Triorganoindium Reagents in Rh-Catalyzed C–H Activation/C–C Cross-Coupling Reactions of 2-Arylpyridines




Abstract
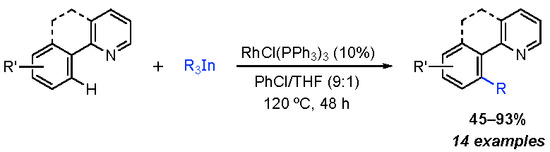
:
1. Introduction
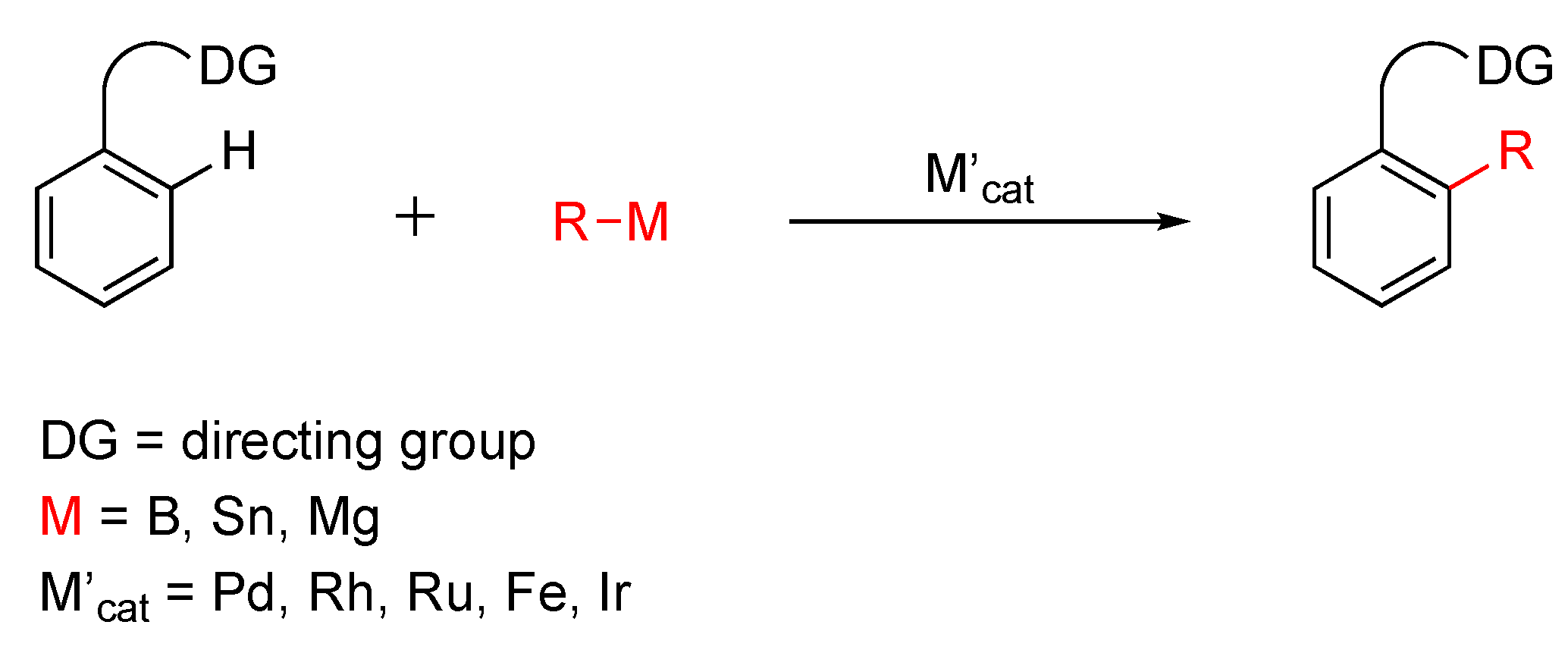
2. Results and Discussion
3. Materials and Methods
3.1. Materials and Reagents
3.2. Preparation of Triorganoindium Reagents
3.3. Preparation of Compounds 4 and 5
3.4. General Procedure C–H Activation/C–C Cross-Coupling with Indium(III) Organometallics under Rhodium Catalysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pérez, I.; Pérez Sestelo, J.; Sarandeses, L.A. Palladium-catalyzed cross-coupling reactions of triorganoindium compounds with vinyl and aryl triflates or iodides. Org. Lett. 1999, 1, 1267–1269. [Google Scholar] [CrossRef]
- Pérez, I.; Pérez Sestelo, J.; Sarandeses, L.A. Atom-efficient metal-catalyzed cross-coupling reaction of indium organometallics with organic electrophiles. J. Am. Chem. Soc. 2001, 123, 4155–4160. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.-L.; Wang, S.-Y.; Chok, Y.-K.; Xu, Y.-H.; Loh, T.-P. Organoindium reagents: The preparation and application in organic synthesis. Chem. Rev. 2013, 113, 271–401. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Shen, L.; Shen, Z.-L.; Loh, T.-P. Transition metal-catalyzed cross-coupling reactions using organoindium reagents. Chem. Soc. Rev. 2017, 46, 586–602. [Google Scholar] [CrossRef] [PubMed]
- Riveiros, R.; Rodríguez, D.; Pérez Sestelo, J.; Sarandeses, L.A. Palladium-catalyzed cross-coupling reaction of triorganoindium reagents with propargylic esters. Org. Lett. 2006, 8, 1403–1406. [Google Scholar] [CrossRef] [PubMed]
- Martínez, M.M.; Peña-López, M.; Pérez Sestelo, J.; Sarandeses, L.A. Synthesis of functionalized thiophenes and oligothiophenes by selective and iterative cross-coupling reactions using indium organometallics. Org. Biomol. Chem. 2012, 10, 3892–3898. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Caaveiro, C.; Pérez Sestelo, J.; Martínez, M.M.; Sarandeses, L.A. Triorganoindium reagents in selective palladium-catalyzed cross-coupling with iodoimidazoles: Synthesis of neurodazine. J. Org. Chem. 2014, 79, 9586–9593. [Google Scholar] [CrossRef] [PubMed]
- Mosquera, Á.; Fernández, M.I.; Canle López, M.; Pérez Sestelo, J.; Sarandeses, L.A. Nonsymmetrical 3,4-dithienylmaleimides by cross-coupling reactions with indium organometallics: Synthesis and photochemical studies. Chem. Eur. J. 2014, 20, 14524–14530. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Lee, J.; Lee, P.H. Highly efficient allyl cross-coupling reactions of allylindiums with organic electrophiles. J. Org. Chem. 2002, 67, 8265–8268. