummersone, M.; Rigoreau, L.; Menear, K.A.; et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol. Cancer Ther. 2009, 8, 2894–2902. [Google Scholar] [CrossRef] [PubMed]
- Velic, D.; Couturier, A.M.; Ferreira, M.T.; Rodrigue, A.; Poirier, G.G.; Fleury, F.; Masson, J.Y. DNA Damage Signalling and Repair Inhibitors: The Long-Sought-After Achilles’ Heel of Cancer. Biomolecules 2015, 5, 3204–3259. [Google Scholar] [CrossRef] [PubMed]
- Degorce, S.L.; Barlaam, B.; Cadogan, E.; Dishington, A.; Ducray, R.; Glossop, S.C.; Hassall, L.A.; Lach, F.; Lau, A.; McGuire, T.M.; et al. Discovery of Novel 3-Quinoline Carboxamides as Potent, Selective, and Orally Bioavailable Inhibitors of Ataxia Telangiectasia Mutated (ATM) Kinase. J. Med. Chem. 2016, 59, 6281–6292. [Google Scholar] [CrossRef] [PubMed]
- Fokas, E.; Prevo, R.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; Cornelissen, B.; Vallis, K.A.; Hammond, E.M.; Olcina, M.M.; Gillies, M.W.; et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012, 3, e441. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Wengner, A.M.; Siemeister, G.; Luecking, U.; Lefranc, J.; Lienau, P.; Deeg, G.; Lagkadinou, E.; Liu, Li.; Golfier, S.; Schatz, C.; et al. Abstract 836: ATR inhibitor BAY 1895344 shows potent anti-tumor efficacy in monotherapy and strong combination potential with the targeted alpha therapy Radium-223 dichloride in preclinical tumor models. Cancer Res. 2017, 77, 836. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F. Ataxia-telangiectasia: From a rare disorder to a paradigm for cell signalling and cancer. Nat. Rev. Mol. Cell Biol. 2008, 9, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y. ATM and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Mao, C.; Wu, J.; Li, S.; Ma, R.; Cao, H.; Ji, M.; Jing, C.; Tang, J. Improved ataxia telangiectasia mutated kinase inhibitor KU60019 provides a promising treatment strategy for non-invasive breast cancer. Oncol. Lett. 2014, 8, 2043–2048. [Google Scholar] [CrossRef] [PubMed]
- Barlaam, B.; Pike, K. Identifying high quality, potent and selective inhibitors of ATM kinase: Discovery of AZD0156. Eur. J. Cancer 2016, 61, S118. [Google Scholar] [CrossRef]
- Brandsma, I.; Fleuren, E.D.G.; Williamson, C.T.; Lord, C.J. Directing the use of DDR kinase inhibitors in cancer treatment. Expert Opin. Investig. Drugs 2017. [Google Scholar] [CrossRef] [PubMed]
- Rainey, M.D.; Charlton, M.E.; Stanton, R.V.; Kastan, M.B. Transient inhibition of ATM kinase is sufficient to enhance cellular sensitivity to ionizing radiation. Cancer Res. 2008, 68, 7466–7474. [Google Scholar] [CrossRef] [PubMed]
- Morgado-Palacin, I.; Day, A.; Murga, M.; Lafarga, V.; Anton, M.E.; Tubbs, A.; Chen, H.T.; Ergan, A.; Anderson, R.; Bhandoola, A.; et al. Targeting the kinase activities of ATR and ATM exhibits antitumoral activity in mouse models of MLL-rearranged AML. Sci. Signal. 2016, 9, ra91. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.B.; Newsome, D.; Wang, Y.; Boucher, D.M.; Eustace, B.; Gu, Y.; Hare, B.; Johnson, M.A.; Milton, S.; Murphy, C.E.; et al. Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget 2014, 5, 5674–5685. [Google Scholar] [CrossRef] [PubMed]
- Nishida, H.; Tatewaki, N.; Nakajima, Y.; Magara, T.; Ko, K.M.; Hamamori, Y.; Konishi, T. Inhibition of ATR protein kinase activity by schisandrin B in DNA damage response. Nucleic Acids Res. 2009, 37, 5678–5689. [Google Scholar] [CrossRef] [PubMed]
- Foote, K.M.; Blades, K.; Cronin, A.; Fillery, S.; Guichard, S.S.; Hassall, L.; Hickson, I.; Jacq, X.; Jewsbury, P.J.; McGuire, T.M.; et al. Discovery of 4-{4-[(3R)-3-Methylmorpholin-4-yl]-6-[1-(methylsulfonyl)cyclopropyl]pyrimidin-2-yl}-1H-indole (AZ20): A potent and selective inhibitor of ATR protein kinase with monotherapy in vivo antitumor activity. J. Med. Chem. 2013, 56, 2125–2138. [Google Scholar] [CrossRef] [PubMed]
- Veith, S.; Mangerich, A. RecQ helicases and PARP1 team up in maintaining genome integrity. Ageing Res. Rev. 2015, 23, 12–28. [Google Scholar] [CrossRef] [PubMed]
- Foote, K.M.; Lau, A.; Nissink, J.W.M. Drugging ATR: Progress in the development of specific inhibitors for the treatment of cancer. Future Med. Chem. 2015, 7, 873–891. [Google Scholar] [CrossRef] [PubMed]
- Kwok, M.; Davies, N.; Agathanggelou, A.; Smith, E.; Petermann, E.; Yates, E.; Brown, J.; Lau, A.; Stankovic, T. Synthetic lethality in chronic lymphocytic leukaemia with DNA damage response defects by targeting the ATR pathway. Lancet Oncol. 2015, 385 (Suppl. 1), S58. [Google Scholar] [CrossRef]
- Peasland, A.; Wang, L.Z.; Rowling, E.; Kyle, S.; Chen, T.; Hopkins, A.; Cliby, W.A.; Sarkaria, J.; Beale, G.; Edmondson, R.J.; et al. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br. J. Cancer 2011, 105, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Brosh, R.M. DNA helicases involved in DNA repair and their roles in cancer. Nat. Rev. Cancer 2013, 13, 542–558. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, K.A.; Gangloff, S.; Rothstein, R. The RecQ DNA helicases in DNA repair. Annu. Rev. Genet. 2010, 44, 393–417. [Google Scholar] [CrossRef] [PubMed]
- Ellis, N.A.; Groden, J.; Ye, T.Z.; Straughen, J.; Lennon, D.J.; Ciocci, S.; Proytcheva, M.; German, J. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell 1995, 83, 655–666. [Google Scholar] [CrossRef]
- Arora, H.; Chacon, A.H.; Choudhary, S.; McLeod, M.P.; Meshkov, L.; Nouri, K.; Izakovic, J. Bloom syndrome. Int. J. Dermatol. 2014, 53, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.E.; Oshima, J.; Fu, Y.H.; Wijsman, E.M.; Hisama, F.; Alisch, R.; Matthews, S.; Nakura, J.; Miki, T.; Ouais, S.; et al. Positional cloning of the Werner’s syndrome gene. Science 1996, 272, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Kitao, S.; Shimamoto, A.; Goto, M.; Miller, R.W.; Smithson, W.A.; Lindor, N.M.; Furuichi, Y. Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nat. Genet. 1999, 22, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Mo, D.; Zhao, Y.; Balajee, A.S. Human RecQL4 helicase plays multifaceted roles in the genomic stability of normal and cancer cells. Cancer Lett. 2018, 413, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Croteau, D.L.; Popuri, V.; Opresko, P.L.; Bohr, V.A. Human RecQ helicases in DNA repair, recombination, and replication. Ann. Rev. Biochem. 2014, 83, 519–552. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, M.; Tadokoro, T.; Rybanska, I.; Ghosh, A.K.; Wersto, R.; May, A.; Kulikowicz, T.; Sykora, P.; Croteau, D.L.; Bohr, V.A. RECQL5 cooperates with Topoisomerase II alpha in DNA decatenation and cell cycle progression. Nucleic Acids Res. 2012, 40, 1621–1635. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Raynard, S.; Sehorn, M.G.; Lu, X.; Bussen, W.; Zheng, L.; Stark, J.M.; Barnes, E.L.; Chi, P.; Janscak, P.; et al. RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev. 2007, 21, 3073–3084. [Google Scholar] [CrossRef] [PubMed]
- Mankouri, H.W.; Hickson, I.D. The RecQ helicase-topoisomerase III-Rmi1 complex: A DNA structure-specific ‘dissolvasome’? Trends Biochem. Sci. 2007, 32, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Manthei, K.A.; Keck, J.L. The BLM dissolvasome in DNA replication and repair. Cell. Mol. Life Sci. 2013, 70, 4067–4084. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Agarwal, H.; Priya, S.; Batra, H.; Modi, P.; Pandey, M.; Saha, D.; Raghavan, S.C.; Sengupta, S. MRN complex-dependent recruitment of ubiquitylated BLM helicase to DSBs negatively regulates DNA repair pathways. Nat. Commun. 2018, 9, 1016. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.H.; Dexheimer, T.S.; Rosenthal, A.S.; Chu, W.K.; Singh, D.K.; Mosedale, G.; Bachrati, C.Z.; Schultz, L.; Sakurai, M.; Savitsky, P.; et al. A small molecule inhibitor of the BLM helicase modulates chromosome stability in human cells. Chem. Biol. 2013, 20, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Banerjee, T.; Sommers, J.A.; Brosh, R.M. Targeting an Achilles’ heel of cancer with a WRN helicase inhibitor. Cell Cycle 2013, 12, 3329–3335. [Google Scholar] [CrossRef] [PubMed]
- Moles, R.; Bai, X.T.; Chaib-Mezrag, H.; Nicot, C. WRN-targeted therapy using inhibitors NSC 19630 and NSC 617145 induce apoptosis in HTLV-1-transformed adult T-cell leukemia cells. J. Hematol. Oncol. 2016, 9, 121. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Sommers, J.A.; Shoemaker, R.H.; Brosh, R.M. Inhibition of helicase activity by a small molecule impairs Werner syndrome helicase (WRN) function in the cellular response to DNA damage or replication stress. Proc. Natl. Acad. Sci. USA 2011, 108, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, N.; Allan, J. Supercoiling in DNA and chromatin. Curr. Opin. Genet. Dev. 2014, 25, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y. Drugging topoisomerases: Lessons and challenges. ACS Chem. Biol. 2013, 8, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Jiang, T.; Li, Q.; Ling, X. Camptothecin (CPT) and its derivatives are known to target topoisomerase I (Top1) as their mechanism of action: Did we miss something in CPT analogue molecular targets for treating human disease such as cancer? Am. J. Cancer Res. 2017, 7, 2350–2394. [Google Scholar] [PubMed]
- Liu, L.F.; Desai, S.D.; Li, T.K.; Mao, Y.; Sun, M.; Sim, S.P. Mechanism of action of camptothecin. Ann. N. Y. Acad. Sci. 2000, 922, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fox, B.M.; Xiao, X.; Antony, S.; Kohlhagen, G.; Pommier, Y.; Staker, B.L.; Stewart, L.; Cushman, M. Design, synthesis, and biological evaluation of cytotoxic 11-alkenylindenoisoquinoline topoisomerase I inhibitors and indenoisoquinoline-camptothecin hybrids. J. Med. Chem. 2003, 46, 3275–3282. [Google Scholar] [CrossRef] [PubMed]
- Mathijssen, R.H.J.; Loos, W.J.; Verweij, J.; Sparreboom, A. Pharmacology of topoisomerase I inhibitors irinotecan (CPT-11) and topotecan. Curr. Cancer Drug Targets 2002, 2, 103–123. [Google Scholar] [CrossRef] [PubMed]
- Kurtzberg, L.S.; Roth, S.; Krumbholz, R.; Crawford, J.; Bormann, C.; Dunham, S.; Yao, M.; Rouleau, C.; Bagley, R.G.; Yu, X.J.; et al. Genz-644282, a novel non-camptothecin topoisomerase I inhibitor for cancer treatment. Clin. Cancer Res. 2011, 17, 2777–2787. [Google Scholar] [CrossRef] [PubMed]
- Schellens, J.H.; Maliepaard, M.; Scheper, R.J.; Scheffer, G.L.; Jonker, J.W.; Smit, J.W.; Beijnen, J.H.; Schinkel, A.H. Transport of topoisomerase I inhibitors by the breast cancer resistance protein. Potential clinical implications. Ann. N. Y. Acad. Sci. 2000, 922, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Beretta, G.L.; Perego, P.; Zunino, F. Mechanisms of cellular resistance to camptothecins. Curr. Med. Chem. 2006, 13, 3291–3305. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Cushman, M. The indenoisoquinoline noncamptothecin topoisomerase I inhibitors: Update and perspectives. Mol. Cancer Ther. 2009, 8, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Antony, S.; Agama, K.K.; Miao, Z.H.; Takagi, K.; Wright, M.H.; Robles, A.I.; Varticovski, L.; Nagarajan, M.; Morrell, A.; Cushman, M.; et al. Novel indenoisoquinolines NSC 725776 and NSC 724998 produce persistent topoisomerase I cleavage complexes and overcome multidrug resistance. Cancer Res. 2007, 67, 10397–10405. [Google Scholar] [CrossRef] [PubMed]
