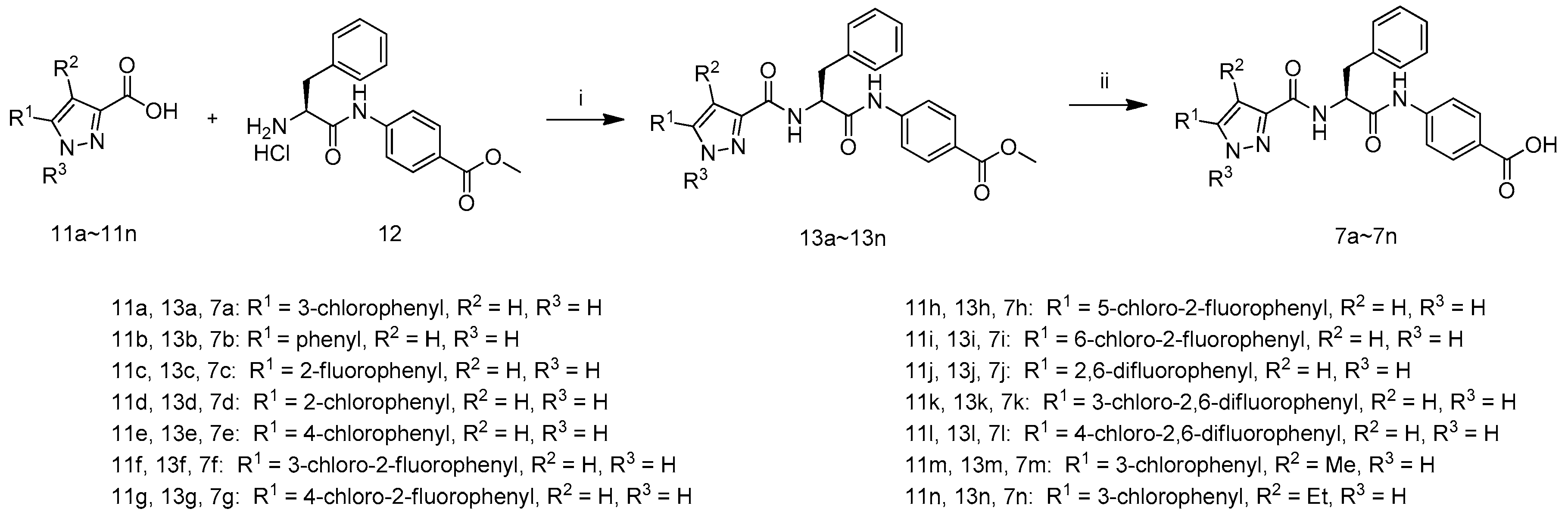
3.2. Chemistry
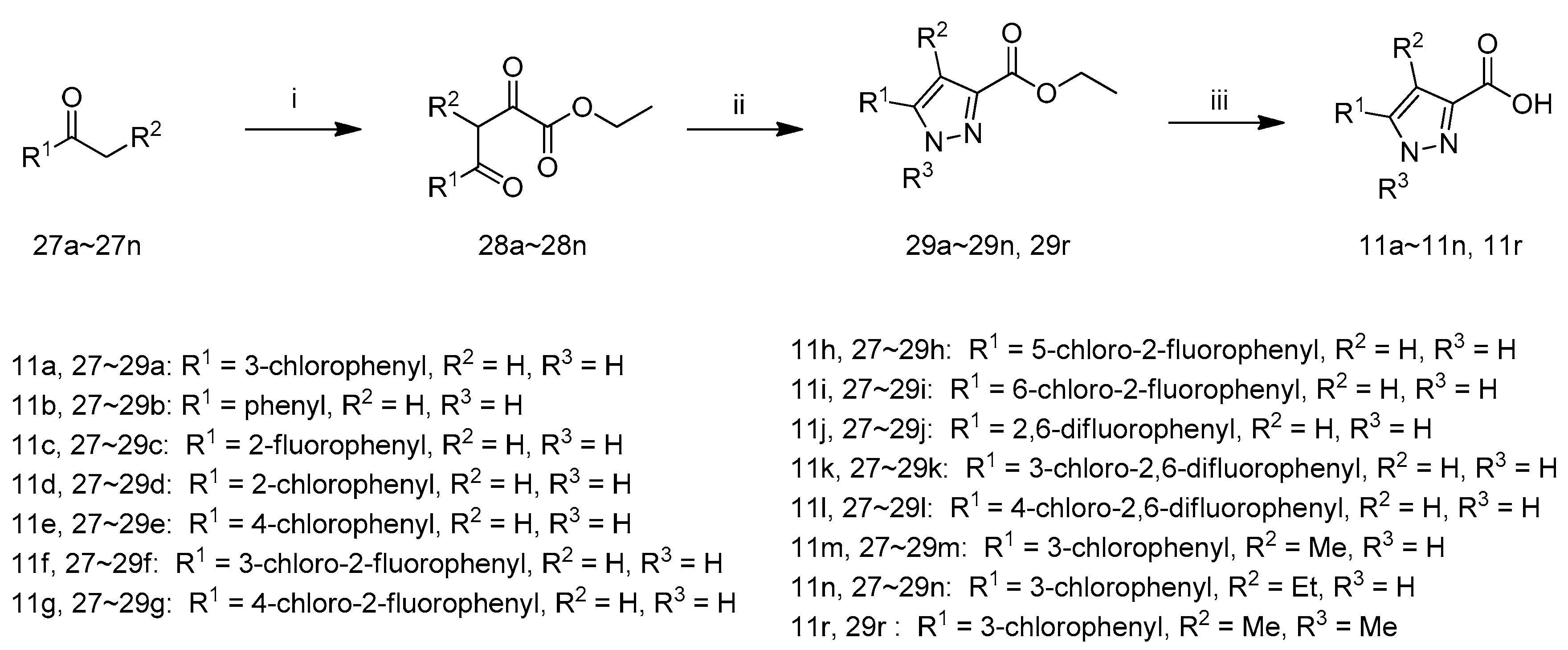
Ethyl 2,4-dioxo-4-phenylbutanoate (28b): To a stirred solution of acetophenone (27b) (2.0 g, 16.5 mmol) in MTBE (30 mL) was added lithium hexamethyldisilazide (1.3 M, 12.7 mL, 16.5 mmol) dropwise at 0 °C; After addition, the reaction mixture was stirred at 0 °C for 0.5 h and diethyl oxalate (3.0 g, 20.8 mmol) was added dropwise. Then, the mixture was stirred at room temperature overnight. TLC analysis showed reaction was complete and the reaction mixture was extracted with H2O (20 mL). The aqueous layer was separated, acidified by hydrochloric acid (1 M) to pH 6 and extracted by ethyl acetate (10 mL × 2). The combined organic layer was concentrated in vacuum to give 28b as yellow oil, which was used for next step without further purification (3.4 g, 92.7% yield).
Ethyl3-phenyl-1H-pyrazole-5-carboxylate (29b): To a solution of 28b (3.4 g, 15.4 mmol) in EtOH (15 mL) was added hydrazine hydrate (1.2 g, 24.0 mmol) and the mixture was stirred at 50 °C for 2 h when TLC analysis indicated completion of reaction. Then the reaction mixture was evaporated to get crude 29b as brown oil, which was used for next step without further purification (2.6 g, 77.9% yield).