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.H.; Lee, S.W.; Seomoon, D. Tetraorganoindates as nucleophilic coupling partners in Pd-catalyzed cross-coupling reactions. Org. Lett. 2003, 5, 4963–4966. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Knochel, P. Preparation of aryl and heteroaryl indium (III) reagents by the direct insertion of indium in the presence of LiCl. Angew. Chem. Int. Ed. 2008, 47, 7648–7651. [Google Scholar] [CrossRef] [PubMed]
- Adak, L.; Yoshikai, N. Cobalt-catalyzed preparation of arylindium reagents from aryl and heteroaryl bromides. J. Org. Chem. 2011, 76, 7563–7568. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.-L.; Knochel, P. Stereoselective preparation of polyfunctional alkenylindium (III) halides and their cross-coupling with unsaturated halides. Chem. Eur. J. 2015, 21, 7061–7065. [Google Scholar] [CrossRef] [PubMed]
- Caeiro, J.; Pérez Sestelo, J.; Sarandeses, L.A. Enantioselective nickel-catalyzed cross-coupling reactions of trialkynylindium reagents with racemic secondary benzyl bromides. Chem. Eur. J. 2008, 14, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Riveiros, R.; Tato, R.; Pérez Sestelo, J.; Sarandeses, L.A. Rhodium-catalyzed allylic substitution reactions with indium (III) organometallics. Eur. J. Org. Chem. 2012, 2012, 3018–3023. [Google Scholar] [CrossRef]
- Tato, R.; Riveiros, R.; Pérez Sestelo, J.; Sarandeses, L.A. Rhodium-catalyzed conjugate addition of arylindium reagents to α,β-unsaturated carbonyl compounds. Tetrahedron 2012, 68, 1606–1611. [Google Scholar] [CrossRef]
- Thapa, S.; Gurung, S.K.; Dickie, D.A.; Giri, R. Copper-catalyzed coupling of triaryl- and trialkylindium reagents with aryl iodides and bromides through consecutive transmetalations. Angew. Chem. Int. Ed. 2014, 53, 11620–11624. [Google Scholar] [CrossRef] [PubMed]
- Dyker, G. (Ed.) Handbook of C–H Transformations; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Yu, J.-Q.; Shi, Z. (Eds.) C–H Activation; Topics in Current Chemistry 292; Springer: Heidelberg, Germany, 2010. [Google Scholar]
- Nareddy, P.; Jordan, F.; Szostak, M. Recent developments in ruthenium-catalyzed C–H arylation: Array of mechanistic manifolds. ACS Catal. 2017, 7, 5721–5745. [Google Scholar] [CrossRef]
- Davies, D.L.; Macgregor, S.A.; McMullin, C.L. Computational studies of carboxylate-assisted C–H activation and functionalization at group 8–10 transition metal centers. Chem. Rev. 2017, 117, 8649–8709. [Google Scholar] [CrossRef] [PubMed]
- Oi, S.; Fukita, S.; Inoue, Y. Rhodium-catalysed direct ortho arylation of 2-arylpyridines with arylstannanes via C–H activation. Chem. Commun. 1998, 2439–2440. [Google Scholar] [CrossRef]
