Lipase Catalysed Kinetic Resolution of Racemic 1,2-Diols Containing a Chiral Quaternary Center


Abstract
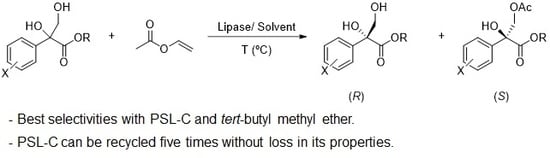
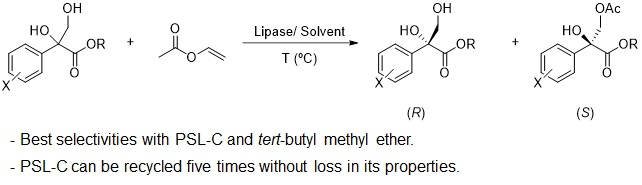
:
1. Introduction
2. Results and Discussion
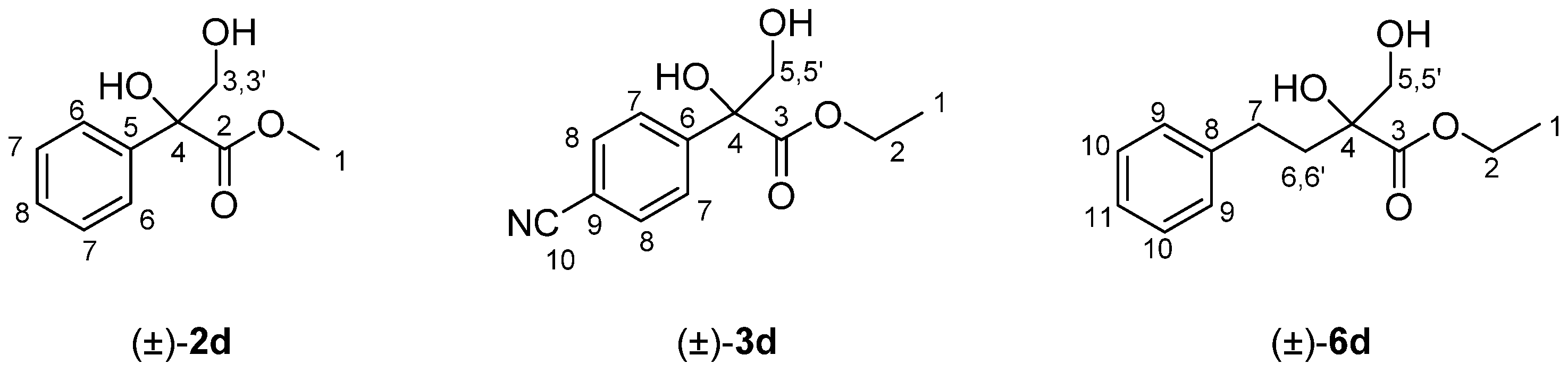
2.1. Preparation of the Racemic 1,2-Diols (±)-1–6d
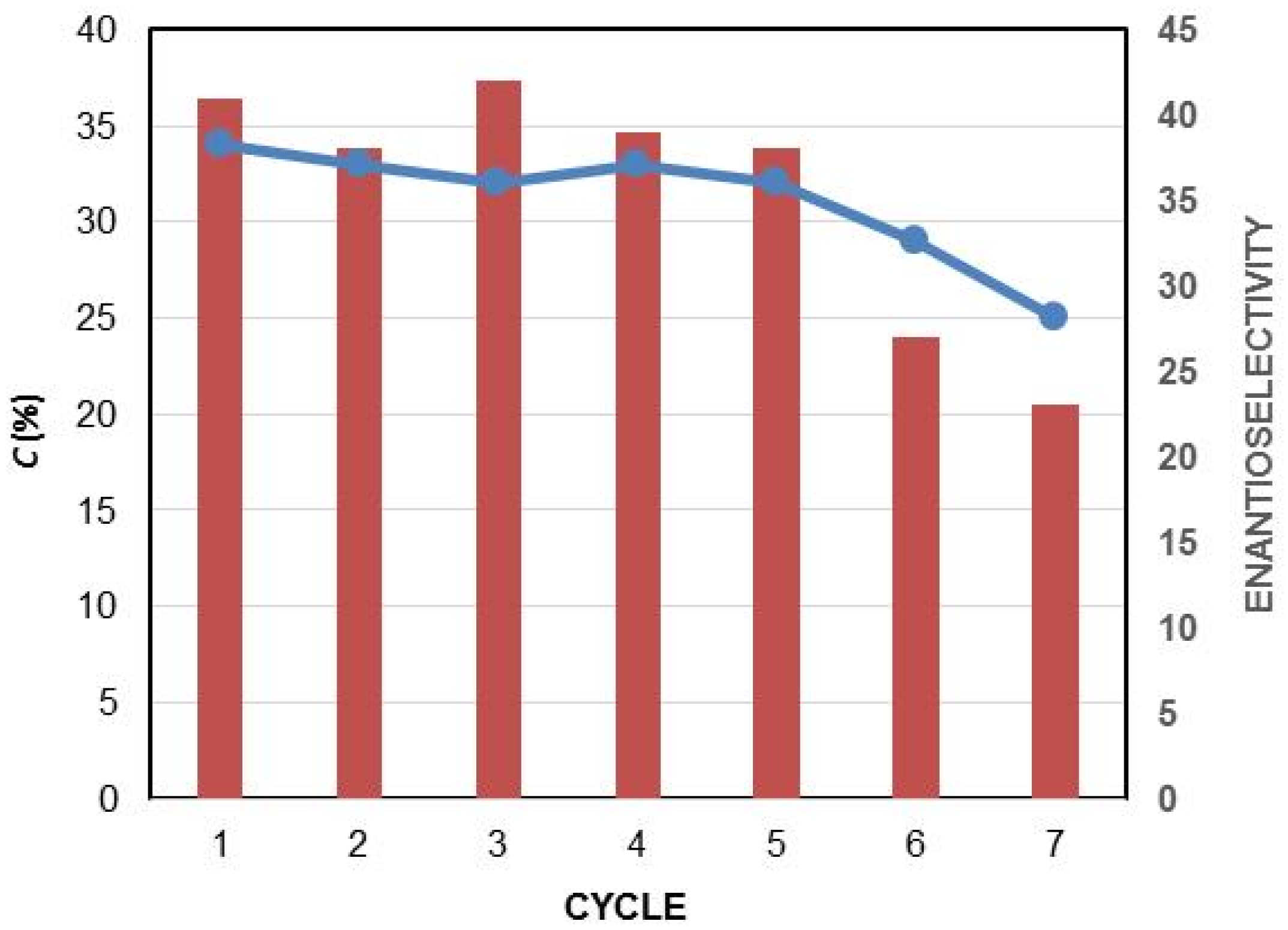
2.2. Kinetic Resolution of Racemic Diols (±)-1–6d
3. Materials and Methods
3.1. General Procedure for the Synthesis of Racemic Azocompounds (±)-1–6b
3.2. General Preparation of the Racemic 1,2-diols (±)-1–6d Starting from Azocompounds (±)-1–6b
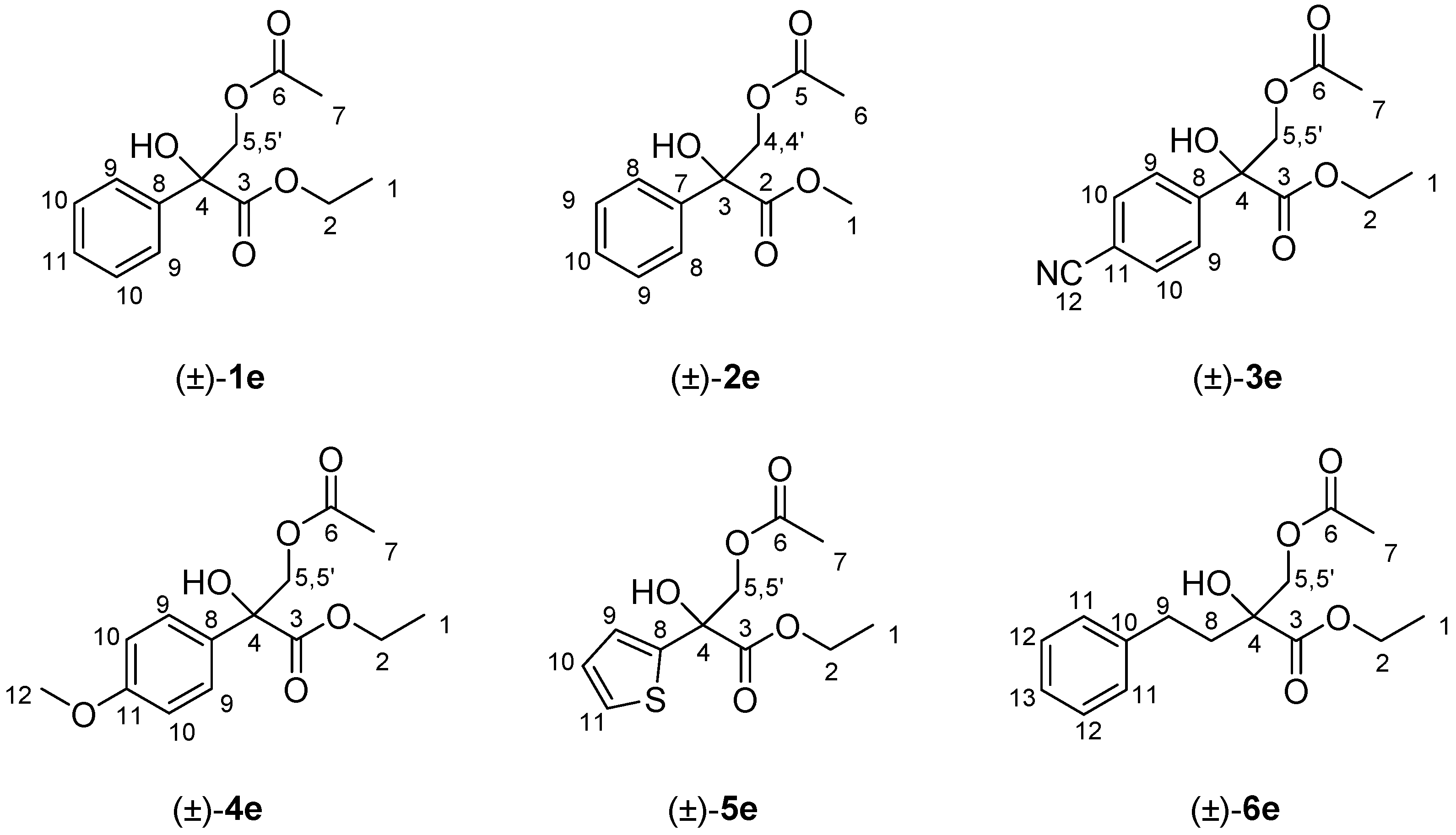
3.3. General Synthesis of the Racemic Acetates (±)-1–6e
3.4. General Procedure for the Biocatalyzed Acylation of the Racemic 1,2-diols (±)-1–6d
4. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Werner, L.; Machara, A.; Hudlicky, T. Short chemoenzymatic azide-free synthesis of oseltamivir (Tamiflu): Approaching the potential for process efficiency. Adv. Synth. Catal. 2010, 352, 195–200. [Google Scholar] [CrossRef]
- Wu, X.; Wang, L.; Wang, S.; Chen, Y. Stereoselective introduction of two chiral centers by a single diketoreductase: An efficient biocatalytic route for the synthesis of statin side chains. Amino Acids 2010, 39, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Omori, A.T.; Finn, K.J.; Leisch, H.; Carroll, R.J.; Hudlicky, T. Chemoenzymatic total synthesis of (+)-codeine by sequential intramolecular Heck cyclizations via C-B-D ring construction. Synlett 2007, 18, 2859–2862. [Google Scholar]
- Kanicha, S.; Montgomery, J. Highly diastereoselective preparation of anti-1,2-diols by catalytic addition of alkynylsilanes to α-silyloxyaldehydes. Org. Lett. 2006, 8, 4441–4443. [Google Scholar]