5-Phenyl-1H-pyrazole-3-carboxylic acid (
11b): To a solution of compound
29b (2.6 g, 9.1 mmol) in MeOH (30 mL) and H
2O (15 mL) was added LiOH·H
2O (0.5 g, 20.0 mmol) and the mixture was stirred at 70 °C for 8 h. The reaction mixture was evaporated and H
2O (15 mL) was added, then acidified by hydrochloric acid (1 M) to pH 3. The suspension was filtered and washed by H
2O (10 mL), dried at 50 °C for 4 h to afford
11b as a white solid (2.1 g, 89.5% yield), m.p.: 227–229 °C, decomposition.
1H-NMR: δ 13.39 (s, 1H), 7.83 (m, 2H), 7.41–7.45 (m, 2H), 7.31–7.35 (m, 1H), 7.18(s, 1H). HRMS (ESI) calcd. For C
10H
8ClN
2O
2+: [M + H]
+ m/
z: 189.0659, found: 189.0659. The
1H-NMR data were in good agreement with those reported [
26].
Compounds 11a, 11r and 11c–11n were synthesized according to the procedure described for the preparation of 11b.
5-(3-Chlorophenyl)-1H-pyrazole-3-carboxylic acid (
11a): white solid product (1.5 g, 47.9% yield), m.p.: 219–221 °C, decomposition.
1H-NMR: δ 13.85–13.15 (m, 2H), 7.90 (m, 1H), 7.82–7.80 (d,
J = 7.6 Hz, 1H), 7.47–7.37 (m, 2H), 7.29 (s, 1H). HRMS (ESI) calcd. For C
10H
9N
2O
2+: [M + H]
+ m/
z: 223.0269, found: 223.0268. The
1H-NMR data were in good agreement with those reported [
26].