- Cortazar, P.; Justice, R.; Johnson, J.; Sridhara, R.; Keegan, P.; Pazdur, R. US Food and Drug Administration approval overview in metastatic breast cancer. J. Clin. Oncol. 2012, 30, 1705–1711. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Moiani, D.; Arvai, A.S.; Perry, J.; Harding, S.M.; Genois, M.M.; Maity, R.; van Rossum-Fikkert, S.; Kertokalio, A.; Romoli, F.; et al. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol. Cell 2014, 53, 7–18. [Google Scholar] [CrossRef] [PubMed]
- McNeil, E.M.; Astell, K.R.; Ritchie, A.M.; Shave, S.; Houston, D.R.; Bakrania, P.; Jones, H.M.; Khurana, P.; Wallace, C.; Chapman, T.; et al. Inhibition of the ERCC1-XPF structure-specific endonuclease to overcome cancer chemoresistance. DNA Repair (Amst.) 2015, 31, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Wijdeven, R.H.; Pang, B.; van der Zanden, S.Y.; Qiao, X.; Blomen, V.; Hoogstraat, M.; Lips, E.H.; Janssen, L.; Wessels, L.; Brummelkamp, T.R.; et al. Genome-Wide Identification and Characterization of Novel Factors Conferring Resistance to Topoisomerase II Poisons in Cancer. Cancer Res. 2015, 75, 4176–4187. [Google Scholar] [CrossRef] [PubMed]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Khasraw, M.; Bell, R.; Dang, C. Epirubicin: Is it like doxorubicin in breast cancer? A clinical review. Breast 2012, 21, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Webb, A.; Cunningham, D.; Scarffe, J.H.; Harper, P.; Norman, A.; Joffe, J.K.; Hughes, M.; Mansi, J.; Findlay, M.; Hill, A.; Oates, J.; et al. Randomized trial comparing epirubicin, cisplatin, and fluorouracil versus fluorouracil, doxorubicin, and methotrexate in advanced esophagogastric cancer. J. Clin. Oncol. 1997, 15, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.D.; Grothe, W.; Haerting, J.; Kleber, G.; Grothey, A.; Fleig, W.E. Chemotherapy in advanced gastric cancer: A systematic review and meta-analysis based on aggregate data. J. Clin. Oncol. 2006, 24, 2903–2909. [Google Scholar] [CrossRef] [PubMed]
- Hande, K.R. Etoposide: Four decades of development of a topoisomerase II inhibitor. Eur. J. Cancer 1998, 34, 1514–1521. [Google Scholar] [CrossRef]
- Wu, C.-C.; Li, T.K.; Farh, L.; Lin, L.Y.; Lin, T.S.; Yu, Y.J.; Yen, T.J.; Chiang, C.W.; Chan, N.L. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 2011, 333, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Capranico, G.; Orr, A.; Kohn, K.W. Distribution of topoisomerase II cleavage sites in simian virus 40 DNA and the effects of drugs. J. Mol. Biol. 1991, 222, 909–924. [Google Scholar] [CrossRef]
- Evison, B.J.; Sleebs, B.E.; Watson, K.G.; Phillips, D.R.; Cutts, S.M. Mitoxantrone, More than Just Another Topoisomerase II Poison. Med. Res. Rev. 2016, 36, 248–299. [Google Scholar] [CrossRef] [PubMed]
- Vavrova, A.; Jansova, H.; Mackova, E.; Machacek, M.; Haskova, P.; Tichotova, L.; Sterba, M.; Simunek, T. Catalytic inhibitors of topoisomerase II differently modulate the toxicity of anthracyclines in cardiac and cancer cells. PLoS ONE 2013, 8, e76676. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.S.; Williams, J.S.; Tainer, J.A. Mre11-Rad50-Nbs1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem. Cell Biol. 2007, 85, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Stracker, T.H.; Petrini, J.H.J. The MRE11 complex: Starting from the ends. Nat. Rev. Mol. Cell Biol. 2011, 12, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Dupré, A.; Boyer-Chatenet, L.; Sattler, R.M.; Modi, A.P.; Lee, J.H.; Nicolette, M.L.; Kopelovich, L.; Jasin, M.; Baer, R.; Paull, T.T.; et al. A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat. Chem. Biol. 2008, 4, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Rass, E.; Grabarz, A.; Plo, I.; Gautier, J.; Bertrand, P.; Lopez, B.S. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat. Struct. Mol. Biol. 2009, 16, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Jordheim, L.P.; Barakat, K.H.; Heinrich-Balard, L.; Matera, E.L.; Cros-Perrial, E.; Bouledrak, K.; El Sabeh, R.; Perez-Pineiro, R.; Wishart, D.S.; Cohen, R.; et al. Small molecule inhibitors of ERCC1-XPF protein-protein interaction synergize alkylating agents in cancer cells. Mol. Pharmacol. 2013, 84, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.; Raghavan, S.C. DNA double-strand break repair inhibitors as cancer therapeutics. Chem. Biol. 2015, 22, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Gavande, N.S.; VanderVere-Carozza, P.S.; Hinshaw, H.D.; Jalal, S.I.; Sears, C.R.; Pawelczak, K.S.; Turchi, J.J. DNA repair targeted therapy: The past or future of cancer treatment? Pharmacol. Ther. 2016, 160, 65–83. [Google Scholar] [CrossRef] [PubMed]
- Jette, N.; Lees-Miller, S.P. The DNA-dependent protein kinase: A multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog. Biophys. Mol. Biol. 2015, 117, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Stiff, T.; O’Driscoll, M.; Rief, N.; Iwabuchi, K.; Löbrich, M.; Jeggo, P.A. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004, 64, 2390–2396. [Google Scholar] [CrossRef] [PubMed]
- Ciszewski, W.M.; Tavecchio, M.; Dastych, J.; Curtin, N.J. DNA-PK inhibition by NU7441 sensitizes breast cancer cells to ionizing radiation and doxorubicin. Breast Cancer Res. Treat. 2014, 143, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhang, X.; Xie, Y.; Tanaka, K.; Wang, B.; Zhang, H. DNA-PKcs inhibition sensitizes cancer cells to carbon-ion irradiation via telomere capping disruption. PLoS ONE 2013, 8, e72641. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Halbrook, J.; Nickoloff, J.A. Interactive competition between homologous recombination and non-homologous end joining. Mol. Cancer Res. 2003, 1, 913–920. [Google Scholar] [PubMed]
- Sarkaria, J.N.; Tibbetts, R.S.; Busby, E.C.; Kennedy, A.P.; Hill, D.E.; Abraham, R.T. Inhibition of phosphoinositide 3-kinase related kinases by the radiosensitizing agent wortmannin. Cancer Res. 1998, 58, 4375–4382. [Google Scholar] [PubMed]
- Vlahos, C.J.; Matter, W.F.; Hui, K.Y.; Brown, R.F. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 1994, 269, 5241–5248. [Google Scholar] [PubMed]
- Collis, S.J.; DeWeese, T.L.; Jeggo, P.A.; Parker, A.R. The life and death of DNA-PK. Oncogene 2005, 24, 949–961. [Google Scholar] [CrossRef] [PubMed]
- Leahy, J.J.; Golding, B.T.; Griffin, RJ.; Hardcastle, I.R.; Richardson, C.; Rigoreau, L.; Smith, G.C. Identification of a highly potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor (NU7441) by screening of chromenone libraries. Bioorg. Med. Chem. Lett. 2004, 14, 6083–6087. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Thomas, H.D.; Batey, M.A.; Cowell, I.G.; Richardson, C.J.; Griffin, R.J.; Calvert, A.H.; Newell, D.R.; Smith, G.C.; Curtin, N.J. Preclinical evaluation of a potent novel DNA-dependent protein kinase inhibitor NU7441. Cancer Res. 2006, 66, 5354–5362. [Google Scholar] [CrossRef] [PubMed]
- Willmore, E.; de Caux, S.; Sunter, N.J.; Tilby, M.J.; Jackson, G.H.; Austin, C.A.; Durkacz, B.W. A novel DNA-dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia. Blood 2004, 103, 4659–4665. [Google Scholar] [CrossRef] [PubMed]
- Tavecchio, M.; Munck, J.M.; Cano, C.; Newell, D.R.; Curtin, N.J. Further characterisation of the cellular activity of the DNA-PK inhibitor, NU7441, reveals potential cross-talk with homologous recombination. Cancer Chemother. Pharmacol. 2012, 69, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Zenke, F.T.; Zimmermann, A.; Sirrenberg, C.; Dahmen, H.; Vassilev, L.; Pehl, U.; Fuchss, T.; Blaukat, A. M3814, a novel investigational DNA-PK inhibitor: Enhancing the effect of fractionated radiotherapy leading to complete regression of tumors in mice. Cancer Ref. 2016, 76, 1658. [Google Scholar] [CrossRef]
- Harnor, S.J.; Brennan, A.; Cano, C. Targeting DNA-Dependent Protein Kinase for Cancer Therapy. ChemMedChem 2017, 12, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Boucher, D.; Newsome, D.; Takemoto, D.; Hillier, S.; Wang, Y.; Arimoto, R.; Maxwell, J.; Charifson, P.; Fields, S.Z.; Tanner, K.; Penney, M.S. Abstract P5-06-05: Preclinical characterization of VX-984, a selective DNA-dependent protein kinase (DNA-PK) inhibitor in combination with doxorubicin in breast and ovarian cancers. Cancer Res. 2017, 77, P5-06-05. [Google Scholar] [CrossRef]
- Boucher, D.; Hillier, S.; Newsome, D.; Wang, Y.; Takemoto, D.; Gu, Y.; Markland, W.; Hoover, R.; Arimoto, R.; Maxwell, J.; et al. Preclinical characterization of the selective DNA-dependent protein kinase (DNA-PK) inhibitor VX-984 in combination with chemotherapy. Ann. Oncol. 2016, 27, vi114–vi135. [Google Scholar] [CrossRef]
- Neal, J.A.; Dang, V.; Douglas, P.; Wold, M.S.; Lees-Miller, S.P.; Meek, K. Inhibition of homologous recombination by DNA-dependent protein kinase requires kinase activity, is titratable, and is modulated by autophosphorylation. Mol. Cell. Biol. 2011, 31, 1719–1733. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.; Nambiar, M.; Sharma, S.; Karki, S.S.; Goldsmith, G.; Hegde, M.; Kumar, S.; Pandey, M.; Singh, R.K.; Ray, P.; et al. An inhibitor of nonhomologous end-joining abrogates double-strand break repair and impedes cancer progression. Cell 2012, 151, 1474–1487. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.E.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev. 2011, 25, 409–433. [Google Scholar] [CrossRef] [PubMed]
- Riballo, E.; Critchlow, S.E.; Teo, S.H.; Doherty, A.J.; Priestley, A.; Broughton, B.; Kysela, B.; Beamish, H.; Plowman, N.; Arlett, C.F.; et al. Identification of a defect in DNA ligase IV in a radiosensitive leukaemia patient. Curr. Biol. CB 1999, 9, 699–702. [Google Scholar] [CrossRef]
- Kondo, N.; Takahashi, A.; Mori, E.; Ohnishi, K.; McKinnon, P.J.; Sakaki, T.; Nakase, H.; Ohnishi, T. DNA ligase IV as a new molecular target for temozolomide. Biochem. Biophys. Res. Commun. 2009, 387, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Greco, G.E.; Matsumoto, Y.; Brooks, R.C.; Lu, Z.; Lieber, M.R.; Tomkinson, A.E. SCR7 is neither a selective nor a potent inhibitor of human DNA ligase IV. DNA Repair (Amst.) 2016, 43, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.H.; Schild, D. Homologous recombinational repair of DNA ensures mammalian chromosome stability. Mutat. Res. 2001, 477, 131–153. [Google Scholar] [CrossRef]
- Li, X.; Heyer, W.-D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Dai, Q.; Park, D.; Deng, X. Targeting DNA Replication Stress for Cancer Therapy. Genes (Basel) 2016, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Mazina, O.M.; Zentner, I.J.; Cocklin, S.; Mazin, A.V. Inhibition of homologous recombination in human cells by targeting RAD51 recombinase. J. Med. Chem. 2012, 55, 3011–3020. [Google Scholar] [CrossRef] [PubMed]