- Bataille, C.J.R.; Donohoe, T.J. Osmium-free direct syn-dihydroxylation of alkenes. Chem. Soc. Rev. 2011, 40, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, K.B.; Akashi, K. Osmium catalyzed vicinal hydroxylation of olefins by tert-butyl hydroperoxide under alkaline conditions. J. Am. Chem. Soc. 1976, 98, 1986–1987. [Google Scholar] [CrossRef]
- Andersson, M.A.; Epple, R.; Fokin, V.V.; Sharpless, K.B. A new approach to osmium-catalyzed asymmetric dihydroxylation and aminohydroxylation of olefins. Angew. Chem. Int. Ed. 2002, 41, 472–475. [Google Scholar] [CrossRef]
- De Gonzalo, G.; Domínguez de María, P. Biocatalysis: An Industrial Perspective, 1st ed.; Royal Society of Chemistry: Cambridge, UK, 2017; ISBN 978-1-78262-619-0. [Google Scholar]
- Sheldon, R.A.; Woodley, J.M. Role of biocatalysis in sustainable chemistry. Chem. Rev. 2018, 118, 801–838. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.N. Green Biocatalysis, 1st ed.; John Wiley & Sons: Hoboken, NJ, USA, 2016; ISBN 978-1-118-82229-6. [Google Scholar]
- Hudlicky, T.; Reed, J.W. Application of biotransformations and biocatalysis to complexity generation in organic synthesis. Chem. Soc. Rev. 2009, 38, 3117–3132. [Google Scholar] [CrossRef] [PubMed]
- Boyd, D.R.; Sharma, N.D.; Bowers, N.I.; Brannigan, I.N.; Groocock, M.R.; Malone, J.E.; McConville, G.; Allen, C.C.R. Biocatalytic asymmetric dihydroxylation of conjugated mono- and poly-alkenes to yield enantiopure cyclic cis-diols. Adv. Synth. Catal. 2005, 347, 1081–1089. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, C.; Wu, X. Dicarbonyl reduction by single enzyme for the preparation of chiral diols. Chem. Soc. Rev. 2012, 41, 1742–1753. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.V.; Li, X.F.; Lou, W.Y.; Zhong, M.H. Cross-linked enzyme aggregates of Mung bean epoxide hydrolases: A highly active, stable and recyclable biocatalyst for asymmetric hydrolysis of epoxides. J. Biotechnol. 