5-(3-Chlorophenyl)-1H-pyrazole-3-carboxylic acid (
11a): white solid product (1.5 g, 47.9% yield), m.p.: 219–221 °C, decomposition.
1H-NMR: δ 13.85–13.15 (m, 2H), 7.90 (m, 1H), 7.82–7.80 (d,
J = 7.6 Hz, 1H), 7.47–7.37 (m, 2H), 7.29 (s, 1H). HRMS (ESI) calcd. For C
10H
9N
2O
2+: [M + H]
+ m/
z: 223.0269, found: 223.0268. The
1H-NMR data were in good agreement with those reported [
26].
3-(2-Fluorophenyl)-1H-pyrazole-5-carboxylic acid (
11c): white solid product (2.3 g, 77.0% yield), m.p.: 232–234 °C, decomposition.
1H-NMR: δ 13.84–13.52 (m, 2H), 7.94–7.91 (m, 1H), 7.43–7.38 (m, 1H), 7.34–7.27 (m, 2H), 7.05–7.04 (d,
J = 3.6 Hz, 1H). HRMS (ESI) calcd. For C
10H
8FN
2O
2+: [M + H]
+ m/
z: 207.0564, found: 207.0563 [
27].
3-(2-Chlorophenyl)-1H-pyrazole-5-carboxylic acid (
11d): white solid product (1.8g, 62.5% yield), m.p.: 229–231 °C, decomposition.
1H-NMR: δ 13.64–13.49 (m, 2H), 7.74 (s, 1H), 7.57–7.55 (m, 1H), 7.44–7.41 (m, 2H), 7.12 (s, 1H). HRMS (ESI) calcd. For C
10H
8ClN
2O
2+: [M + H]
+ m/
z: 223.0269, found: 223.0268 [
27].
3-(4-Chlorophenyl)-1H-pyrazole-5-carboxylic acid (
11e): white solid product (1.5 g, 52.1% yield), m.p.: 240–242 °C, decomposition.
1H-NMR: δ 13.86–13.64 (m, 2H), 7.86–7.84 (d,
J = 8.4 Hz, 2H), 7.49–7.47 (d,
J = 8.4 Hz, 2H), 7.04 (s, 1H). HRMS (ESI) calcd. For C
10H
8ClN
2O
2+: [M + H]
+ m/
z: 223.0269, found: 223.0269. The
1H-NMR data were in good agreement with those reported [
26].
3-(3-Chloro-2-fluorophenyl)-1H-pyrazole-5-carboxylic acid (11f): white solid product (2.0 g, 71.7% yield), m.p.: 236–238 °C, decomposition. 1H-NMR: δ 13.77 (m, 1H), 7.92–7.88 (m, 1H), 7.56–7.52 (m, 1H), 7.30–7.26 (m, 1H), 6.98–6.97 (d, J = 4.0 Hz, 1H). HRMS (ESI) calcd. For C10H7ClFN2O2+: [M + H]+ m/z: 241.0175, found: 241.0173.
3-(4-Chloro-2-fluorophenyl)-1H-pyrazole-5-carboxylic acid (11g): white solid product (1.9 g, 68.1% yield), m.p.: 234–236 °C, decomposition. 1H-NMR: δ 13.07 (s, 1H), 7.98–7.93 (t, J = 8.4 Hz, 2H), 7.47–7.44 (qd, J = 2 Hz, 1H), 7.31–7.28 (qd, J = 2 Hz, 1H), 6.66–6.65 (d, J = 4.4 Hz, 1H). HRMS (ESI) calcd. For C10H7ClFN2O2+: [M + H]+ m/z: 241.0175, found: 241.0172.
3-(5-Chloro-2-fluorophenyl)-1H-pyrazole-5-carboxylic acid (11h): white solid product (1.5 g, 53.8% yield), m.p.: 229–231 °C, decomposition. 1H-NMR: δ 14.11–13.42 (m, 2H), 7.97–7.95 (m, 1H), 7.47–7.36 (m, 2H), 7.10–7.05 (m, 1H). HRMS (ESI) calcd. For C10H7ClFN2O2+: [M + H]+ m/z: 241.0175, found: 241.0172.
3-(2-Chloro-6-fluorophenyl)-1H-pyrazole-5-carboxylic acid (
11i): white solid product (1.6 g, 57.4% yield), m.p.: 182–184 °C, decomposition.