- Vogler, T.; Studer, A. Oxidative coupling of arylboronic acids with arenes via Rh-catalyzed direct C−H arylation. Org. Lett. 2008, 10, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, F.; Kan, S.; Igi, K.; Chatani, N.; Murai, S. A Ruthenium-catalyzed reaction of aromatic ketones with arylboronates: A new method for the arylation of aromatic compounds via C−H bond cleavage. J. Am. Chem. Soc. 2003, 125, 1698–1699. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, L.; Vicente, R.; Kapdi, A.R. Transition-metal-catalyzed direct arylation of (Hetero)Arenes by C–H bond cleavage. Angew. Chem. Int. Ed. 2009, 48, 9792–9826. [Google Scholar] [CrossRef] [PubMed]
- Giri, R.; Thapa, S.; Kafle, A. Palladium-catalysed, directed C–H coupling with organometallics. Adv. Synth. Catal. 2014, 356, 1395–1411. [Google Scholar] [CrossRef]
- Zhu, R.-Y.; Farmer, M.E.; Chen, Y.-Q.; Yu, J.-Q. A simple and versatile amide directing group for C−H functionalizations. Angew. Chem. Int. Ed. 2016, 55, 10578–10599. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, B.; Zhang, J.; Yu, W.; Liu, Z.; Zhang, Y. Transition metal-catalyzed C–H bond functionalizations by the use of diverse directing groups. Org. Chem. Front. 2015, 2, 1107–1295. [Google Scholar] [CrossRef]
- Shang, R.; Ilies, L.; Nakamura, E. Iron-catalyzed C–H bond activation. Chem. Rev. 2017, 117, 9086–9139. [Google Scholar] [CrossRef] [PubMed]
- Pototschnig, G.; Maulide, N.; Schnürch, M. Direct functionalization of C−H bonds by iron, nickel, and cobalt catalysis. Chem. Eur. J. 2017, 23, 9206–9232. [Google Scholar] [CrossRef] [PubMed]
- Kalyani, D.; Deprez, N.R.; Desai, L.V.; Sanford, M.S. Oxidative C–H activation/C–C bond forming reactions: Synthetic scope and mechanistic insights. J. Am. Chem. Soc. 2005, 127, 7330–7331. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yin, Z.; Jiang, X.; Sun, P. Palladium-catalyzed direct ortho C–H arylation of 2-arylpyridine derivatives with aryltrimethoxysilane. J. Org. Chem. 2011, 76, 8543–8548. [Google Scholar] [CrossRef] [PubMed]
- Yoshikai, N.; Asako, S.; Yamakawa, T.; Ilies, L.; Nakamura, E. Iron-catalyzed C–H bond activation for the ortho-arylation of aryl pyridines and imines with Grignard reagents. Chem. Asian J. 2011, 6, 3059–3065. [Google Scholar] [CrossRef] [PubMed]
- Miyamura, S.; Tsurugi, H.; Satoh, T.; Miura, M. Rhodium-catalyzed regioselective arylation of phenylazoles and related compounds with arylboron reagents via C–H bond cleavage. J. Organomet. Chem. 2008, 693, 2438–2442. [Google Scholar] [CrossRef]
- Shuai, Q.; Yang, L.; Guo, X.; Baslé, O.; Li, C.-J. Rhodium-catalyzed oxidative C–H arylation of 2-arylpyridine derivatives via decarbonylation of aromatic aldehydes. J. Am. Chem. Soc. 2010, 132, 12212–12213. [Google Scholar] [CrossRef] [PubMed]