- Budke, B.; Logan, H.L.; Kalin, J.H.; Zelivianskaia, A.S.; Cameron, M.W.; Miller, L.L.; Stark, J.M.; Kozikowski, A.P.; Bishop, D.K.; Connell, P.P. RI-1: A chemical inhibitor of RAD51 that disrupts homologous recombination in human cells. Nucleic Acids Res. 2012, 40, 7347–7357. [Google Scholar] [CrossRef] [PubMed]
- Jayathilaka, K.; Sheridan, S.D.; Bold, T.D.; Bochenska, K.; Logan, H.L.; Weichselbaum, R.R.; Bishop, D.K.; Connell, P.P. A chemical compound that stimulates the human homologous recombination protein RAD51. Proc. Natl. Acad. Sci. USA 2008, 105, 15848–15853. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Chen, H.; Guo, X.E.; Qiu, X.L.; Hu, C.M.; Chamberlin, A.R.; Lee, W.H. Synthesis, molecular modeling, and biological evaluation of novel RAD51 inhibitors. European J. Med. Chem. 2015, 96, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Goyal, N.; Sullivan, K.; Hanamshet, K.; Patel, M.; Mazina, O.M.; Wang, C.X.; An, W.F.; Spoonamore, J.; Metkar, S.; et al. Targeting BRCA1- and BRCA2-deficient cells with RAD52 small molecule inhibitors. Nucleic Acids Res. 2016, 44, 4189–4199. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, K.; Cramer-Morales, K.; McElroy, D.L.; Ostrov, D.A.; Haas, K.; Childers, W.; Hromas, R.; Skorski, T. Identification of a Small Molecule Inhibitor of RAD52 by Structure-Based Selection. PLoS ONE 2016, 11, e0147230. [Google Scholar] [CrossRef] [PubMed]
- Hengel, S.R.; Malacaria, E.; Folly da Silva Constantino, L.; Bain, F.E.; Diaz, A.; Koch, B.G.; Yu, L.; Wu, M.; Pichierri, P.; Spies, M.A.; et al. Small-molecule inhibitors identify the RAD52-ssDNA interaction as critical for recovery from replication stress and for survival of BRCA2 deficient cells. eLife Sci. 2016, 5, e14740. [Google Scholar] [CrossRef] [PubMed]
- Chandramouly, G.; McDevitt, S.; Sullivan, K.; Kent, T.; Luz, A.; Glickman, J.F.; Andrake, M.; Skorski, T.; Pomerantz, R.T. Small-Molecule Disruption of RAD52 Rings as a Mechanism for Precision Medicine in BRCA-Deficient Cancers. Chem. Biol. 2015, 22, 1491–1504. [Google Scholar] [CrossRef] [PubMed]
- Deakyne, J.S.; Huang, F.; Negri, J.; Tolliday, N.; Cocklin, S.; Mazin, A.V. Analysis of the activities of RAD54, a SWI2/SNF2 protein, using a specific small-molecule inhibitor. J. Biol. Chem. 2013, 288, 31567–31580. [Google Scholar] [CrossRef] [PubMed]
- Ambaye, N.; Chen, C.-H.; Khanna, S.; Li, Y.-J.; Chen, Y. Streptonigrin Inhibits SENP1 and Reduces the Protein Level of Hypoxia-Inducible Factor 1α (HIF1α) in Cells. Biochemistry 2018, 57, 1807–1813. [Google Scholar] [CrossRef] [PubMed]
- Petermann, E.; Orta, M.L.; Issaeva, N.; Schultz, N.; Helleday, T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell 2010, 37, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Klein, H.L. The consequences of Rad51 overexpression for normal and tumor cells. DNA Repair (Amst.) 2008, 7, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.; Stark, J.M.; Ommundsen, M.; Jasin, M. Rad51 overexpression promotes alternative double-strand break repair pathways and genome instability. Oncogene 2004, 23, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Raderschall, E.; Stout, K.; Freier, S.; Suckow, V.; Schweiger, S.; Haaf, T. Elevated levels of Rad51 recombination protein in tumor cells. Cancer Res. 2002, 62, 219–225. [Google Scholar] [PubMed]
- Huang, F.; Motlekar, N.A.; Burgwin, C.M.; Napper, A.D.; Diamond, S.L.; Mazin, A.V. Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chem. Biol. 2011, 6, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Mazin, A.V. A small molecule inhibitor of human RAD51 potentiates breast cancer cell killing by therapeutic agents in mouse xenografts. PLoS ONE 2014, 9, e100993. [Google Scholar] [CrossRef] [PubMed]
- Alagpulinsa, D.A.; Ayyadevara, S.; Shmookler Reis, R.J. A Small-Molecule Inhibitor of RAD51 Reduces Homologous Recombination and Sensitizes Multiple Myeloma Cells to Doxorubicin. Front. Oncol. 2014, 4, 289. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.M.; Logan, H.L.; Budke, B.; Wu, M.; Pawlowski, M.; Weichselbaum, R.R.; Kozikowski, A.P.; Bishop, D.K.; Connell, P.P. The RAD51-Stimulatory Compound RS-1 Can Exploit the RAD51 Overexpression That Exists in Cancer Cells and Tumors. Cancer Res. 2014, 74, 3546–3555. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Miki, Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci. 2004, 95, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, L.; Yu, D.S.; Lo, T.; Anand, S.; Lee, M.; Blundell, T.L.; Venkitaraman, A.R. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature 2002, 420, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Carreira, A.; Kowalczykowski, S.C. Two classes of BRC repeats in BRCA2 promote RAD51 nucleoprotein filament function by distinct mechanisms. Proc. Natl. Acad. Sci. USA 2011, 108, 10448–10453. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhou, L.; Wu, G.; Konig, H.; Lin, X.; Li, G.; Qiu, X.L.; Chen, C.F.; Hu, C.M.; Goldblatt, E.; et al. A novel small molecule RAD51 inactivator overcomes imatinib-resistance in chronic myeloid leukaemia. EMBO Mol. Med. 2013, 5, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Mazina, O.M.; Keskin, H.; Hanamshet, K.; Storici, F.; Mazin, A.V. Rad52 Inverse Strand Exchange Drives RNA-Templated DNA Double-Strand Break Repair. Mol. Cell 2017, 67, 19–29.e3. [Google Scholar] [CrossRef] [PubMed]
- Lok, B.H.; Carley, A.C.; Tchang, B.; Powell, S.N. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene 2013, 32, 3552–3558. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Scott, S.P.; Bussen, W.; Sharma, G.G.; Guo, G.; Pandita, T.K.; Powell, S.N. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Pomerantz, R.T. Abstract B39: Small Molecule Disruption of RAD52 Rings as a Mechanism for Precision Medicine in BRCA Deficient Cancers. Am. Assoc. Cancer Res. 2017, 16, B39. [Google Scholar]
- Mazin, A.V.; Mazina, O.M.; Bugreev, D.V.; Rossi, M.J. Rad54, the motor of homologous recombination. DNA Repair (Amst.) 2010, 9, 286–302. [Google Scholar] [CrossRef] [PubMed]
- Bolzán, A.D.; Bianchi, M.S. Genotoxicity of streptonigrin: A review. Mutat. Res. 2001, 488, 25–37. [Google Scholar] [CrossRef]
- Jiang, C.; Pugh, B.F. Nucleosome positioning and gene regulation: Advances through genomics. Nat. Rev. Genet. 2009, 10, 161–172. [Google Scholar] [CrossRef] [PubMed]
- McGinty, R.K.; Tan, S. Nucleosome structure and function. Chem. Rev. 2015, 115, 2255–2273. [Google Scholar] [CrossRef] [PubMed]
- McAnena, P.; Brown, J.A.; Kerin, M.J. Circulating Nucleosomes and Nucleosome Modifications as Biomarkers in Cancer. Cancers (Basal) 2017, 9, 908–929. [Google Scholar] [CrossRef] [PubMed]
- Shogren-Knaak, M.; Ishii, H.; Sun, J.M.; Pazin, M.J.; Davie, J.R.; Peterson, C.L. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 2006, 311, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Zentner, G.E.; Henikoff, S. Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol. 2013, 20, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, T.; Chakravarti, D. A peek into the complex realm of histone phosphorylation. Mol. Cell. Biol. 2011, 31, 4858–4873. [Google Scholar] [CrossRef] [PubMed]
- Brownell, J.E.; Allis, C.D. Special HATs for special occasions: Linking histone acetylation to chromatin assembly and gene activation. Curr. Opin. Genet. Dev. 1996, 6, 176–184. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Price, B.D.; D’Andrea, A.D. Chromatin remodeling at DNA double-strand breaks. Cell 2013, 152, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Esteller, M.; Rountree, M.R.; Bachman, K.E.; Schuebel, K.; Herman, J.G. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum. Mol. Genet. 2001, 10, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Hunt, C.R.; Ramnarain, D.; Horikoshi, N.; Iyengar, P.; Pandita, R.K.; Shay, J.W.; Pandita, T.K. Histone modifications and DNA double-strand break repair after exposure to ionizing radiations. Radiat. Res. 2013, 179, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Corpet, A.; Almouzni, G. A histone code for the DNA damage response in mammalian cells? EMBO J. 2009, 28, 1828–1830. [Google Scholar] [CrossRef] [PubMed]
- Bennetzen, M.V.; Larsen, D.H.; Dinant, C.; Watanabe, S.; Bartek, J.; Lukas, J.; Andersen, J.S. Acetylation dynamics of human nuclear proteins during the ionizing radiation-induced DNA damage response. Cell Cycle 2013, 12, 1688–1695. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.L.; Bourke, E.; Eriksson, L.A.; Kerin, M.J. Targeting cancer using KAT inhibitors to mimic lethal knockouts. Biochem. Soc. Trans. 2016, 44, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.; Altucci, L. Molecular pathways: The complexity of the epigenome in cancer and recent clinical advances. Clin. Cancer Res. 2012, 18, 5526–5534. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.; Chiu, L.-Y.; Miller, K.M. Acetylation Reader Proteins: Linking Acetylation Signaling to Genome Maintenance and Cancer. PLoS Genet. 2016, 12, e1006272. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, P. Inside HDAC with HDAC inhibitors. Eur. J. Med. Chem. 2010, 45, 2095–2116. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef] [PubMed]
- De Ruijter, A.J.M.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B.P. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Inche, A.G.; La Thangue, N.B. Chromatin control and cancer-drug discovery: Realizing the promise. Drug Discov. Today 2006, 11, 97–109. [Google Scholar] [CrossRef]
- Miller, T.A.; Witter, D.J.; Belvedere, S. Histone deacetylase inhibitors. J. Med. Chem. 2003, 46, 5097–5116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Richon, V.; Ni, X.; Talpur, R.; Duvic, M. Selective induction of apoptosis by histone deacetylase inhibitor SAHA in cutaneous T-cell lymphoma cells: Relevance to mechanism of therapeutic action. J. Investig. Dermatol. 2005, 125, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef] [PubMed]
- Madsen, A.S.; Kristensen, H.M.E.; Lanz, G.; Olsen, C.A. The effect of various zinc binding groups on inhibition of histone deacetylases 1-11. ChemMedChem 2014, 9, 614–626. [Google Scholar] [CrossRef] [PubMed]
- Carey, N.; La Thangue, N.B. Histone deacetylase inhibitors: Gathering pace. Curr. Opin. Pharmacol. 2006, 6, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Khan, O.; La Thangue, N.B. HDAC inhibitors in cancer biology: Emerging mechanisms and clinical applications. Immunol. Cell Biol. 2012, 90, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Kelly, W.K.; Marks, P.A. Drug insight: Histone deacetylase inhibitors—Development of the new targeted anticancer agent suberoylanilide hydroxamic acid. Nat. Clin. Pract. Oncol. 2005, 2, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Zagni, C.; Floresta, G.; Monciino, G.; Rescifina, A. The Search for Potent, Small-Molecule HDACIs in Cancer Treatment: A Decade after Vorinostat. Med. Res. Rev. 2017, 37, 1373–1428. [Google Scholar] [CrossRef] [PubMed]