2013, 166, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Han, B.; Huang, Z.; Miller, M.; Huang, H.; Malashock, D.S.; Zhu, Z.; Milan, A.; Robertson, D.E.; Weiner, D.P.; et al. Epoxide hydrolase-catalyzed synthesis of chiral 1,2-diols via desymmetrization of meso-epoxides. J. Am. Chem. Soc. 2004, 126, 11156–11157. [Google Scholar] [CrossRef] [PubMed]
- Gotor-Fernández, V.; Gotor, V. Aminolysis and ammonolysis of carboxylic acid derivatives. In Asymmetric Organic Synthesis with Enzymes; Gotor, V., Alfonso, I., García-Urdiales, E., Eds.; Wiley-VCH: Weinheim, Germany, 2008; pp. 171–228. ISBN 978-3-527-31825-4. [Google Scholar]
- Ghanem, A. Trends in lipase-catalyzed asymmetric access to enantiomerically pure/enriched compounds. Tetrahedron 2007, 63, 1721–1754. [Google Scholar] [CrossRef]
- Bornscheuer, U.T.; Kazlauskas, R.J. Hydrolases in Organic Synthesis: Regio- and Stereoselective Biotransformations, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2005; ISBN 978-3-527-60712-9. [Google Scholar]
- Mathpati, A.C.; Vyas, V.K.; Bhanage, B.M. Kinetic resolution of 1,2-diols using immobilized Burkholderia cepacea: A combined experimental and molecular dynamics investigation. J. Biotechnol. 2017, 262, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Parve, J.; Reile, I.; Aid, T.; Kudrjasova, M.; Muurisepp, A.M.; Vallikivi, I.; Villo, L.; Aav, R.; Pehk, T.; Vares, L.; et al. Lipase-catalyzed stereoresolution of long-chain 1,2-alkanediols: A screening of preferable reaction conditions. J. Mol. Catal. B: Enzym. 2015, 116, 60–69. [Google Scholar] [CrossRef]