- Pena, M.A.; Pérez Sestelo, J.; Sarandeses, L.A. Palladium-catalyzed aryl–aryl cross-coupling reaction using ortho-substituted arylindium reagents. J. Org. Chem. 2007, 72, 1271–1275. [Google Scholar] [CrossRef] [PubMed]
- Núñez, A.; Sánchez, A.; Burgos, C.; Alvarez-Builla, J. Synthesis of carbo- and heterobiaryls by intermolecular radical addition of aryl bromides onto aromatic solvents. Tetrahedron 2004, 60, 6217–6224. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Song, B.; Bin, X. Palladium-catalyzed regioselective C–H bond ortho-acetoxylation of arylpyrimidines. Eur. J. Org. Chem. 2010, 2010, 4376–4380. [Google Scholar] [CrossRef]
- Chen, X.; Goodhue, C.E.; Yu, J.-Q. Palladium-catalyzed alkylation of sp2 and sp3 C–H bonds with methylboroxine and alkylboronic acids: Two distinct C–H activation pathways. J. Am. Chem. Soc. 2006, 128, 12634–12635. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Feng, J.; Li, C.-J. Palladium-catalyzed methylation of aryl C–H bonds by using peroxides. J. Am. Chem. Soc. 2008, 130, 2900–2901. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.-Y.; Sit, W.N.; Zhou, Z.; Chan, A.S.C. Palladium-catalyzed decarboxylative arylation of C–H bonds by aryl acylperoxides. Org. Lett. 2009, 11, 3174–3177. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yu, Z. Rhodium-catalyzed regioselective C–H functionalization via decarbonylation of acid chlorides and C–H bond activation under phosphine-free conditions. J. Am. Chem. Soc. 2008, 130, 8136–8137. [Google Scholar] [CrossRef] [PubMed]
- So, C.M.; Lau, C.P.; Kwong, F.Y. Easily accessible and highly tunable indolyl phosphine ligands for Suzuki–Miyaura coupling of aryl chlorides. Org. Lett. 2007, 9, 2795–2798. [Google Scholar] [CrossRef] [PubMed]
- Oi, S.; Fukita, S.; Hirata, N.; Watanuki, N.; Miyano, S.; Inoue, Y. Ruthenium complex-catalyzed direct ortho arylation and alkenylation of 2-arylpyridines with organic halides. Org. Lett. 2001, 3, 2579–2581. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.; Liu, B.; Chen, W. Room-temperature Kumada cross-coupling of unactivated aryl chlorides catalyzed by N-heterocylic carbene-based nickel (II) complexes. J. Org. Chem. 2008, 73, 3954–3957. [Google Scholar] [CrossRef] [PubMed]
- Norinder, J.; Matsumoto, A.; Yoshikai, N.; Nakamura, E. Iron-catalyzed direct arylation through directed C–H bond activation. J. Am. Chem. Soc. 2008, 130, 5858–5859. [Google Scholar] [CrossRef] [PubMed]
- Szadkowska, A.; Gstrein, X.; Burtscher, D.; Jarzembska, K.; Woźniak, K.; Slugovc, C.; Grela, K. Latent thermo-switchable olefin metathesis initiators bearing a pyridyl-functionalized chelating carbene: Influence of the leaving group’s rigidity on the catalyst’s performance. Organometallics 2010, 29, 117–124. [Google Scholar] [CrossRef]