- Mai, A.; Altucci, L. Epi-drugs to fight cancer: From chemistry to cancer treatment, the road ahead. Int. J. Biochem. Cell Biol. 2009, 41, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Curtin, M.L.; Garland, R.B.; Heyman, H.R.; Frey, R.R.; Michaelides, M.R.; Li, J.; Pease, L.J.; Glaser, K.B.; Marcotte, P.A.; Davidsen, S.K. Succinimide hydroxamic acids as potent inhibitors of histone deacetylase (HDAC). Bioorg. Med. Chem. Lett. 2002, 12, 2919–2923. [Google Scholar] [CrossRef]
- Marks, P.A. Discovery and development of SAHA as an anticancer agent. Oncogene 2007, 26, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Marchenko, N.D.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.D.; Issa, J.-P.J. The promise of epigenetic therapy: Reprogramming the cancer epigenome. Curr. Opin. Genet. Dev. 2017, 42, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.L.; Licht, J.D. Targeting Epigenetics in Cancer. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 187–207. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, K.; Fradet-Turcotte, A.; Avvakumov, N.; Lambert, J.P.; Roques, C.; Pandita, R.K.; Paquet, E.; Herst, P.; Gingras, A.C.; Pandita, T.K.; et al. The TIP60 Complex Regulates Bivalent Chromatin Recognition by 53BP1 through Direct H4K20me Binding and H2AK15 Acetylation. Mol. Cell 2016, 62, 409–421. [Google Scholar] [CrossRef] [PubMed]
- VanderMolen, K.M.; McCulloch, W.; Pearce, C.J.; Oberlies, N.H. Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): A natural product recently approved for cutaneous T-cell lymphoma. J. Antibiot. 2011, 64, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Guo, J.; Zhao, Y.; Fei, C.; Zheng, Q.; Li, X.; Chang, C. Chidamide, a novel histone deacetylase inhibitor, inhibits the viability of MDS and AML cells by suppressing JAK2/STAT3 signaling. Am. J. Transl. Res. 2016, 8, 3169–3178. [Google Scholar] [PubMed]
- Shi, Y.; Dong, M.; Hong, X.; Zhang, W.; Feng, J.; Zhu, J.; Yu, L.; Ke, X.; Huang, H.; Shen, Z.; et al. Results from a multicenter, open-label, pivotal phase II study of chidamide in relapsed or refractory peripheral T-cell lymphoma. Ann. Oncol. 2015, 26, 1766–1771. [Google Scholar] [CrossRef] [PubMed]
- Yardley, D.A.; Ismail-Khan, R.R.; Melichar, B.; Lichinitser, M.; Munster, P.N.; Klein, P.M.; Cruickshank, S.; Miller, K.D.; Lee, M.J.; Trepel, J.B. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J. Clin. Oncol. 2013, 31, 2128–2135. [Google Scholar] [PubMed]
- Damaskos, C.; Garmpis, N.; Valsami, S.; Kontos, M.; Spartalis, E.; Kalampokas, T.; Kalampokas, E.; Athanasiou, A.; Moris, D.; Daskalopoulou, A.; et al. Histone Deacetylase Inhibitors: An Attractive Therapeutic Strategy against Breast Cancer. Anticancer Res. 2017, 37, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Gottlicher, M.; Minucci, S.; Zhu, P.; Krämer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.M.; Hackanson, B.; Lübbert, M.; Jung, M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin. Epigenet. 2010, 1, 117–136. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Li, S.Y.; Chen, X.H.; et al. FDA Approval: Belinostat for the Treatment of Patients with Relapsed or Refractory Peripheral T-cell Lymphoma. Clin. Cancer Res. 2015, 21, 2666–2670. [Google Scholar] [CrossRef] [PubMed]
- Atadja, P. Development of the pan-DAC inhibitor panobinostat (LBH589): Successes and challenges. Cancer Lett. 2009, 280, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Barbarotta, L.; Hurley, K. Romidepsin for the Treatment of Peripheral T-Cell Lymphoma. J. Adv. Pract. Oncol. 2015, 6, 22–36. [Google Scholar] [PubMed]
- Chan, T.; Tse, E.; Kwong, Y.-L. Chidamide in the treatment of peripheral T-cell lymphoma. OTT 2017, 10, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.-C.; Jung, M.G.; Lee, Y.H.; Yoon, J.C.; Kwon, S.H.; Kang, H.B.; Kim, M.J.; Cha, J.H.; Kim, Y.J.; Jun, W.J.; et al. Epigallocatechin-3-gallate, a histone acetyltransferase inhibitor, inhibits EBV-induced B lymphocyte transformation via suppression of RelA acetylation. Cancer Res. 2009, 69, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-H.; Kwak, J.; Choi, H.K.; Choi, K.C.; Kim, S.; Lee, J.; Jun, W.; Park, H.J.; Yoon, H.G. EGCG suppresses prostate cancer cell growth modulating acetylation of androgen receptor by anti-histone acetyltransferase activity. Int. J. Mol. Med. 2012, 30, 69–74. [Google Scholar] [PubMed]
- Balasubramanyam, K.; Varier, R.A.; Altaf, M.; Swaminathan, V.; Siddappa, N.B.; Ranga, U.; Kundu, T.K. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J. Biol. Chem. 2004, 279, 51163–51171. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Bourke, E.; Scobie, M.; Famme, M.A.; Koolmeister, T.; Helleday, T.; Eriksson, L.A.; Lowndes, N.F.; Brown, J.A. Rational design and validation of a Tip60 histone acetyltransferase inhibitor. Sci. Rep. 2014, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Coffey, K.; Blackburn, T.J.; Cook, S.; Golding, B.T.; Griffin, R.J.; Hardcastle, I.R.; Hewitt, L.; Huberman, K.; McNeill, H.V.; Newell, D.R.; et al. Characterisation of a Tip60 Specific Inhibitor, NU9056, in Prostate Cancer. PLoS ONE 2012, 7, e45539. [Google Scholar] [CrossRef] [PubMed]
- Gajer, J.M.; Furdas, S.D.; Gründer, A.; Gothwal, M.; Heinicke, U.; Keller, K.; Colland, F.; Fulda, S.; Pahl, H.L.; Fichtner, I.; et al. Histone acetyltransferase inhibitors block neuroblastoma cell growth in vivo. Oncogenesis 2015, 4, e137. [Google Scholar] [CrossRef] [PubMed]
- Trisciuoglio, D.; Ragazzoni, Y.; Pelosi, A.; Desideri, M.; Carradori, S.; Gabellini, C.; Maresca, G.; Nescatelli, R.; Secci, D.; Bolasco, A.; et al. CPTH6, a thiazole derivative, induces histone hypoacetylation and apoptosis in human leukemia cells. Clin. Cancer Res. 2012, 18, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Ravindra, K.C.; Selvi, B.R.; Arif, M.; Reddy, B.A.; Thanuja, G.R.; Agrawal, S.; Pradhan, S.K.; Nagashayana, N.; Dasgupta, D.; Kundu, T.K. Inhibition of lysine acetyltransferase KAT3B/p300 activity by a naturally occurring hydroxynaphthoquinone, plumbagin. J. Biol. Chem. 2009, 284, 24453–24464. [Google Scholar] [CrossRef] [PubMed]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Basavapathruni, A.; Jin, L.; Boriack-Sjodin, P.A.; Allain, C.J.; Klaus, C.R.; Raimondi, A.; Scott, M.P.; et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Vedadi, M.; Barsyte-Lovejoy, D.; Liu, F.; Rival-Gervier, S.; Allali-Hassani, A.; Labrie, V.; Wigle, T.J.; Dimaggio, P.A.; Wasney, G.A.; Siarheyeva, A.; et al. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat. Chem. Biol. 2011, 7, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Knutson, S.K.; Kawano, S.; Minoshima, Y.; Warholic, N.M.; Huang, K.C.; Xiao, Y.; Kadowaki, T.; Uesugi, M.; Kuznetsov, G.; Kumar, N.; et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol. Cancer Ther. 2014, 13, 842–854. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, A.D.; Larsen, N.A.; Howard, T.; Pollard, H.; Green, I.; Grande, C.; Cheung, T.; Garcia-Arenas, R.; Cowen, S.; Wu, J.; et al. Structural basis of substrate methylation and inhibition of SMYD2. Structure 2011, 19, 1262–1273. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.E.; Dalisay, D.S.; Li, F.; Amphlett, J.; Maneerat, W.; Chavez, M.A.; Wang, Y.A.; Matainaho, T.; Yu, W.; Brown, P.J.; et al. Nahuoic acid A produced by a Streptomyces sp. isolated from a marine sediment is a selective SAM-competitive inhibitor of the histone methyltransferase SETD8. Org. Lett. 2013, 15, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Judge, R.A.; Zhu, H.; Upadhyay, A.K.; Bodelle, P.M.; Hutchins, C.W.; Torrent, M.; Marin, V.L.; Yu, W.; Vedadi, M.; Li, F.; et al. Turning a Substrate Peptide into a Potent Inhibitor for the Histone Methyltransferase SETD8. ACS Med. Chem. Lett. 2016, 7, 1102–1106. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.M.Z.; McCafferty, D.G. trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry 2007, 46, 4408–4416. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, H.P.; Kruger, R.G. Antitumor activity of LSD1 inhibitors in lung cancer. Mol. Cell Oncol. 2016, 3, e1117700. [Google Scholar] [CrossRef] [PubMed]
- Carrozza, M.J.; Utley, R.T.; Workman, J.L.; Cote, J. The diverse functions of histone acetyltransferase complexes. Trends Genet. 2003, 19, 321–329. [Google Scholar] [CrossRef]
- Farria, A.; Li, W.; Dent, S.Y.R. KATs in cancer: Functions and therapies. Oncogene 2015, 34, 4901–4913. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Xie, N.; Wu, Z.; Zhang, Y.; Zheng, Y.G. Bisubstrate Inhibitors of the MYST HATs Esa1 and Tip60. Bioorg. Med. Chem. 2009, 17, 1381–1386. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.P.; Robaa, D.; Alhalabi, Z.; Sippl, W.; Jung, M. KATching-Up on Small Molecule Modulators of Lysine Acetyltransferases. J. Med. Chem. 2016, 59, 1249–1270. [Google Scholar] [CrossRef] [PubMed]
- Koeller, K.M.; Haggarty, S.J.; Perkins, B.D.; Leykin, I.; Wong, J.C.; Kao, M.C.; Schreiber, S.L. Chemical genetic modifier screens: Small molecule trichostatin suppressors as probes of intracellular histone and tubulin acetylation. Chem. Biol. 2003, 10, 397–410. [Google Scholar] [CrossRef]
- Ghizzoni, M.; Wu, J.; Gao, T.; Haisma, H.J.; Dekker, F.J.; Zheng, Y.G. 6-alkylsalicylates are selective Tip60 inhibitors and target the acetyl-CoA binding site. Eur. J. Med. Chem. 2012, 47, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Furdas, S.D.; Kannan, S.; Sippl, W.; Jung, M. Small molecule inhibitors of histone acetyltransferases as epigenetic tools and drug candidates. Arch. Pharm. (Weinh.) 2012, 345, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A. The cancer epigenome: Concepts, challenges, and therapeutic opportunities. Science 2017, 355, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Di Martile, M.; Desideri, M.; De Luca, T.; Gabellini, C.; Buglioni, S.; Eramo, A.; Sette, G.; Milella, M.; Rotili, D.; Mai, A.; et al. Histone acetyltransferase inhibitor CPTH6 preferentially targets lung cancer stem-like cells. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Murray, K. The Occurrence Of Epsilon-N-Methyl Lysine In Histones. Biochemistry 1964, 3, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bedford, M.T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 2013, 13, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.; Miller, K.M. Histone methylation and the DNA damage response. Mutat. Res./Rev. Mutat. Res. 2017. [Google Scholar] [CrossRef]