1H-NMR: δ 14.13–14.06 (m, 1H), 13.72–13.48 (m, 1H), 7.49–7.46 (m, 2H), 7.35 (m, 1H), 6.91 (s, 1H). HRMS (ESI) calcd. For C
10H
7ClFN
2O
2+: [M + H]
+ m/
z: 241.0175, found: 241.0170 [
28].
3-(2,6-Difluorophenyl)-1H-pyrazole-5-carboxylic acid (11j): white solid product (1.6 g, 55.7% yield), m.p.: 232–334 °C, decomposition. 1H-NMR: δ 14.08–13.05 (m, 2H), 7.53–7.46 (m, 1H), 7.25–7.21 (m, 2H), 6.98 (s, 1H). HRMS (ESI) calcd. For C10H7F2N2O2+: [M + H]+ m/z: 225.0470, found: 241.0465.
3-(3-Chloro-2,6-difluorophenyl)-1H-pyrazole-5-carboxylic acid (11k): white solid product (1.2 g, 44.2% yield), m.p.: 233–235 °C, decomposition. 1H-NMR: δ 14.17 (s, 1H), 7.72–7.67 (m, 1H), 7.33–7.29 (m, 1H), 7.02 (s, 1H). HRMS (ESI) calcd. For C10H6ClF2N2O2+: [M + H]+ m/z: 259.0080, found: 259.0074.
3-(4-Chloro-2,6-difluorophenyl)-1H-pyrazole-5-carboxylic acid (11l): white solid product (1.6 g, 58.9% yield), m.p.: 238–240 °C, decomposition. 1H-NMR: δ 13.64 (s, 1H), 7.51–7.49 (d, J = 8.0 Hz, 2H), 6.99 (s, 1H). HRMS (ESI) calcd. For C10H6ClF2N2O2+: [M + H]+ m/z: 259.0080, found: 259.0078.
5-(3-Chlorophenyl)-4-methyl-1H-pyrazole-3-carboxylic acid (11m): white solid product (1.1 g, 39.2% yield), m.p.: 226–228 °C, decomposition. 1H-NMR: δ 13.64–13.57 (m, 2H), 7.66 (s, 1H), 7.63–7.57 (m, 1H), 7.51–7.42 (m, 2H), 2.39(s, 3H). HRMS (ESI) calcd. For C11H10ClN2O2+: [M + H]+ m/z: 237.0425, found: 237.0424.
5-(3-Chlorophenyl)-4-ethyl-1H-pyrazole-3-carboxylic acid (11n): white solid product (0.2 g, 7.3% yield), m.p.: 236–238 °C, decomposition. 1H-NMR: δ 13.43 (s, 1H), 7.58 (s, 1H), 7.53–7.43 (m, 3H), 2.82–2.76 (qd, J = 7.2 Hz, 2H), 1.12–1.08 (m, 3H). HRMS (ESI) calcd. For C12H12ClN2O2+: [M + H]+ m/z: 251.0582, found: 251.0577.
5-(3-Chlorophenyl)-1,4-dimethyl-1H-pyrazole-3-carboxylic acid (11r): white solid product (1.1 g, 37.0% yield), m.p.: 147–149 °C, decomposition. 1H-NMR: δ 12.52 (s, 1H), 7.56–7.54 (m, 3H), 7.41–7.39 (m, 1H), 3.74 (s, 3H), 2.10 (s, 3H). The structure was confirmed by NOESY.HRMS (ESI) calcd. For C12H12ClN2O2+: [M + H]+ m/z: 251.0582, found: 251.0575.
(S)-Methyl-4-(2-((tert-butoxycarbonyl)amino)-3-phenylpropanamido)benzoate (32): To a stirred mixture of Boc-Phe-OH (30) (10.0 g, 37.7mmol), methyl 4-aminobenzoate (31) (5.7 g, 37.7 mmol) and pyridine (10 mL) in CH2Cl2 (100 mL) at −10 °C was added POCl3 (5.8 g, 37.7 mmol) dropwise.After addition, the reaction mixture was stirred at 0 °C for 2 h when TLC analysis indicated completion of reaction, then H2O (20 mL) was added and the organic layer was separated, washed by hydrochloric acid (1 M, 20 mL), dried by Na2SO4, filtered. The filtrate was evaporated in vacuum to get crude 32 as a yellow solid, which was used for next step without further purification (11.6 g, 77.3% yield).