- Hamberg, A.; Magnusson, A.O.; Hu, F.J.; Hult, K. Selective monoacylation of diols by substrate assisted catalysis in T40A Candida antarctica Lipase B. ChemCatChem 2013, 5, 743–747. [Google Scholar] [CrossRef]
- Bencze, L.C.; Paizs, C.; Tosa, M.I.; Irimie, F.D. Sequential use of regio- and stereoselective lipases for the efficient kinetic resolution of racemic 1-(5-phenylfuran-2-yl)ethane-1,2-diols. Tetrahedron Asymmetry 2011, 22, 675–683. [Google Scholar] [CrossRef]
- Jew, S.; Roh, E.; Baek, E.; Mireille, L.; Kim, H.; Jeong, B.; Park, M.; Park, H. Asymmetric synthesis of (R)-(+)-etomoxir via enzymatic resolution. Tetrahedron Asymmetry 2000, 11, 3395–3401. [Google Scholar] [CrossRef]
- Carmona, J.A.; de Gonzalo, G.; Serrano, I.; Crespo-Peña, A.; Simek, M.; Monge, D.; Fernández, R.; Lassaletta, J.M. Asymmetric organocatalytic synthesis of tertiary azomethyl alcohols: Key intermediates towards azoxy compounds and α-hydroxy-β-amino esters. Org. Biomol. Chem. 2017, 15, 2993–3005. [Google Scholar] [CrossRef] [PubMed]
- Serrano, I.; Monge, D.; Álvarez, E.; Fernández, R.; Lassaletta, J.M. Asymmetric organocatalytic synthesis of quaternary α-hydroxy phosphonates: En route to α-aryl phosphaisoserines. Chem. Commun. 2015, 51, 4077–4080. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Peña, A.; Monge, D.; Martín-Zamora, E.; Álvarez, E.; Fernández, R.; Lassaletta, J.M. Asymmetric Formal Carbonyl-Ene Reactions of Formaldehyde tert-Butyl Hydrazone with α-Keto Esters: Dual Activation by Bis-urea Catalysts. J. Am. Chem. Soc. 2012, 134, 12912–12915. [Google Scholar] [CrossRef] [PubMed]
- Lehn, J.-S.M.; Javed, S.; Hoffman, D.M. Synthesis of zirconium, hafnium, and tantalum complexes with sterically demanding hydrazide ligands. Inorg. Chem. 2007, 46, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, P.E.; Wittkopp, A. H-Bonding additives act like Lewis acid catalysts. Org. Lett. 2002, 4, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Peña, A. Metodologías y Estrategias Para la Formilación Selectiva de Compuestos Carbonílicos. Ph.D. Thesis, University of Sevilla, Sevilla, Spain, September 2013. [Google Scholar]