- Kim, M.; Kwak, J.; Chang, S. Rhodium/N-heterocyclic carbene catalyzed direct intermolecular arylation of sp2 and sp3 C–H bonds with chelation assistance. Angew. Chem. Int. Ed. 2009, 48, 8935–8939. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 2a, 2b, 7a, 7b, 8a, 8b, 9a, 9b, 10a, 10b are available from the authors. |


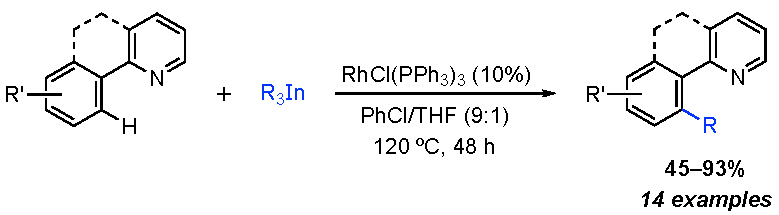{kind=link}
{kind=link}

| Entry | R (mol % of R3In) | Catalyst | Solvent | Yield (%) 2 |
|---|---|---|---|---|
| 1 | Me (100) | Rh(PPh3)3Cl | THF | 16 |
| 2 | Ph (100) | Rh(PPh3)3Cl | THF | 17 |
| 3 | Me (100) | Rh(cod)(acac) | THF | 14 |
| 4 | Me (100) | Rh(nbd)(acac) | THF | 15 |
| 5 3 | Me (100) | [Rh(cod)Cl]2, Ph3P | THF | 14 |
| 6 3 | Me (100) | [Rh(cod)Cl]2, Cy3P | THF | 15 |
| 7 | Ph (100) | Rh(PPh3)3Cl | Toluene/THF (9:1) | 18 |
| 8 | Ph (100) | Rh(PPh3)3Cl | Cl2CHCHCl2/THF (9:1) | 27 (4) |
| 9 | Ph (150) | Rh(PPh3)3Cl | Cl2CHCHCl2/THF (9:1) | 42 (16) |
| 10 | Me (150) | Rh(PPh3)3Cl | Cl2CHCHCl2/THF (9:1) | 60 (34) |
| 11 | Me (150) | Rh(PPh3)3Cl | PhCl/THF (9:1) | 80 (12) |
| 12 | Ph (150) | Rh(PPh3)3Cl | PhCl/THF (9:1) | 47 (6) |

| Entry | 2-Arylpyridine | R | Product | Yield (%) 2 |
|---|---|---|---|---|
| 1 |  | Me |  | 80 (12) |
| 2 | Ph |  | 47 (6) | |
| 3 |  | Me |  | 84 (7) |
| 4 | Ph |  | 53 (6) | |
| 5 |  | Me |  | 60 |
| 6 | Ph |  | 57 | |
| 7 |  | Me |  | 62 |
| 8 | Ph |  | 57 (7) | |
| 9 |  | Me |  | 93 |
| 10 | Ph |  | 72 | |
| 11 | CH2=CH |  | 25 3 | |
| 12 | 4-CF3C6H4 |  | 20 3 | |
| 13 | 4-FC6H4 |  | 45 3 | |
| 14 | 3-FC6H4 |  | 30 3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riveiros, R.; Tato, R.; Pérez Sestelo, J.; Sarandeses, L.A. Triorganoindium Reagents in Rh-Catalyzed C–H Activation/C–C Cross-Coupling Reactions of 2-Arylpyridines. Molecules 2018, 23, 1582. https://doi.org/10.3390/molecules23071582
Riveiros R, Tato R, Pérez Sestelo J, Sarandeses LA. Triorganoindium Reagents in Rh-Catalyzed C–H Activation/C–C Cross-Coupling Reactions of 2-Arylpyridines. Molecules. 2018; 23(7):1582. https://doi.org/10.3390/molecules23071582
Chicago/Turabian StyleRiveiros, Ricardo, Rubén Tato, José Pérez Sestelo, and Luis A. Sarandeses. 2018. "Triorganoindium Reagents in Rh-Catalyzed C–H Activation/C–C Cross-Coupling Reactions of 2-Arylpyridines" Molecules 23, no. 7: 1582. https://doi.org/10.3390/molecules23071582
APA StyleRiveiros, R., Tato, R., Pérez Sestelo, J., & Sarandeses, L. A. (2018). Triorganoindium Reagents in Rh-Catalyzed C–H Activation/C–C Cross-Coupling Reactions of 2-Arylpyridines. Molecules, 23(7), 1582. https://doi.org/10.3390/molecules23071582