(S)-Methyl-4-(2-amino-3-phenylpropanamido)benzoate hydrochloride (12): To a solution of compound 32 (11.5 g, 28.9 mmol) in ethyl acetate (50 mL) was added hydrochloric/ethyl acetate (50 mL, saturated solution), and the mixture was stirred at room temperature overnight. Then TLC analysis showed reaction was complete. The suspension was filtered and the filter cake was washed by ethyl acetate (20 mL), dried at 50 °C for 4 h to afford 12 as a pink solid (8.2 g, 84.3% yield), m.p.: 127–129 °C. 1H-NMR: δ 11.27 (s, 1H), 8.45 (s, 3H), 7.93–7.91 (d, J = 8.4 Hz, 2H), 7.73–7.71 (d, J = 8.8 Hz, 2H), 7.32–7.23 (m, 5H), 4.34 (m, 1H), 3.81 (s, 3H), 3.24–3.12 (m, 2H). ESI-MS (m/z) = 299.09 [M +H]+.
(S)-Ethyl 5-(2-((tert-butoxycarbonyl)amino)-3-phenylpropanamido)-1H-indole-2-carboxylate (35a): To a mixture of Boc-Phe-OH (33a) (5 g, 18.9 mmol), ethyl 5-amino-1H-indole-2-carboxylate (34) (3.9 g, 18.9 mmol) and pyridine (5 mL) in CH2Cl2 (50 mL) at −10 °C was added POCl3 (2.9 g, 18.9 mmol) dropwise. After addition, the reaction mixture was stirred at −10 °C for 2 h when TLC analysis indicated completion of reaction, then H2O (10 mL) was added and the organic layer was separated, washed by hydrochloric acid (1 M, 10 mL), dried by Na2SO4, filtered. The filtrate was evaporated in vacuum to get crude 35a as a brown solid, which was used for next step without further purification (6.6 g, 77.6% yield).
(S)-Ethyl 5-(2-amino-3-phenylpropanamido)-1H-indole-2-carboxylate hydrochloride (
14a): To a solution of compound
35a (6.5 g, 14.4 mmol) in ethyl acetate (15 mL) was added hydrochloric/ethyl acetate (30 mL, saturated solution), and the mixture was stirred at room temperature overnight. Then TLC analysis showed reaction was complete. The suspension was filtered and the filter cake was washed by ethyl acetate (20 mL) and dried at 50 °C for 4 h to afford
14a as a grey solid (4.9 g, 87.7% yield), m.p.: 135–137 °C, decomposition.
1H-NMR: δ 11.86 (s, 1H), 10.46 (s, 1H), 8.36–8.32 (m, 4H), 7.91 (s, 1H), 7.33–7.12 (m, 8H), 4.35–4.29 (m, 2H), 4.19 (m, 1H), 3.13–3.08 (m, 2H), 1.34–1.31 (m, 3H). ESI-MS (
m/
z) = 352.10 [M + 1]
+ [
18].
Compounds 14b~14f were synthesized according to the procedure described for the preparation of 14a.
(S)-Ethyl5-(2-amino-3-(4-fluorophenyl)propanamido)-1H-indole-2-carboxylatehydrochloride (
14b): grey solid product (4.0 g, 52.3% yield), m.p.: 243–245 °C, decomposition.
1H-NMR: δ 11.85 (s, 1H), 10.84 (s, 1H), 8.45 (s, 2H), 7.96 (s, 1H), 7.41–7.36 (m, 4H), 7.15–7.11 (m, 3H), 4.36–4.29 (m, 2H), 3.15–3.09 (m, 2H), 1.34–1.30 (m, 3H). ESI-MS (
m/
z) = 370.06 [M + 1]
+ [
29].
(S)-Ethyl 5-(2-amino-3-(3-fluorophenyl)propanamido)-1H-indole-2-carboxylatehydrochloride (14c): grey solid product (3.7 g, 48.6% yield), m.p.: 154–156 °C, decomposition. 1H-NMR: δ 11.86 (s, 1H), 10.77 (s, 1H), 8.43 (s, 3H), 7.95 (s, 1H), 7.41–7.32 (m, 3H), 7.22–7.05 (m, 4H), 4.35–4.02 (m, 3H), 3.37–3.11 (m, 2H), 1.35–1.31 (t, J = 6.8 Hz, 3H). ESI-MS (m/z) = 370.02 [M + 1]+, HRMS (ESI) calcd. For C20H21FN3O3+: [M + H]+ m/z: 370.1561, found: 370.1553.