- Bakkenist, C.J.; Kastan, M.B. Chromatin perturbations during the DNA damage response in higher eukaryotes. DNA Repair (Amst.) 2015. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Højfeldt, J.W.; Agger, K.; Helin, K. Histone lysine demethylases as targets for anticancer therapy. Nat. Rev. Drug Discov. 2013, 12, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Chen, P.; Diao, J.; Cheng, G.; Deng, L.; Anglin, J.L.; Prasad, B.V.; Song, Y. Selective inhibitors of histone methyltransferase DOT1L: Design, synthesis, and crystallographic studies. J. Am. Chem. Soc. 2011, 133, 16746–16749. [Google Scholar] [CrossRef] [PubMed]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Majer, C.R.; Sneeringer, C.J.; Song, J.; Johnston, L.D.; Scott, M.P.; Smith, J.J.; Xiao, Y.; et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 2011, 20, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Bernt, K.M.; Zhu, N.; Sinha, A.U.; Vempati, S.; Faber, J.; Krivtsov, A.V.; Feng, Z.; Punt, N.; Daigle, A.; Bullinger, L.; et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 2011, 20, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, L.; Chen, Y.; Liu, J.; Xiao, S.; Lian, F.; Zhang, N.; Ding, H.; Zhang, Y.; Chen, K.; et al. Discovery of potent DOT1L inhibitors by AlphaLISA based High Throughput Screening assay. Bioorg. Med. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Dillon, S.C.; Zhang, X.; Trievel, R.C.; Cheng, X. The SET-domain protein superfamily: Protein lysine methyltransferases. Genome Biol. 2005, 6, 227. [Google Scholar] [CrossRef] [PubMed]
- Botuyan, M.V.; Lee, J.; Ward, I.M.; Kim, J.E.; Thompson, J.R.; Chen, J.; Mer, G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 2006, 127, 1361–1373. [Google Scholar] [CrossRef] [PubMed]
- Takawa, M.; Cho, H.S.; Hayami, S.; Toyokawa, G.; Kogure, M.; Yamane, Y.; Iwai, Y.; Maejima, K.; Ueda, K.; Masuda, A.; et al. Histone lysine methyltransferase SETD8 promotes carcinogenesis by deregulating PCNA expression. Cancer Res. 2012, 72, 3217–3227. [Google Scholar] [CrossRef] [PubMed]
- Ma, A.; Yu, W.; Li, F.; Bleich, R.M.; Herold, J.M.; Butler, K.V.; Norris, J.L.; Korboukh, V.; Tripathy, A.; Janzen, W.P.; et al. Discovery of a selective, substrate-competitive inhibitor of the lysine methyltransferase SETD8. J. Med. Chem. 2014, 57, 6822–6833. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.-C.; Yu, B.; Chen, Z.-S.; Liu, Y.; Liu, H.-M. TCPs: Privileged scaffolds for identifying potent LSD1 inhibitors for cancer therapy. Epigenomics 2016, 8, 651–666. [Google Scholar] [CrossRef] [PubMed]
- Ferrari-Amorotti, G.; Chiodoni, C.; Shen, F.; Cattelani, S.; Soliera, A.R.; Manzotti, G.; Grisendi, G.; Dominici, M.; Rivasi, F.; Colombo, M.P.; et al. Suppression of invasion and metastasis of triple-negative breast cancer lines by pharmacological or genetic inhibition of slug activity. Neoplasia 2014, 16, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, A.; Minucci, S. A comprehensive review of lysine-specific demethylase 1 and its roles in cancer. Epigenomics 2017, 9, 1123–1142. [Google Scholar] [CrossRef] [PubMed]





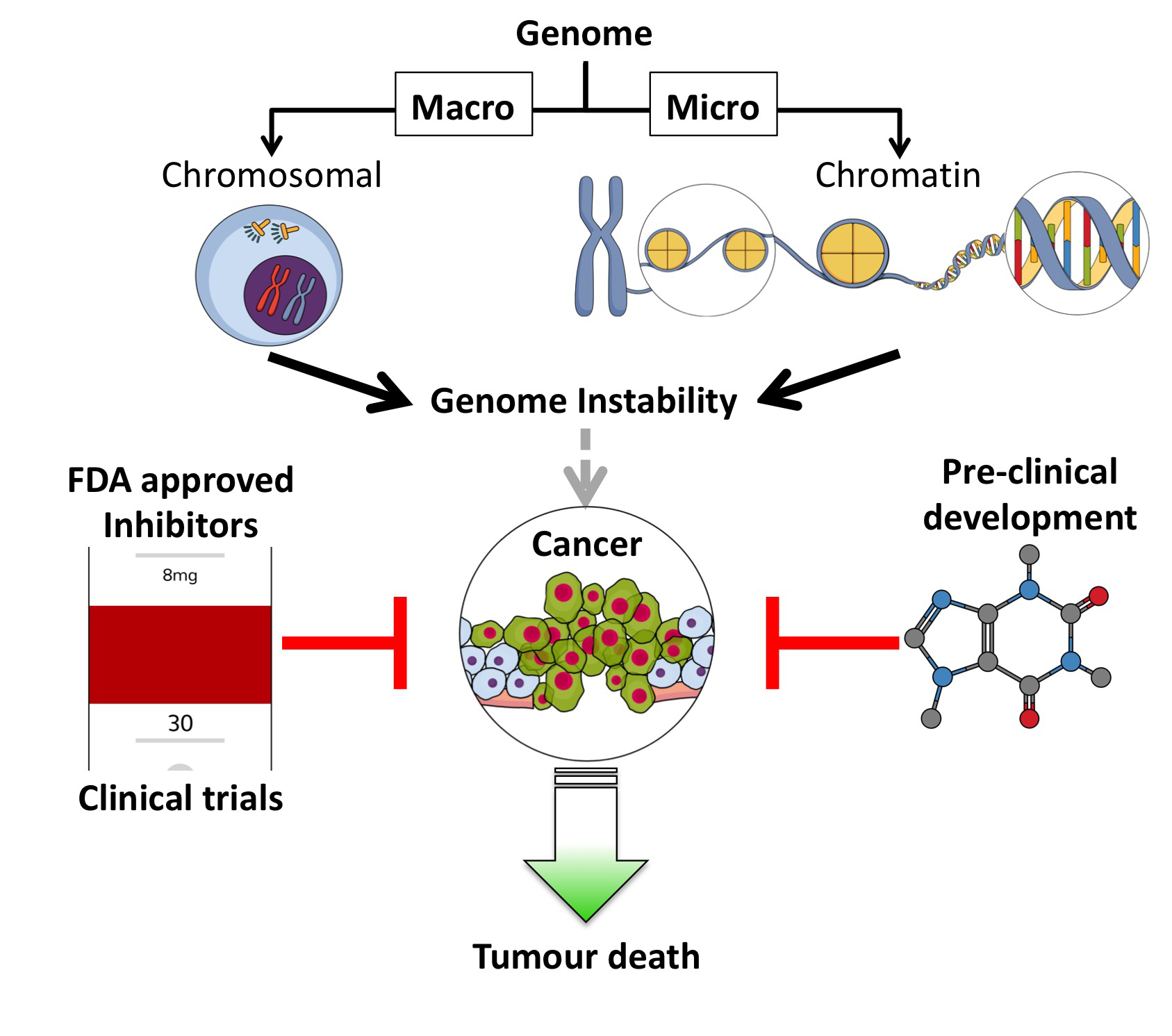{kind=link}
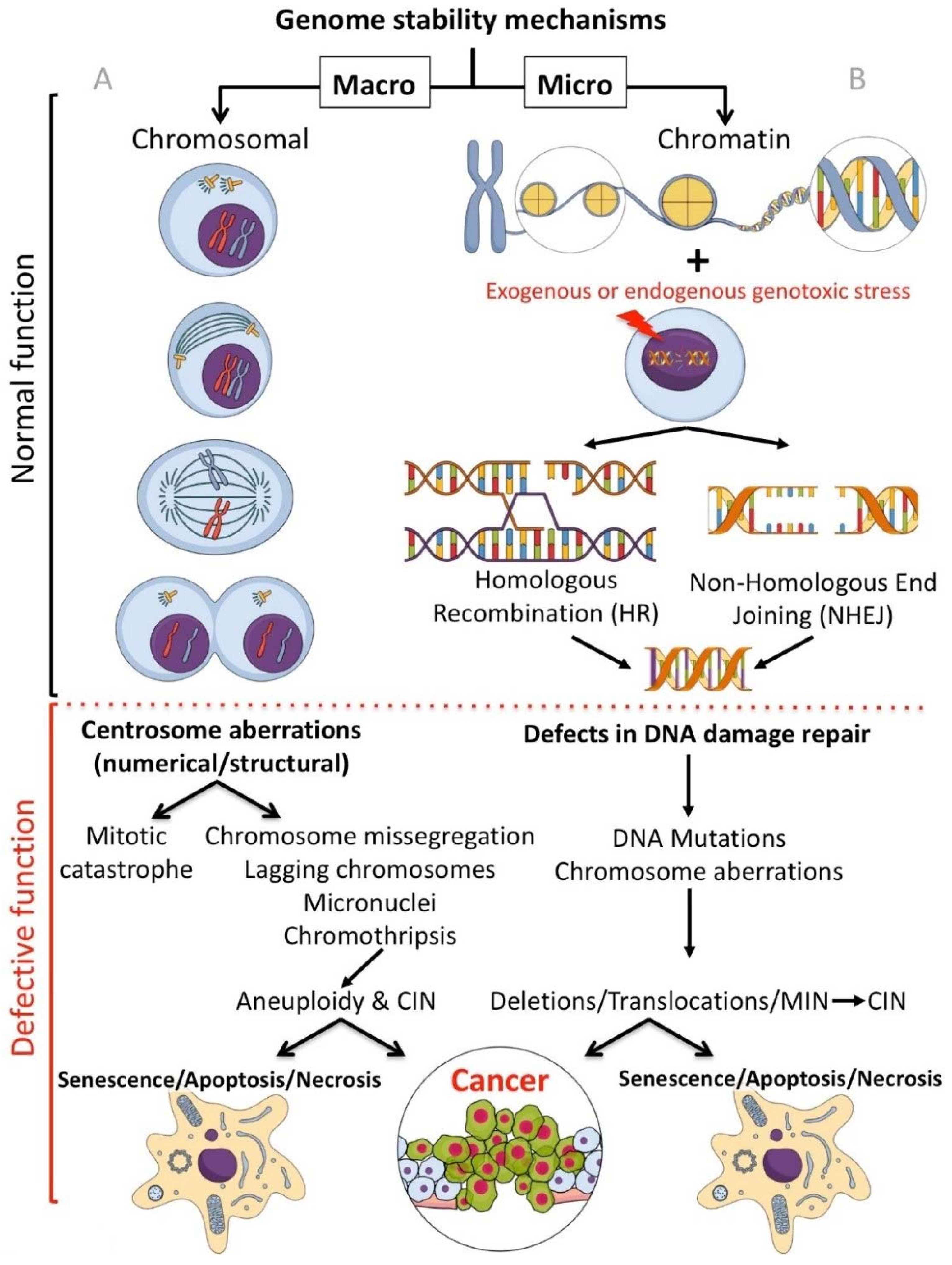{kind=link}
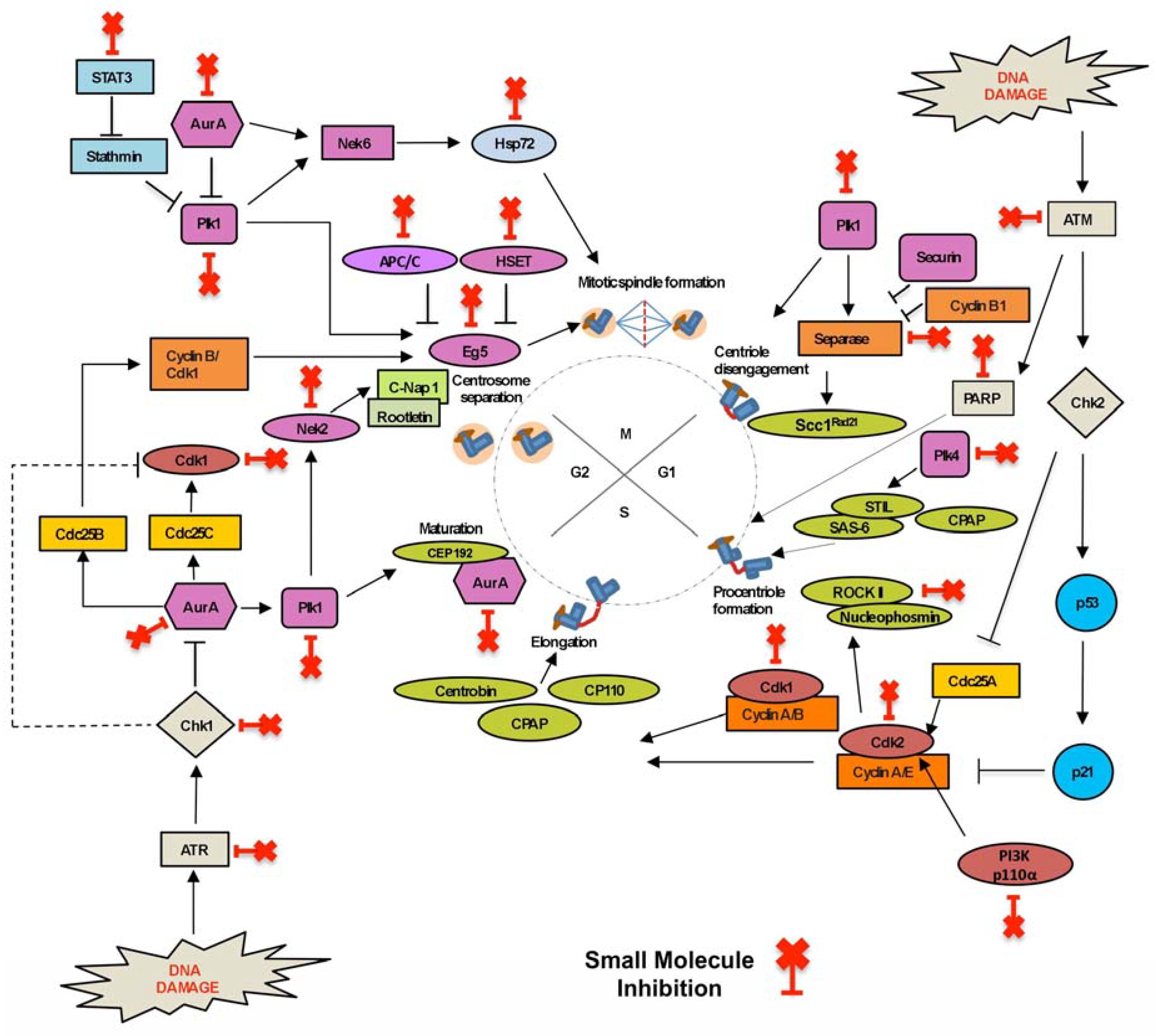{kind=link}
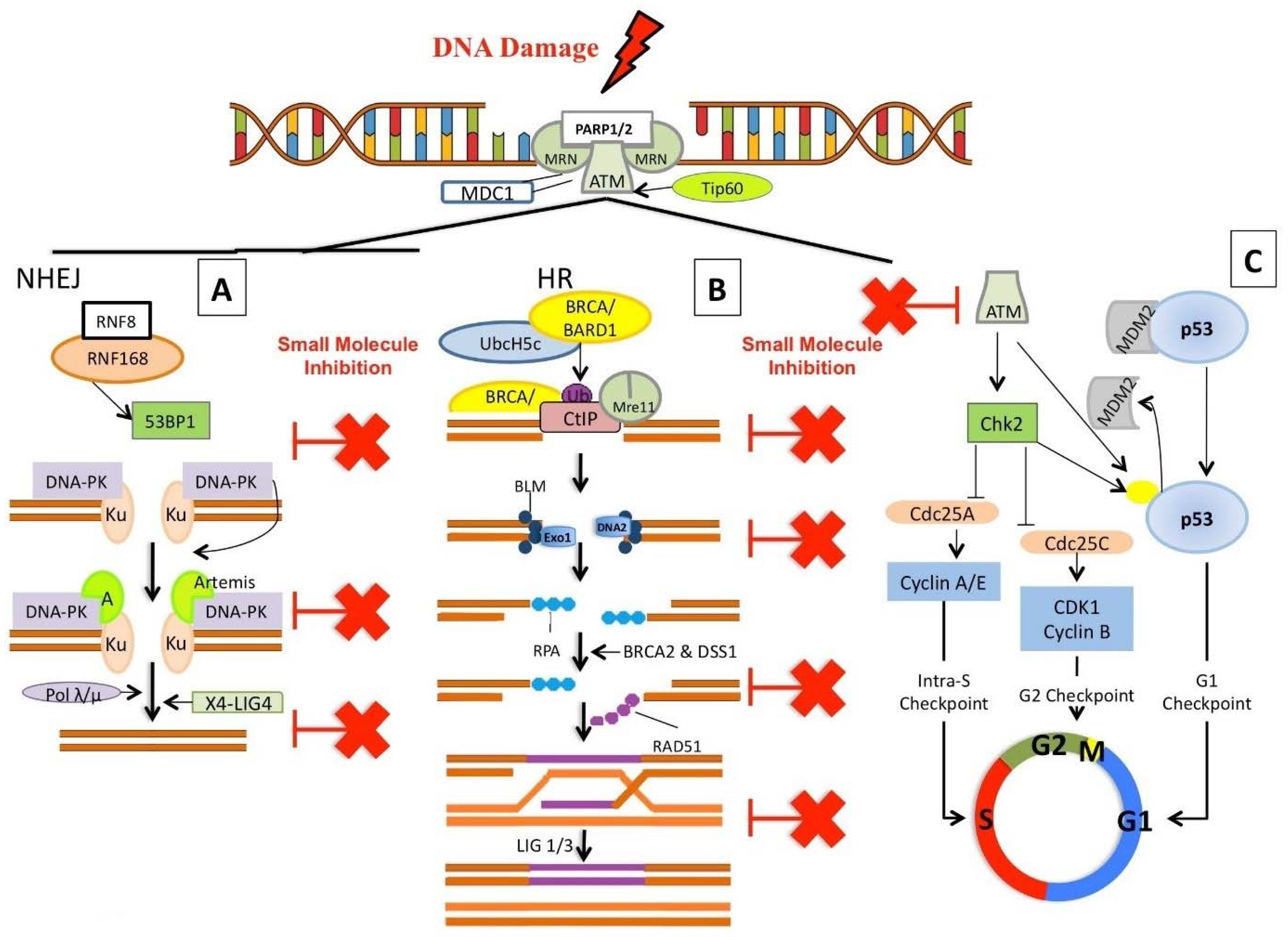{kind=link}
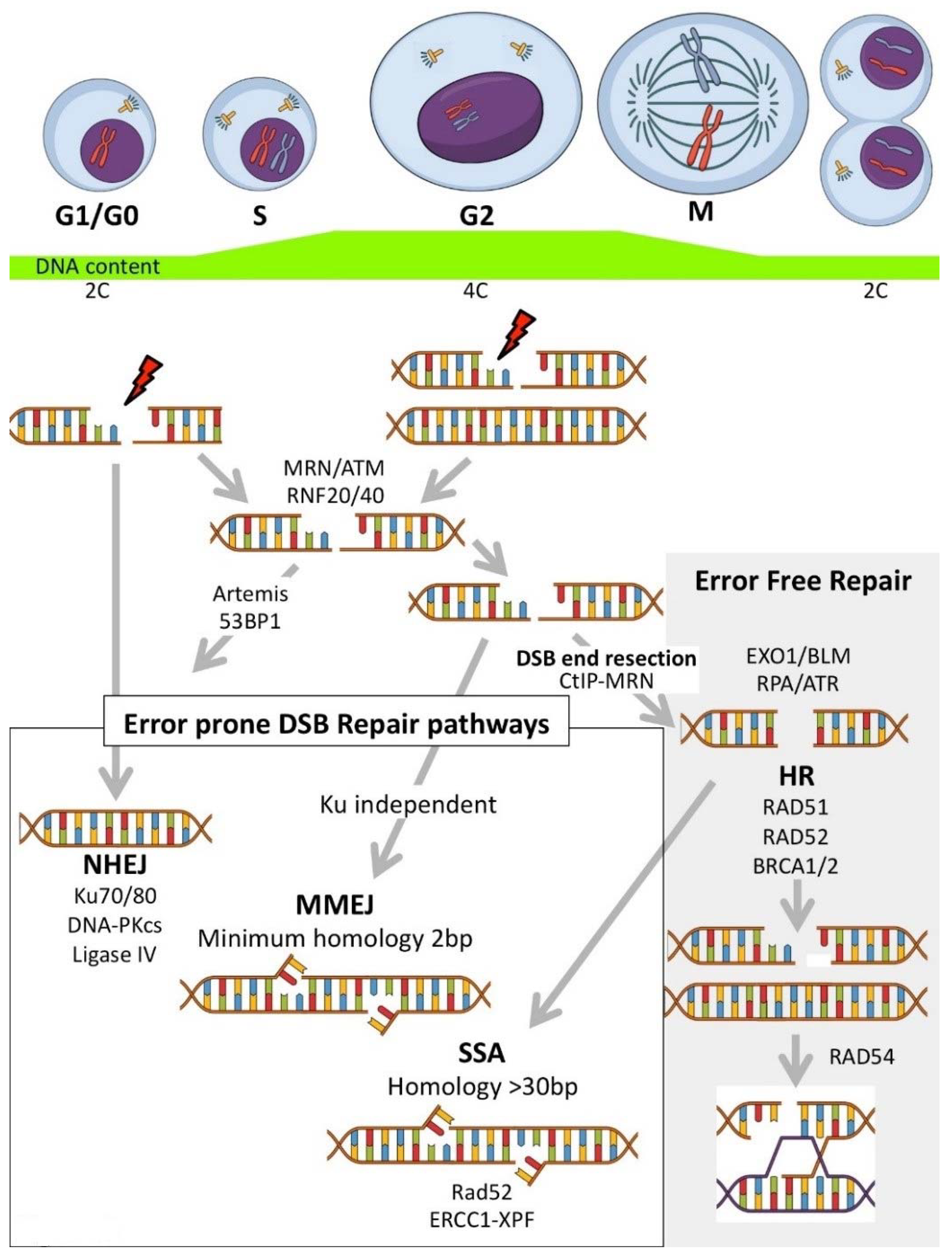{kind=link}
| Inhibitors Targeting | Enzymes | Pathways | Protein Target | Inhibitor | Mechanism of Action of Inhibitors | Clinical Trial |
|---|---|---|---|---|---|---|
| Centriole disengagement | Rad21 | Centrosome duplication | Separase | Sepin-1 | Inhibits Rad21 cleavage by activated separase [16,17]. | Pre-clinical development [17]. |
| Centriole disengagement and centrosome maturation | Plk1 | Centrosome duplication | Plk1 | BI 2536 | 2-aminopyrimidine-containing ATP-competitive kinase inhibitor. Causes disruption of the spindle assembly leading to mitotic arrest and apoptosis [18]. | Phase II clinical trials completed in patients with prostatic and pancreatic neoplasms, breast cancer, endometrial cancer, head and neck cancer, non-small cell lung cancer and SCLC. https://clinicaltrials.gov/ct2/results?cond=Cancer&term=BI2536&cntry=&state=&city=&dist= |
| Volasertib (BI 6727) | Blocks cell division by competitively binding to the ATP-binding pocket of Plk1 [18,19]. | Phase II clinical trial completed in patients with ovarian cancer and urothelial cancer. https://clinicaltrials.gov/ct2/results?cond=Cancer&term=BI+6727&cntry=&state=&city=&dist= Phase III clinical trial in combination with low dose cytarabine in AML patients: NCT01721876. | ||||
| GSK461364 | ATP-competitive Plk1 inhibitor caused mitotic arrest through G2/M arrest [18]. | Phase I clinical trial completed in non-Hodgkins lymphoma: NCT00536835. | ||||
| ZK-thiazolidinone (TAL) | ATP-competitive inhibitor specifically inhibits Plk1 and causes prometaphase-like mitotic arrest [18]. | Pre-clinical development [20]. | ||||
| Centriole disengagement and centrosome duplication | CDK2 | Cell cycle (G1/S transition) | CDK2 | Milciclib (PHA-848125 AC) | ATP-competitive inhibitor of cyclin-dependent kinases (Cdks) that potently inhibits Cdk2/cyclin A (IC50 = 45 nM) [21]. | Phase I clinical trial completed in patients with advanced/metastatic solid tumours: NCT01300468. Phase II clinical trial in patients with malignant thymoma, thymic carcinoma and hepatocellular carcinoma. https://clinicaltrials.gov/ct2/results?cond=Cancer&term=PHA-848125+AC&cntry=&state=&city=&dist= |
| SU9516 | Inhibits pRb phosphorylation causing enhanced pRB/E2F complex formation and induces G1 and G2-M cell cycle arrest [22]. | Pre-clinical development. | ||||
| CDK2 and CDK1 | Butyrolactone I | An ATP-competitive inhibitor of CDKs which inhibits phosphorylation of pRB and transcription factor EF-1 and prevents cell cycle progression both at G1/S and G2/M transitions [23,24]. | Pre-clinical development. | |||
| RNA polymerase II | Cell cycle progression | Pan Cdk | Flavopiridol (Alvocidib or HMR 1275) | Causes downregulation of cyclin D1, c-MYC, and MCL-1 and induces apoptosis in tumour cells [25]. | Phase II clinical trial completed in prostate cancer, kidney cancer and endometrial cancer and phase II drug combination studies completed in esophageal cancer, liver cancer pancreatic cancer, breast cancer, head and neck cancer, germ cell tumours and ovarian epithelial cancer. https://clinicaltrials.gov/ct2/results?term=Flavopiridol&cond=cancer&age_v=&gndr=&type=&rslt=&phase=1&phase=2&phase=3&Search=Apply | |
| pRb phosphorylation | R-547 (RG547) | ATP-competitive CDK inhibitor, inhibits retinoblastoma protein phosphorylation in tumour cells and induces apoptosis [25,26]. | Phase I clinical trial completed in patients with advanced solid tumours: NCT00400296. | |||
| Mcl-1 transcription | CYC-202 (R-roscovitine/Seliciclib) | Competes with ATP for its binding site on CDKs, reduces retinoblastoma protein phosphorylation and arrests cell cycle at G1, S, and G2-M phases [23,25,27,28]. | Phase I clinical trial of CYC-202 in sequential or concomitant combination therapies in patients with breast cancer (NCT01333423), non-small cell lung cancer (NCT00372073) and advanced solid tumours (NCT00999401). | |||
| polymerase (RNA Pol) II | SNS-032 | Reversibly inhibits phosphorylation of RNA polymerase II and causes RNA synthesis inhibition [29]. | Phase I clinical trial completed in patients with selected advanced solid tumours (NCT00292864) and advanced B-lymphoid malignancies (NCT00446342). | |||
| Centriole elongation and centrosome duplication | Plk4 | Centrosome duplication | Plk4 | CFI- 400945 | ATP competitive inhibitor and inhibits autophosphorylation of PLK4 at serine 305 [30]. | Phase I clinical trial in patients with advanced cancer (NCT01954316), and in relapsed or refractory AML or myelodysplastic syndrome (NCT03187288). |
| Centrosome maturation and separation | Aurora-A | Cell cycle (mitotic regulator) and centrosome maturation | Aurora-A | Alisertib (MLN8237) | Binds and inhibits Aurora-A, causing delayed mitotic entry and cell cycle arrest, leading to the formation of cells with tetraploid DNA content [31]. | Phase II clinical trial completed in prostate cancer, childhood solid tumours or leukaemia (NCT01799278), and adult advanced non-haematological malignancies (NCT01045421). Phase II combination studies with Paclitaxel in small cell lung cancer, breast cancer and combination therapy in malignant rhabdoid tumours. https://clinicaltrials.gov/ct2/results?term=MLN8237&cond=cancer+and+neoplasia&age_v=&gndr=&type=&rslt=&phase=1&phase=2&phase=3&Search=Apply Phase III clinical trial evaluating Alisertib, compared with Pralatrexate or Gemcitabine or Romidepsin, in patients with relapsed or refractory peripheral T-Cell Lymphoma: NCT01482962. |
| ENMD-2076 | Causes G2-M arrest and a decrease in the intercentrosomal distance, with induction of monopolar spindles and apoptosis [32]. | Phase II clinical trial completed in patients with ovarian cancer, ovarian clear cell carcinomas, haematological malignancies, hepatocellular, sarcoma and triple negative breast cancer. https://clinicaltrials.gov/ct2/results?cond=Cancer&term=ENMD-2076&cntry=&state=&city=&dist= | ||||
| MK-5108 (VX-689) | Inhibits Aurora-A kinase by competitively binding to the ATP binding site [33]. | Phase I clinical trial completed in patients with advanced solid tumours: NCT00543387. | ||||
| KW-2449 | A multikinase inhibitor which causes downregulation of Aurora kinases and leads to G2/M arrest [34]. | Phase I clinical trial in patients with acute myelogenous leukaemia (AML) myelodysplastic syndromes and chronic myelogenous leukaemia (NCT00779480, NCT00346632). | ||||
| XL228 | Multi-tyrosine kinases inhibitor which inhibits Aurora-A, the T315I mutant form of the Abl protein, IGF1R, Src tyrosine kinase. It prevents tumor angiogenesis, cell proliferation, and metastasis [35]. | Phase I clinical trial in patients with advanced malignancies (NCT00526838), and chronic myeloid leukaemia or philadelphia-chromosome-positive acute lymphocytic leukaemia (NCT00464113). | ||||
| MLN8054 | An ATP-competitive inhibitor which targets Aurora-A and causes monopolar, acentriolar bipolar, and multipolar spindles, leading to chromosomal segregation defects, aneuploidy and cell death [36]. | Phase I clinical trials in patients with advanced malignancies (NCT00652158) and breast, colon, pancreatic and bladder tumours (NCT00249301). | ||||
| Aurora-B | Cell cycle (sister chromatid cohesion) | Aurora-B | Barasertib (AZD1152-HQPA) | Competitively blocks the ATP-binding pocket of Aurora-B kinase [31,33,37]. | Phase I clinical trial completed in patients with AML (NCT01019161, NCT00926731). Phase I/II clinical trial in in patients with relapsed acute myeloid leukaemia/high-risk myelodysplastic syndrome (NCT03217838). | |
| BI811283 | Inhibits by binding to the ATP binding pocket of Aurora-B [33]. | Phase I clinical trial completed in patients with various solid tumours: NCT00701324. Phase I/IIa combination study with Cytarabine completed in patients with previously untreated acute myeloid leukaemia: NCT00701324. | ||||
| Aurora-A/B/C | Cell cycle | (Pan Aurora) Aurora-A/B/C | Danusertib (PHA-739358) | ATP competitive pan-Aurora kinase inhibitor that inhibits the catalytic domain of Aurora kinases [31,33]. | Phase II study completed in patients with metastatic hormone refractory prostate cancer (NCT00766324), multiple myeloma (NCT00872300) and leukaemia (NCT00335868). | |