Sample Availability: Samples of the compounds (±)-1–6d and (±)-1–6e are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | R1 | R2 | n | Yield (±)-1–6b a | Yield (±)-1–6d a |
|---|---|---|---|---|---|
| 1 | Ph | Et | 0 | (±)-1b, b 87 | (±)-1d, 47 |
| 2 | Ph | Me | 0 | (±)-2b, 80 | (±)-2d, 49 |
| 3 | 4-CN-Ph | Et | 0 | (±)-3b, 92 | (±)-3d, 55 |
| 4 | 4-OMe-Ph | Et | 0 | (±)-4b, 85 | (±)-4d, 45 |
| 5 | 2-Thiophenyl | Et | 0 | (±)-5b, 90 | (±)-5d, 41 |
| 6 | H | Et | 2 | (±)-6b, 85 | (±)-6d, 57 |

| Entry | Lipase | Solvent | T (°C) | t (h) | c (%) a | ee 1d (%) b | ee 1e (%) c | E d |
|---|---|---|---|---|---|---|---|---|
| 1 | CalB | Toluene | 30 | 4 | 23 | 11 | 37 | 2 |
| 2 | CalA | Toluene | 30 | 12 | 32 | 37 | 77 | 12 |
| 3 | PSL-C | Toluene | 30 | 20 | 32 | 40 | 86 | 20 |
| 4 | PSF | Toluene | 30 | 20 | 41 | 35 | 50 | 4 |
| 5 | BSL | Toluene | 30 | 20 | 38 | 42 | 69 | 8 |
| 6 | R. oryzae | Toluene | 30 | 20 | 23 | 22 | 75 | 9 |
| 7 | A. oryzae | Toluene | 30 | 20 | 16 | 11 | 57 | 4 |
| 8 | PPL | Toluene | 30 | 24 | 48 | 67 | 73 | 13 |
| 9 | M. miehei | Toluene | 20 | 24 | 13 | 12 | 78 | 9 |
| 10 | PSL-C | TBME | 30 | 12 | 42 | 62 | 91 | 41 |
| 11 | CalA | TBME | 30 | 6 | 41 | 54 | 79 | 17 |
| 12 | PPL | TBME | 30 | 12 | 38 | 50 | 77 | 13 |
| 13 | PSL-C | 1,4-Dioxane | 30 | 20 | 23 | 25 | 86 | 17 |
| 14 | PSL-C | THF | 30 | 20 | 6 | 6 | 87 | 15 |
| 15 | PSL-C | DIPE | 30 | 12 | 45 | 67 | 83 | 22 |
| 16 | PSL-C | TBME | 15 | 24 | 43 | 67 | 91 | 43 |
| 17 | PSL-C e | TBME | 30 | 24 | 36 | 52 | 91 | 37 |