(S)-Ethyl 5-(2-amino-3-(pyridin-4-yl)propanamido)-1H-indole-2-carboxylatehydrochloride (14d): grey solid product (0.5 g, 6.8%), m.p.: 171–173 °C, decomposition. 1H-NMR: δ 11.87 (s, 1H), 11.32 (s, 1H), 8.88–8.86 (d, J = 6.4 Hz, 2H), 8.59 (s, 3H), 8.13–8.11 (d, J = 5.6 Hz, 2H), 8.03 (s, 1H), 7.47–7.40 (m, 2H), 7.12 (s, 1H), 4.57 (s, 1H), 4.35–4.29 (qd, J = 6.8 Hz and 7.2 Hz, 2H), 3.65–3.62 (m, 1H), 3.41–3.35 (m, 1H), 1.34–1.30 (t, J = 7.2 Hz, 3H). ESI-MS (m/z) = 353.00 [M + 1]+, HRMS (ESI) calcd. For C19H21N4O3+: [M + H]+ m/z: 353.1608, found: 353.1602.
(S)-Ethyl 5-(2-amino-3-(pyridin-3-yl)propanamido)-1H-indole-2-carboxylatehydrochloride (14e): grey solid product (1.5 g, 19.2%). 1H-NMR: δ 11.90 (s, 1H), 11.07 (s, 1H), 8.89 (s, 1H), 8.79–8.77 (d, J = 5.6 Hz, 1H), 8.48–8.43 (m, 4H), 8.00 (s, 1H), 7.91–7.88 (t, J = 6.4 Hz, 1H), 7.40 (s, 1H), 7.13 (s, 1H), 4.47 (s, 1H), 4.35–4.29 (m, 2H), 3.52–3.47 (m, 1H), 3.32–3.26 (m, 1H), 1.34–1.30 (m, 3H). ESI-MS (m/z) = 353.15 [M + 1]+.
(S)-Ethyl 5-(2-amino-3-(pyridin-2-yl)propanamido)-1H-indole-2-carboxylatehydrochloride (14f): grey solid product (1.8 g, 24.6%), m.p.: 75–77 °C, decomposition. 1H-NMR: δ 11.86 (s, 1H), 10.89 (s, 1H), 8.76–8.75 (d, J = 5.2 Hz, 1H), 8.65 (s, 3H), 8.21–8.19 (m, 1H), 7.97 (s, 1H), 7.81–7.79 (d, J = 7.6 Hz, 1H), 7.69–7.67 (m, 1H), 7.42–7.38 (m, 2H), 7.11 (s, 1H), 4.60 (s, 1H), 4.35–4.29 (m, 2H), 3.63–3.54 (m, 2H), 1.34–1.30 (t, J = 7.2 Hz, 3H). ESI-MS (m/z) = 353.05 [M + 1]+, HRMS (ESI) calcd. For C19H21N4O3+: [M + H]+ m/z: 353.1608, found: 353.1604.
(S)-Methyl 4-(2-(5-(3-chlorophenyl)-1H-pyrazole-3-carboxamido)-3-phenylpropanamido)benzoate (13a): To a mixture of 3-(3-chlorophenyl)-1H-pyrazole-5-carboxylic acid (14a, 133 mg, 0.60 mmol) inDMF(4 mL) was added (S)-methyl-4-(2-amino-3-phenylpropanamido)benzoate hydrochloride (12, 200 mg, 0.60 mmol), N,N-diisopropyl-ethylamine (232 mg, 1.80 mmol), 1-hydroxybenzotriazole(161 mg, 1.20 mmol) and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (229 mg, 1.20 mmol) and the reaction mixture was stirred at room temperature overnight. Then TLC analysis indicated reaction was complete, and H2O (40 mL) was added. The mixture was stirred for 10 min and filtered to get crude product 13a as a yellow solid, which was used for next step without further purification.
(S)-4-(2-(5-(3-Chlorophenyl)-1H-pyrazole-3-carboxamido)-3-phenylpropanamido)benzoic acid (7a): To a mixture of compound 13a (289 mg, 0.60 mmol) in MeOH (3 mL) and H2O (1.5 mL) was added LiOH·H2O (100 mg, 2.40 mmol) and the reaction mixture was stirred at 40 °C for 6 h when TLC analysis indicated completion of reaction. The mixture was evaporated in vacuum and H2O (2 mL) was added, extracted by MTBE (2 mL) and acidified by hydrochloric acid (1 M) to pH 3–4. After stirred for 0.5 h, the suspension was filtered, and the filter cake washed by H2O (5 mL) and dried at 50 °C for 2 h to afford 7a as a white solid (185 mg, 63.4% yield for 2 steps); m.p.: 147–149 °C, decomposition.1H-NMR: δ 13.68 (s, 1H), 12.65 (s, 1H), 10.57 (s, 1H), 7.93–7.88 (m, 3H), 7.73–7.71 (m, 2H), 7.60–7.15 (m, 8H),4.93–4.88 (m, 1H), 3.31–3.15 (m, 2H).13C-NMR: δ 170.64, 167.03, 142.99, 137.61, 133.84, 130.88, 130.43, 129.34, 129.20, 128.20, 127.94, 126.51, 125.55, 124.88, 123.91, 118.78, 103.51, 55.29, 37.46. ESI-MS (m/z) = 488.90 [M + H]+, HRMS (ESI) calcd. For C26H22ClN4O4+: [M + H]+ m/z: 489.1324, found: 489.1317.