| Aurora-A/B | Aurora-A/B | PF-03814735 | ATP competitive reversible inhibitor which blocks cytokinesis, resulting in decreased cell proliferation and the appearance of polyploid multinucleate cells [31,33,38]. | Phase I clinical trial completed in patients with advanced solid tumours: NCT00424632. | ||
| Aurora-A/B/C | Aurora-A/B/C | AMG 900 | ATP-competitive inhibitor of Aurora kinases, causing inhibition of autophosphorylation of Aurora-A and -B [31,33]. | Phase I clinical trial completed in active study in advanced malignancy and solid tumours (NCT00858377) and acute myeloid leukaemia (NCT01380756). | ||
| Linker dissociation | NIMA Related Kinase 2 (Nek2) | Centrosome duplication | Nek2 | JH295 | Irreversibly inhibits Nek2 via alkylation of residue Cys22 without affecting the mitotic kinases, Cdk1, Aurora-B, or Plk1 [39]. | Pre-clinical development. |
| NCL 00017509 | Binds to ATP domain of Nek2 and causes irreversible inhibition [40]. | Pre-clinical development. | ||||
| Centrosome separation and spindle fibre formation | Cdk1 | Cell cycle (G2/M transition) | CDK1/cyclin B1 | RO-3306 | Binds to ATP binding pocket and inhibits CDK1/cyclin B1 [41,42]. | Pre-clinical development. |
| CDK1 | CGP 74514A | Binds to ATP binding pocket and reduces Akt phosphorylation, increasing mitochondrial damage and inducing apoptosis [43,44]. | Pre-clinical development. | |||
| KSP/Eg5 | (Cell cycle) mitotic spindle pole separation | KSP/Eg5 | Monastrol | ATP non-competitive reversible inhibitor which binds to the Eg5-ADP complex and prevents force generation and kinesin motility [45]. | Pre-clinical development. | |
| Ispinesib (SB-715992) | ATP non-competitive reversible inhibitor which binds to the Eg5-ADP complex and prevents force generation and kinesin motility [46,47]. | Phase II clinical trial completed in patients with breast cancer, prostate cancer, ovarian cancer, non-small cell lung cancer, liver cancer, kidney cancer, colorectal cancer and melanoma. https://clinicaltrials.gov/ct2/results?cond=cancer&term=Ispinesib+SB-715992&cntry=&state=&city=&dist= | ||||
| MK-0731 | ATP non-competitive reversible inhibitor which binds to the Eg5-ADP complex and prevents force generation and kinesin motility [45]. | Phase I clinical trial completed in patients with advanced solid tumours: NCT00104364. | ||||
| KSP/Eg5 mcl-1 | KSP/Eg5 mcl-1 | Filanesib (ARRY-520) | Non-ATP competitive inhibitor which binds to Eg5 at the same site as monastrol and induces cell cycle arrest at mitosis, leading to apoptosis [45,48]. | Phase I/II clinical trial completed in patients with advanced solid tumours and haematological malignancies. https://clinicaltrials.gov/ct2/results?cond=cancer&term=ARRY-520&cntry=&state=&city=&dist= |
| Inhibitors Targeting | Enzymes | Pathways | Protein Target | Inhibitor | Mechanism of Action of Inhibitors | Clinical Trial |
|---|---|---|---|---|---|---|
| Centrosome amplification (CA) | ATM/ATR kinases | DNA damage | ATM/ATR kinases | Caffeine | Induces G1/S arrest and abrogates the G1/S and G2/M checkpoint delay periods [84]. | Phase I–IV studies in a wide range of solid tumours (lymphoma, small cell lung cancer, melanoma, kidney, pancreatic, ovarian and leukaemia). https://clinicaltrials.gov/ct2/results?cond=Cancer&term=caffeine&cntry=&state=&city=&dist= |
| Chk1 | Chk1 | UCN-01 | Binds the ATP-binding pocket of Chk1, resulting in accumulation of cells in G1 phase and induction of apoptosis [92]. | Phase II clinical trials in lymphoma, small-cell lung cancer, melanoma, kidney, pancreatic, ovarian and leukaemia patients. https://clinicaltrials.gov/ct2/results?term=UCN-01&age_v=&gndr=&type=&rslt=&phase=1&Search=Apply | ||
| MK-8776 (SCH 900776) | Radiosensitizes tumor cells by causing abrogation of the G2 block and DSB repair [93]. | Phase II clinical trial completed in patients with relapsed acute myeloid leukaemia: NCT01870596. | ||||
| poly (ADP-ribose) polymerase (PARP) | PARP-1 | 3-Aminobenzamide (3-AB) | 1st generation PARP inhibitor: shows structural similarity with nicotinimide and binds PARP preventing it from depleting NAD+. PARP-1 inhibition causes the uncoupling of DNA and centrosome duplication cycles leading to CA [87]. | Pre-clinical development [89]. | ||
| Rucaparib (AG14361) | Hydrogen bonds with the Gly863, Ser904, and Glu988 residues of the PARP-1 protein [91]. | Phase I/II/III trials in a range of human malignancies alone and in combination with other agents. https://clinicaltrials.gov/ct2/results?cond=Cancer&term=rucaparib&cntry=&state=&city=&dist= | ||||
| NU1025 | Hydrogen bonds with the Gly863, Ser904, and Glu988 residues of the PARP-1 protein [91]. | Pre-clinical development [88]. | ||||
| Phosphoinositide 3-kinase (PI3K) | PI3K/Akt | PI3K/p110α | LY294002 | Reversibly inhibits PI3K by competing with ATP for the active site of catalytic subunit p110 [94]. | Phase I clinical trial in patients with relapsed or refractory neuroblastoma: NCT02337309. | |
| GDC-0941 Pictrelisib | Selectively binds to PI3K isoforms in an ATP-competitive manner and inhibits the production of the secondary messenger phosphatidylinositol-3,4,5-trisphosphate (PIP3) [95]. | Phase II clinical study in completed in breast cancer and non-small cell lung cancer. https://clinicaltrials.gov/ct2/results?term=GDC-0941&cond=cancer+and+neoplasia&age_v=&gndr=&type=&rslt=&phase=1&Search=Apply | ||||
| Wortmannin | Binds to the ATP-binding site of p110 by forming a covalent bond between C20 of the wortmannin furan ring and K802 of p110a [94]. | Pre-clinical development. | ||||
| A66 | Blocks insulin signalling to Akt/PKB by inhibiting p110α and reducing cell growth [96]. | Pre-clinical development [97]. | ||||
| Akt | Akt | MK-2206 | Non-ATP competitive inhibitor of the PI3K/Akt signalling pathway causing decreased cell proliferation and induction of apoptosis [96]. | Clinical trials in advanced breast cancer, metastatic neuroendocrine tumors (NET), advanced colorectal carcinoma, ovarian cancer, endometrial cancer and non-small cell lung cancer. https://clinicaltrials.gov/ct2/results?cond=cancer&term=MK-2206&cntry=&state=&city=&dist=&Search=Search | ||
| Mtor p70S6 kinase | Akti X | Inhibits phosphorylation of mTOR, p70S6 kinase and S6 ribosomal protein resulting in apoptosis [96]. | Phase I clinical trial in cancer patients with metastatic melanoma: NCT02489266. | |||
| PI3K /Mtor | PF-04691502 | ATP-competitive PI3K/mTOR dual inhibitor, which potently inhibits recombinant class I PI3K and mTOR [98]. | Phase II clinical trial in patients with breast cancer (NCT01658176, NCT01430585) and endometrial cancer (NCT01420081). | |||
| ROCK1 and ROCK2 | RhoA/ROCK | ROCK | Y27632 | Y-27632 inhibits both ROCK1 and ROCK2 by competing with ATP for binding to the catalytic site [96,99]. | Pre-clinical trials [99]. | |
| H1152 | ATP-competitive inhibitor of G-protein Rho-associated [96,99]. | Pre-clinical trials [99]. | ||||
| Plk4 | Centriole duplication | Plk4 | CFI-400945 | ATP competitive inhibitor, inhibits autophosphorylation of Plk4 at serine 305 [30]. | Phase I clinical trial in patients with advanced cancer (NCT01954316) and phase I study in patients with relapsed or refractory acute myeloid leukaemia or myelodysplastic syndrome (NCT03187288). |
| Inhibitors Targeting | Enzymes | Pathways | Protein Target | Inhibitor | Mechanism of Action of Inhibitors | Clinical Trial |
|---|---|---|---|---|---|---|
| Centrosome Clustering (CC) | Stathmin | STAT3–Stathmin | STAT3 | Stattic | Blocks STAT3 to inhibit stathmin depolymerase function, allowing stathmin to remain active to depolymerize microtubules [112]. | Pre-clinical development. |
| Napabucasin(BBI-608) | Blocks STAT3 to inhibit stathmin depolymerase function, allowing stathmin to remain active to depolymerize microtubules [112]. | Phase I-III clinical trials including trials in combination with other compounds in patients with pancreatic, metastatic colorectal cancer, gastric, gastro-esophageal junction cancer and non-squamous, non-small cell lung cancer. https://clinicaltrials.gov/ct2/results?cond=cancer&term=BBI-608&cntry=&state=&city=&dist=&Search=Search | ||||
| APC/C Cdc20/CDH1 | Mitotic progression | Anaphase-promoting Complex | ProTAME | Disrupts APC3–Cdc20 IR-tail binding interaction and prevents its activation by Cdc20 and Cdh1 [119,120]. | Pre-clinical development. | |
| APC/C Cdc20 | Apcin | Disrupts D-box interaction between Cdc20 and the substrate [120]. | Pre-clinical development [122]. | |||
| HSET | Mitotic spindle assembly | HSET | CW069 | Binds to loop 5 cleft of HSET motor domain causing selective allosteric inhibition of HSET [45,123]. | Pre-clinical development. | |
| AZ82 | Blocks the ATP binding pocket and binds specifically to the KIFC1/microtubule (MT) binary complex, inhibiting the MT-stimulated KIFC1 enzymatic activity [45]. | Pre-clinical development [124,125]. | ||||
| α/β Tubulin | microtubule dynamics | Tubulin | Griseofulvin | Decreases the dynamicity of microtubules, leading to multipolar spindles and inducing mitotic arrest [126]. | Preclinical development [127]. | |
| GF-15 | Disrupts microtubule dynamics, leading to multipolar spindles [128]. | Pre-clinical development [128,129]. | ||||
| Poly (ADP-ribose) polymerase | DNA damage | PARP5a (TNKS1) PARP1, 2, 6 | AZ0108 | NAD+ competitive inhibitor [130]. | Pre-clinical development [130]. | |
| PARP-1 | PJ-34 | Induces G2/M arrest in cancer cells via p21 gene activation and subsequent cell death via centrosome declustering [124]. | Pre-clinical development [131,132]. | |||
| PARP-1, 2 | Veliparib (ABT-888) | Inhibits PARP catalytic activity and, to a lesser extent, exerts PARP trapping activity [133]. | Phase I/II/III trials in a range of human malignancies, alone and in combination with other agents. https://clinicaltrials.gov/ct2/results?term=veliparib&cond=Cancer&age_v=&gndr=&type=&rslt=&phase=2&phase=3&Search=Apply | |||
| Non-specific PARP inhibitor/cysteine-containing proteins | Iniparib (BSI-201) | A non-selective modifier of cysteine-containing proteins (protein-reactive compound), rather than a bona fide PARP inhibitor [134,135]. | Phase I/II/III trials in breast, ovarian, uterine, lung and advanced solid tumours, both alone and in combination with other agents. https://clinicaltrials.gov/ct2/results?cond=Cancer&term=Iniparib&cntry=&state=&city=&dist= | |||
| Coiled-Coil Containing Protein 3 (TACC3) Colonic and Hepatic Tumour Overexpressed gene (ch-TOG) | Actin and mitotic microtubule organization | Integrin-linked Kinase (ILK) | QLT-0267 | Inhibits kinase activity of ILK in an ATP-competitive manner and disrupts TACC3 phosphorylation [129]. | Pre-clinical development [136]. | |
| Hsp70 | Nek6–Hsp72 | Hsp70 Hsp72 | VER-155008 | ATP competitive inhibitor of Hsp 70 which blocks the nucleotide-binding domain and prevents substrate binding [137]. | Pre-clinical development [138]. | |