| 18 | PSL-C f | TBME | 30 | 48 | 34 | 47 | 91 | 34 |

| Entry | Substrate | Lipase | R1 | R2 | n | t (h) | c (%) a | ee 2-6d (%) b | ee 2-6e (%) c | E d |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | (±)-2d | PSL-C | Ph | Me | 0 | 12 | 41 | 63 | 91 | 42 |
| 2 | (±)-2d | CalA | Ph | Me | 0 | 8 | 42 | 59 | 80 | 16 |
| 3 | (±)-3d | PSL-C | 4-CN-Ph | Et | 0 | 12 | 44 | 71 | 90 | 40 |
| 4 | (±)-3d | CalA | 4-CN-Ph | Et | 0 | 8 | 47 | 77 | 86 | 30 |
| 5 | (±)-4d | PSL-C | 4-OMe-Ph | Et | 0 | 24 | 41 | 62 | 89 | 32 |
| 6 | (±)-4d | CalA | 4-OMe-Ph | Et | 0 | 16 | 36 | 42 | 72 | 11 |
| 7 | (±)-5d | PSL-C | 2-Thiophenyl | Et | 0 | 24 | 45 | 73 | 88 | 33 |
| 8 | (±)-5d | CalA | 2-Thiophenyl | Et | 0 | 16 | 38 | 47 | 83 | 17 |
| 9 | (±)-6d | PSL-C | Ph | Et | 2 | 2 | 45 | 50 | 62 | 7 |
| 10 | (±)-6d | CalA | Ph | Et | 2 | 6 | 17 | 17 | 82 | 12 |
| 11 e | (±)-6d | PSL-C | Ph | Et | 2 | 6 | 15 | 14 | 80 | 10 |
| 12 f | (±)-6d | PSL-C | Ph | Et | 2 | 4 | 17 | 18 | 86 | 16 |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Gonzalo, G. Lipase Catalysed Kinetic Resolution of Racemic 1,2-Diols Containing a Chiral Quaternary Center. Molecules 2018, 23, 1585. https://doi.org/10.3390/molecules23071585
De Gonzalo G. Lipase Catalysed Kinetic Resolution of Racemic 1,2-Diols Containing a Chiral Quaternary Center. Molecules. 2018; 23(7):1585. https://doi.org/10.3390/molecules23071585
Chicago/Turabian StyleDe Gonzalo, Gonzalo. 2018. "Lipase Catalysed Kinetic Resolution of Racemic 1,2-Diols Containing a Chiral Quaternary Center" Molecules 23, no. 7: 1585. https://doi.org/10.3390/molecules23071585
APA StyleDe Gonzalo, G. (2018). Lipase Catalysed Kinetic Resolution of Racemic 1,2-Diols Containing a Chiral Quaternary Center. Molecules, 23(7), 1585. https://doi.org/10.3390/molecules23071585