Compounds 7b~7n were synthesized according to the procedure described for the preparation of 7a.
(S)-4-(3-Phenyl-2-(5-phenyl-1H-pyrazole-3-carboxamido)propanamido)benzoic acid (7b): white solid product (183 mg, 67.4% yield), m.p.: 137–139 °C, decomposition.1H-NMR: δ 13.68–13.64 (m, 1H), 12.76–12.70 (m, 1H), 10.52 (s, 1H), 8.11–8.09 (m, 1H), 7.91–7.88 (d, J = 8.4 Hz, 2H), 7.77–7.70 (m, 4H), 7.46–7.43 (m, 2H), 7.34–7.19 (m, 5H), 7.17–7.09 (m, 1H), 7.06 (s, 1H), 4.93–4.87 (m, 1H), 3.16–3.15 (m, 2H). 13C-NMR: δ 171.25, 167.67, 161.28, 143.49, 138.15, 131.10, 129.92, 129.82, 129.62, 128.84, 127.16, 126.24, 125.92, 119.42, 103.42, 55.67, 38.12. ESI-MS (m/z) = 454.99 [M + H]+, HRMS (ESI) calcd. For C26H23N4O4+: [M + H]+ m/z: 455.1714, found: 455.1703.
(S)-4-(2-(5-(2-Fluorophenyl)-1H-pyrazole-3-carboxamido)-3-phenylpropanamido)benzoic acid (7c): white solid product (220 mg, 77.9% yield), m.p.: 86–88 °C, decomposition. 1H-NMR: δ 13.76–13.69 (m, 1H), 12.79–12.68 (m, 1H), 10.57 (s, 1H), 9.02–8.97 (m, 1H), 7.90–7.88 (d, J = 8.8 Hz, 2H), 7.73–7.71 (d, J = 8.4 Hz, 2H), 7.39–7.17 (m, 8H), 7.15–7.07 (m, 1H), 7.00 (s, 1H), 4.93–4.87 (m, 1H), 3.23–3.15 (m, 2H). ESI-MS (m/z) = 472.99 [M + H]+. 13C-NMR: δ 171.38, 167.65, 160.65, 143.73, 138.34, 130.98, 130.70, 130.01, 129.79, 128.75, 127.08, 126.09, 125.49, 119.39, 116.81, 106.49, 56.03, 37.90. HRMS (ESI) calcd. For C26H22FN4O4+: [M + H]+ m/z: 473.1620, found: 473.1600.
(S)-4-(2-(5-(2-Chlorophenyl)-1H-pyrazole-3-carboxamido)-3-phenylpropanamido)benzoic acid (7d): white solid product (221 mg, 75.7% yield), m.p.: 92–94 °C, decomposition. 1H-NMR: δ 13.31 (s, 1H), 10.95 (s, 1H), 8.68 (s, 1H), 7.94–7.92 (d, J = 8.8 Hz, 2H), 7.84–7.82 (d, J = 8.4 Hz, 2H), 7.74–7.72 (d, J = 6.8 Hz, 1H), 7.55–7.53 (d, J = 7.2 Hz, 1H), 7.42–7.35 (m, 4H), 7.31–7.22 (m, 3H), 7.17–7.14 (m, 1H), 5.01–4.95 (m, 1H), 3.28–3.11 (m, 2H). 13C-NMR: δ 172.94, 170.79, 167.03, 143.12, 137.74, 131.15, 130.57, 129.94, 129.40, 129.19, 128.30, 127.53, 126.54, 125.47, 118.82, 106.57, 55.39, 36.41. ESI-MS (m/z) = 489.01 [M + H]+, HRMS (ESI) calcd. For C26H22ClN4O4+: [M + H]+ m/z: 489.1324, found: 489.1309.