| N/A | N/A | N/A | CCCI-01 | Inhibits centrosome clustering, promoting spindle multipolarity and cell death selectively in cancer cells [139]. | Pre-clinical development [139]. | |
| End Binding Protein-1 (EB1), cytoplasmic linker protein-170 (CLIP-170) | Microtubule dynamics | EB1, CLIP-170 | EM011 | Disrupts microtubule dynamicity and induces G2/M arrest in cancer cells followed by apoptotic cell death [140]. | Pre-clinical development [141]. | |
| Cofilin | Actin destabilization | Cofilin Platelet-derived Growth Factor Receptor (PDGFR)-a and -b and FLT3 | Crenolanib (CP-868596) | An ATP competitive inhibitor which induces spindle multipolarity in cells with CA by activating cofilin [142]. | Phase I–III clinical trials in D842V PDGFRA gene-mutated tumours, advanced gastrointestinal stromal tumours and acute myeloid leukaemia. https://clinicaltrials.gov/ct2/results?cond=cancer&term=Crenolanib&cntry=&state=&city=&dist=&Search=Search | |
| Cofilin | Actin destabilization | Cofilin PDGFR-b | CP-673451 | An ATP competitive inhibitor which induces spindle multipolarity in cells with CA by activating cofilin [142]. | Pre-clinical development [143]. |
| Inhibitors Targeting | Enzymes | Pathways | Protein Target | Inhibitor | Mechanism of Action | Clinical Trial |
|---|---|---|---|---|---|---|
| ADP- ribosylation | PARP | DDR | PARP1, PARP2 and PARP3 | Olaparib | Binds within the nicotinamide-binding pocket in the ADP-ribosyl transferase catalytic site [177]. | 2014: FDA approved for the treatment of adult patients with deleterious or suspected deleterious germline BRCA-mutated advanced ovarian cancer [183]. 2017: FDA approved for the maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer, who are in a complete or partial response to platinum-based chemotherapy. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm572143.htm 2018: FDA approved for the treatment of adult patients with metastatic breast cancer who have a BRCA gene mutation. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm592347.htm |
| Rucaparib | Binds within the nicotinamide-binding pocket in the ADP-ribosyl transferase catalytic site [177]. | 2016: FDA approved for treatment of adults patients with germline and/or somatic BRCA-mutated advanced ovarian cancer. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm533891.htm | ||||
| PARP1 and PARP2 | Niraparib | Binds within the nicotinamide-binding pocket in the ADP-ribosyl transferase catalytic site and makes contact with the regulatory subdomains. Efficiently traps PARP1 on the damage-containing DNA [177]. | 2017: FDA approved for treatment of adult patients with recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer who are in complete or partial response to platinum-based chemotherapy. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm548487.htm | |||
| Veliparib | Binds within nicotinamide-binding pocket in the ADP-ribosyl transferase catalytic site and makes contact with the regulatory subdomains. Efficiently traps PARP1 on the damage-containing DNA [177]. | Phase I-III clinical trials including patients with previously untreated advanced or metastatic squamous non-small cell lung cancer; patients receiving first cytotoxic chemotherapy for metastatic or advanced non-squamous, non-small cell lung cancer; patients with ovarian cancer; triple negative breast cancer; glioblastoma. Mostly in combination with chemotherapy. https://clinicaltrials.gov/ct2/results?term=Veliparib&age_v=&gndr=&type=&rslt=&phase=0&phase=1&phase=2&phase=3&Search=Apply | ||||
| Talazoparib | Binds within nicotinamide-binding pocket in the ADP-ribosyl transferase catalytic site and makes contact with the regulatory subdomains. Potent PARP trapping [177]. | Phase I–III clinical trials including phase III patients with advanced and/or metastatic breast cancer with germline BRCA (breast cancer susceptibility gene) mutations and squamous cell lung carcinoma. https://clinicaltrials.gov/ct2/results?term=Talazoparib&age_v=&gndr=&type=&rslt=&phase=0&phase=1&phase=2&phase=3&Search=Apply | ||||
| CEP-9722 | Binds within the nicotinamide-binding pocket in the ADP-ribosyl transferase catalytic site [177,179]. | Phase I/II trial in patients with advanced or metastatic solid tumours and documented deficiencies of DNA repair pathways, such as BRCA1/2 (NCT01311713, NCT01345357, NCT00920595). | ||||
| Phosphorylation | PIKK | DSB, cell cycle | ATM | KU-55933 | Binds to the ATP binding pocket of ATM, blocking its kinase function and ATM-mediated signalling [170]. | Pre-clinical development. |
| KU-60019 | Binds to the ATP binding pocket of ATM, blocking its kinase function and ATM-mediated signalling [184]. | Pre-clinical development. | ||||
| KU-59403 | Binds to the ATP binding pocket of ATM, blocking its kinase function and ATM-mediated signalling [185]. | Pre-clinical development. | ||||
| CP466722 | Binds to the ATP binding pocket of ATM, blocking its kinase function and ATM-mediated signalling [185]. | Pre-clinical development. | ||||
| AZD0156 | Binds to the ATP binding pocket of ATM, blocking its kinase function and ATM-mediated signalling [186]. | Phase I trial of AZD0156 in combination with olaparib in patients with locally advanced/metastatic cancer: NCT02588105. | ||||
| SSB, Cell Cycle | ATR | VE-822/VX-970 | Selectively inhibits ATR kinase activity and prevents ATR-mediated signalling in the ATR-checkpoint kinase 1 (Chk1) signalling pathway [187]. | Phase I/II trial of VX970 and topotecan treating small cell lung cancer: NCT02487095. Phase I trial of VX970 in combination with veliparib and cisplatin in patients with advanced refractory solid tumours: NCT02723864 Phase I trial of VX970 and irinotecan hydrochloride in treating patients with metastatic cancer: NCT02595931. | ||
| AZD6738 | Selectively inhibits ATR kinase activity and prevents ATR-mediated signalling [188]. | Phase I/II trial for AZD6738 in combination with acalabrutinib in subjects with relapse or refractory high-risk chronic lymphocytic leukaemia (CLL): NCT03328273. Phase I trial for AZD6738 in combination with palliative radiotherapy or chemotherapy in patients with advanced solid tumours: NCT02223923. | ||||
| BAY-1895344 | Selectively inhibits ATR kinase activity and prevents ATR-mediated signalling in the ATR-checkpoint kinase 1 (Chk1) signalling pathway [189]. | Phase I trial of BAY1895344 monotherapy in patients with advanced solid tumours and lymphomas: NCT03188965. | ||||
| ATR/CDK2 | NU6027 | Low micromolar inhibitor of ATR kinase activity and prevents ATR-mediated signalling in the ATR-checkpoint kinase 1 (Chk1) signalling pathway. | Pre-clinical development. |
| Inhibitors Targeting | Enzymes | Pathways | Protein Target | Inhibitor | Mechanism of Action | Clinical Trial |
|---|---|---|---|---|---|---|
| DNA processing | RecQ DNA Helicases | DDR | BLM | ML216 | Inhibits helicase activity of BLM. ML216 competes with ATP binding, the driving force behind its DNA unwinding, or by preventing BLM from binding to DNA [218]. | Pre-clinical development. |
| WRN | NSC 617145 | Inhibits WRN helicase activity, but not its nuclease activity. Thought to trap WRN on the DNA substrate [219]. | Pre-clinical development. | |||
| NSC 19630 | Inhibits helicase activity, but not nuclease activity [221]. | Pre-clinical development. | ||||
| Topoisomerases | DDR, DNA replication | TOP1 | Irinotecan | Prevents religation of the DNA strand by binding to topoisomerase I-DNA complex [226]. | FDA approved (1996) for treatment of colorectal cancer when disease has recurred following initial fluorouracil treatment. FDA approved (2015) in combination with fluorouracil and leucovorin, in patients with advanced (metastatic) pancreatic cancer previously treated with gemcitabine-based chemotherapy. https://www.accessdata.fda.gov/scripts/cder/ob/search_product.cfm | |
| Topotecan | Binds to the topoisomerase I-DNA complex and prevents re-ligation of single strand breaks [226]. | FDA approved (2006) in combination with cisplatin for the treatment of stage IVB recurrent or persistent cervical cancer that is not amenable to curative treatment with surgery and/or radiotherapy. https://www.cancer.gov/about-cancer/treatment/drugs/fda-topotecan-hydrochloride Phase III trial in combination with radiotherapy in patients with brain metastases from non-small cell lung cancer: NCT00390806. | ||||
| Indotecan (LMP400) | Binds to the topoisomerase I-DNA covalent cleavage complexes, and inhibits repair of single-strand breaks [223]. | Phase I trial of LMP400 in subjects with solid tumours or lymphomas that have not responded to treatment: NCT01794104. | ||||
| Indimitecan (LMP776) | Preferential Top1-DNA trapping at unique sites [232]. | Phase I trial in adults with relapsed solid tumors and lymphomas: NCT01051635. | ||||
| GENZ-644282 | Binds to the topoisomerase I-DNA covalent cleavage complexes, and inhibits repair of single-strand breaks [228]. | Phase I trial of Genz-644282 in patients with advanced malignant, solid tumours: NCT00942799. | ||||
| TOP2 | Doxorubicin | Intercalates into DNA and targets the topoisomerase II cleavage complexes, thereby inhibiting DNA religation [223]. | FDA approved (1974) and currently used for treatment for acute lymphoblastic leukaemia, acute myeloblastic leukaemia, Wilms’ tumour, neuroblastoma, breast carcinoma, ovarian carcinoma, transitional cell bladder carcinoma, thyroid carcinoma, gastric carcinoma, Hodgkin’s disease, malignant lymphoma and bronchogenic carcinoma [233]. | |||
| Etoposide | Intercalates into DNA and poisons the topoisomerase II cleavage complexes, thereby inhibiting DNA re-ligation [223]. | FDA approved (1983) and currently used in combination with other chemotherapeutic drugs for treatment of patients with refractory testicular tumours, small lung cancer, ovarian cancer, leukaemia and lymphoma. https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&applno=074290 | ||||
| Mitoxantrone | Intercalates into DNA, causing crosslinks and strand breaks and targets the topoisomerase II cleavage complexes, thereby inhibiting DNA re-ligation [223]. | FDA approved (1987) and currently used with other drugs to treat acute myeloid leukaemia and prostate cancer. https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&applno=077356 https://www.cancer.gov/about-cancer/treatment/drugs/mitoxantronehydrochloride | ||||
| Aclarubicin | Intercalates into DNA and targets the topoisomerase II cleavage complexes, inhibiting DNA re-ligation [223]. | Phase II–IV trials in combination with other drugs in patients with acute myeloid leukaemia (AML). https://clinicaltrials.gov/ct2/results?cond=cancer&term=Aclarubicin&cntry=&state=&city=&dist= | ||||
| Dexrazoxane (ICRF-187) | Catalytic TOP2 inhibitor. | Phase III–IV trials against multiple cancers. https://clinicaltrials.gov/ct2/results?cond=Cancer&term=Dexrazoxane&cntry=&state=&city=&dist=&Search=Search | ||||
| DNA repair signalling | MRN complex | DDR | Mre11 | Mirin | Binds in the active site of Mre11 blocking DNA phosphate backbone rotation; inhibits exonuclease activity of Mre11 and MRN/DSB-mediated ATM activation without affecting ATM protein kinase activity [234]. | Pre-clinical development. |
| PFM01 | Binds near the dimer interface blocking the ssDNA-binding path towards the catalytic metal ions and disrupts endonuclease activity [185]. | Pre-clinical development. | ||||
| PFM39 | Binds in the active site of Mre11 inhibiting its exonuclease activity [185]. | Pre-clinical development. | ||||
| DNA repair | Nucleotide Excision Repair (NER) | ERCC1–XPF | E-X AS5-4 | Targets the ERCC1–XPF interaction domain for heterodimerisation [235]. | Pre-clinical development. | |
| E-X AS7 | Targets the XPF active site itself [235]. | Pre-clinical development. |
| Inhibitors Targeting | Enzymes | Pathways | Protein Target | Inhibitor | Mechanism of Action | Clinical Trial |
|---|---|---|---|---|---|---|
| Phosphorylation | PIKK | NHEJ | DNA-PKcs | NU7026 | A potent inhibitor of DNA-PK, exhibiting ATP-competitive inhibitor kinetics [264]. | Pre-clinical development. |
| NU7441 | A highly potent and selective DNA-PK inhibitor, exhibiting ATP-competitive inhibition kinetics [263]. | Pre-clinical development. | ||||
| VX-984/M9831 | ATP-competitive inhibitor of DNA-PKcs [269]. | Phase 1 clinical trial as a single agent and in combination with doxorubicin or pegylated liposomal doxorubicin (PLD): NCT02644278. | ||||
| MSC-2490484A/M3814 | Binds to DNA-PK and inhibits its kinase activity and prevents (at least partially) the NHEJ pathway [266]. | Phase I trial of MSC2490484A monotherapy in subjects with advanced solid tumors or chronic lymphocytic leukaemia (CLL) likely to have alterations in DNA repair mechanisms, such as the BRCA and ATM pathways: NCT02316197. Phase I trial of MSC2490484A in combination with radiotherapy and in combination with chemoradiotherapy (radiotherapy and cisplatin) in patients with advanced solid tumours: NCT02516813. | ||||
| DNA processing | DNA ligase | Ligases I, III and IV | L189 | Equally inhibits ligases I, III and IV by blocking DNA binding [253]. | Pre-clinical development. | |
| Ligase-IV (DNA ligases I and III) | SCR7 | Binds to the DNA-binding domain of ligase-IV, thus preventing the binding of ligase IV to DNA ends [271]. | Pre-clinical development. |
| Inhibitors Targeting | Enzymes | Pathways | Protein Target | Inhibitor | Mechanism of Action | Clinical Trial |
|---|---|---|---|---|---|---|
| DNA processing | RecA-like NTPases | HR | RAD51 | B02 | Inhibits RAD51 by disrupting RAD51 ability to bind ssDNA [280]. | Pre-clinical development. |
| RI-1 | Contains a chloromaleimide group that reacts with cysteine 319 of RAD51 at the monomer–monomer interface near the ATP active site [281]. | Pre-clinical development. | ||||
| RS-1 | Stabilizes the RAD51 nucleoprotein filament and stimulates RAD51 biochemical activities [282]. | Pre-clinical development. | ||||
| IBR120 | Inhibits RAD51 by mimicking the effect of BRC repeat binding to RAD51 [283]. | Pre-clinical development. | ||||
| RAD52 | RAD52 | D-103 | Inhibits RAD52-mediated ssDNA annealing and inhibits D-loop formation [284]. | Pre-clinical development. | ||
| D-G23 | Inhibits RAD52-mediated ssDNA annealing and inhibits D-loop formation [284]. | Pre-clinical development. | ||||
| AICAR | Disrupts the RAD52-ssDNA interaction [285]. | Pre-clinical development. | ||||
| (−)-Epigallocatechin) | Inhibits RAD52 ssDNA binding [286]. | Pre-clinical development. | ||||
| 6-HidroxyDL-dopa | Disrupts RAD52 recruitment and recombination activity [287]. | Pre-clinical development. | ||||
| RAD54 | RAD54/SENP1 | Streptonigrin (STN) | An antitumor antibiotic that binds RAD54 ATPase domain and inactivates it by generating reactive oxygen species [288]. Recently found to bind to and inhibit SUMO-specific protease, SENP1 [289]. | Used to treat multiple cancer types (since the 1960s); however, induces severe and prolonged bone marrow depression. |
| Inhibitors Targeting | Enzymes | Pathways | Protein Target | Inhibitor | Mechanism of Action | Clinical Trial |
|---|---|---|---|---|---|---|
| Acetylation | HDAC | Chromatin modification and DDR | Histone deacetylases (HDACs) I, IIa, IIb, IV | Vorinostat/SAHA | Inhibits HDAC by binding the zinc-activated catalytic site [352]. | FDA approved (2006) for the treatment of cutaneous manifestations of T-cell lymphoma [352]. Phase III trial of vorinostat in the treatment of advanced malignant pleural mesothelioma and multiple myeloma: NCT00128102. Phase III trial in combination with chemotherapy for the treatment of advanced non-small cell lung cancer patients: NCT00473889. |
| HDACs I, II | Belinostat | Inhibits HDAC by binding to the zinc-activated catalytic site [353]. | FDA approved (2014) for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma [353]. Phase I–II clinical trials in the treatment of a range of solid tumours, acute myeloid leukaemia, cutaneous T-cell lymphoma, lung and liver cancer and non-Hodgkins lymphoma and other haematological malignancies. https://clinicaltrials.gov/ct2/results?term=belinostat&age_v=&gndr=&type=&rslt=&Search=Apply | |||
| HDACs I, II | Panobinostat | Inhibits HDAC by binding to the zinc-activated catalytic site [354]. | FDA approved (2015) for the treatment of patients with multiple myeloma. https://www.accessdata.fda.gov/scripts/cder/ob/search_product.cfm Phase I–III trials in the treatment of a range of cancers, including pancreatic, breast, lung, liver, prostate, thyroid, renal, colon, brain, gastric, skin, and haematological malignancies. https://clinicaltrials.gov/ct2/results?term=panobinostat+AND+Cancer+AND+Neoplasms&phase=0123 | |||
| HDACs 1,2,4,6 | Romidepsin | inhibits HDAC by binding to the zinc-activated catalytic site [345]. | FDA approved (2009) for the treatment of cutaneous T-cell lymphoma in patients who have received at least one prior systemic therapy. 2011: FDA approved for the treatment of peripheral T-cell lymphoma in patients who have received at least one prior therapy [355]. | |||
| HDACs 1,2,3,10 | Chidamide | Inhibits HDAC by binding to the zinc-activated catalytic site [356]. | Phase III trial in combination with exemestane for the treatment of hormone-receptor positive advanced breast cancer: NCT02482753. Phase III trial in combination with chemotherapy for the treatment of peripheral T-cell lymphoma: NCT03023358. | |||
| HDACs I, IIa | Valproic acid (VPA) | In vivo and in vitro induces differentiation of transformed cells and can delay growth in primary tumours [350,351,357,358]. | Enrolled in >80 clinical cancer trials, including five in phase III (for multiple tumour types). https://clinicaltrials.gov/ct2/results?cond=Cancer&term=Valproic+acid&cntry=&state=&city=&dist= | |||
| histone acetyl transferases (HAT) | p300/CBP, PCAF, Tip60 | Curcumin | Inhibits p300/CBP by decreasing the binding efficiency of both histones and acetyl CoA to p300 [359]. | Phase I–III clinical trials for the treatment of multiple tumour types. https://clinicaltrials.gov/ct2/results?cond=Cancer&term=Curcumin+&cntry=&state=&city=&dist= | ||
| p300, CBP, Tip60, PCAF | EGCG | Did not appear bind to the HAT domain, potentially binds another site on the protein [357]. | Phase I–IV clinical trials in a range of tumours including breast, prostate, colon, lung, pancreas. https://clinicaltrials.gov/ct2/results?cond=Cancer&term=EGCG&cntry=&state=&city=&dist= | |||
| Tip60 | TH1834 | Binds into the AcCoA binding pocket [360]. | Pre-clinical development. | |||
| Tip60 | NU9056 | Binds into the AcCoA binding pocket [361]. | Pre-clinical development. | |||
| PCAF, Gcn5, p300 CREB | PU139 | Predicted to bind at the catalytic site binding pocket [362]. | Pre-clinical development. | |||
| CBP, p300 | PU141 | Predicted to bind at the catalytic site binding pocket [362]. | Pre-clinical development. | |||
| PCAF, Gcn5 | CPTH6 | Competes with Acetyl-CoA to bind at the catalytic site [363]. | Pre-clinical development. | |||
| p300 | RTK1 | Through its hydroxyl group, possibly forms a specific interaction with lysine residue (Lys-1358) in the p300 HAT domain [364]. | Pre-clinical development. | |||
| Methylation | KMT | DOT1-L | EPZ-5676 | Occupies the S-adenosyl methionine (SAM) binding pocket of DOT1-L [365]. | Phase I trial for the treatment of acute myeloid leukaemia (AML) and acute lymphoblastic leukaemia (ALL): NCT02141828. | |
| G9a | UNC0638 | Occupies the histone peptide-binding channel and interacts with the lysine-binding pocket [366]. | Pre-clinical development. | |||
| EZH2 | EPZ-6438 (tazemetostat) | Occupies the S-adenosyl methionine (SAM) binding pocket of EZH2 [367]. | Phase I–II clinical trials for the treatment of recurrent ovarian, primary peritoneal, or endometrial cancer, different types of lymphomas, sarcomas and advanced solid tumours. https://clinicaltrials.gov/ct2/results?term=EPZ-6438&age_v=&gndr=&type=&rslt=&phase=0&phase=1&phase=2&phase=3&Search=Apply | |||
| SMYD2 | AZ505 | Inhibits though its benzooxazinone group, which is positioned within the lysine-binding channel of the substrate [368]. | Pre-clinical development. | |||
| SETD8 | Nahuoic acid A | Occupies the S-adenosyl methionine (SAM) binding pocket [369]. | Pre-clinical development. | |||
| SETD8 | Peptide based inhibitors | Selective norleucine containing peptide inhibitor [370] | Pre-clinical development. | |||
| KDM | LSD1 | TCP (tranylcypromine) | Inhibits LSD1 by forming a covalent adduct with the FAD cofactor [371]. | Phase I/II trial in combination with ATRA (all-trans-retinoic acid) for the treatment of acute myeloid leukaemia or myelodysplastic syndrome (NCT02717884, NCT02273102). | ||
| GSK2879552 | Inhibits LSD1 by forming a covalent adduct with the FAD cofactor, leading to homolytic cleavage of the cyclopropyl ring [372]. | Phase I trial for the treatment of myelocytic leukaemia (NCT02177812) and small cell carcinoma (NCT02034123). Phase II trial in combination with azacitidine for the treatment of myelodysplastic syndrome: NCT02929498. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prakash, A.; Garcia-Moreno, J.F.; Brown, J.A.L.; Bourke, E. Clinically Applicable Inhibitors Impacting Genome Stability. Molecules 2018, 23, 1166. https://doi.org/10.3390/molecules23051166
Prakash A, Garcia-Moreno JF, Brown JAL, Bourke E. Clinically Applicable Inhibitors Impacting Genome Stability. Molecules. 2018; 23(5):1166. https://doi.org/10.3390/molecules23051166
Chicago/Turabian StylePrakash, Anu, Juan F. Garcia-Moreno, James A. L. Brown, and Emer Bourke. 2018. "Clinically Applicable Inhibitors Impacting Genome Stability" Molecules 23, no. 5: 1166. https://doi.org/10.3390/molecules23051166
APA StylePrakash, A., Garcia-Moreno, J. F., Brown, J. A. L., & Bourke, E. (2018). Clinically Applicable Inhibitors Impacting Genome Stability. Molecules, 23(5), 1166. https://doi.org/10.3390/molecules23051166