(S)-4-(2-(5-(4-Chlorophenyl)-1H-pyrazole-3-carboxamido)-3-phenylpropanamido)benzoic acid (7e): white solid product (177 mg, 60.3% yield), m.p.: 174–176 °C, decomposition. 1H-NMR: δ 13.74–13.63 (m, 1H), 12.66 (s, 1H), 10.49 (s, 1H), 10.37 (s, 1H), 7.91–7.86 (t, J = 8.8 Hz, 2H), 7.80–7.78 (d, J = 8.0 Hz, 1H), 7.72–7.67 (m, 2H), 7.51 (s, 2H), 7.27–7.15 (m, 6H), 7.08 (s, 1H), 4.90–4.89 (m, 1H), 3.17–3.15 (m, 2H). 13C-NMR: δ 170.59, 168.12, 160.16, 142.37, 137.66, 132.74, 130.36, 129.35, 129.02, 128.23, 127.97, 127.58, 127.04, 126.54, 118.72, 103.22, 55.37, 37.50. ESI-MS (m/z) = 488.89 [M + H]+, HRMS (ESI) calcd. For C26H22ClN4O4+: [M + H]+ m/z: 489.1324, found: 489.1311.
(S)-4-(2-(5-(3-Chloro-2-fluorophenyl)-1H-pyrazole-3-carboxamido)-3-phenylpropanamido)benzoic acid (7f): white solid product (258 mg, 84.8% yield), m.p.: 185–187 °C, decomposition. 1H-NMR: δ 13.86 (s, 1H,), 12.69 (s, 1H), 10.54 (s, 1H), 10.38 (s, 1H), 8.31–8.29 (d, J = 8.0 Hz, 1H), 7.91–7.86 (m, 2H), 7.73–7.67 (m, 2H), 7.57 (s, 1H), 7.35–7.19 (m, 5H), 7.17–7.15 (m, 1H), 4.91–4.89 (m, 1H), 3.16–2.99 (m, 1H), 2.86–2.81 (m, 1H). 13C-NMR: δ 170.70, 168.35, 159.87, 155.44, 142.28, 137.77, 130.47, 129.42, 128.36, 128.00, 127.03, 126.68, 125.87, 121.01, 118.85, 106.21, 55.55, 37.46. ESI-MS (m/z) = 506.93 [M + H]+, HRMS (ESI) calcd. For C26H21ClFN4O4+: [M + H]+ m/z: 507.1230, found: 507.1220.
(S)-4-(2-(5-(4-Chloro-2-fluorophenyl)-1H-pyrazole-3-carboxamido)-3-phenylpropanamido)benzoic acid (7g): white solid product (220 mg, 72.6% yield), m.p.: 143–145 °C, decomposition. 1H-NMR: δ 13.82 (s, 1H), 12.70 (s, 1H), 10.55 (s, 1H), 9.00 (s, 1H), 7.94 (s, 1H), 7.90–7.88 (d, J = 8.4 Hz, 2H), 7.73–7.71 (d, J = 8.4 Hz, 2H), 7.58–7.56 (m, 1H), 7.35–7.27 (m, 3H), 7.26–7.24 (m, 2H), 7.18–7.15 (m, 1H), 4.93–4.87 (m, 1H), 3.18–3.11 (m, 2H). 13C-NMR: δ 171.27, 167.62, 160.54, 158.03, 143.55, 138.26, 133.94, 131.04, 129.91, 128.78, 127.11, 126.15, 125.80, 119.39, 117.65, 106.48, 55.92, 37.90. HRMS (ESI) calcd. For C26H21ClFN4O4+: [M + H]+ m/z: 507.1230, found: 507.1215.
(S)-4-(2-(5-(5-Chloro-2-fluorophenyl)-1H-pyrazole-3-carboxamido)-3-phenylpropanamido)benzoic acid (7h): grey solid product (176 mg, 57.9% yield), m.p.: 179–181 °C, decomposition. 1H-NMR: δ 13.89–13.83 (m, 1H), 12.78 (s, 1H), 10.55 (s, 1H), 8.78–8.77 (m, 1H), 7.96–7.94 (m, 1H), 7.90–7.88 (d, J = 8.8 Hz, 2H), 7.71–7.69 (d, J = 8.4 Hz, 2H), 7.47–7.18 (m, 6H), 7.16–7.15 (m, 1H), 4.92–4.87 (m, 1H), 3.19–3.07 (m, 2H). 13C-NMR): δ 171.21, 168.05, 159.40, 143.20, 138.31, 130.98, 129.88, 129.5, 128.80, 127.73, 127.11, 119.32, 119.13, 118.89, 106.79, 55.95, 37.90. ESI-MS (m/z) = 506.87 [M + H]+, HRMS (ESI) calcd. For C26H21ClFN4O4+: [M + H]+ m/z: 507.1230, found: 507.1221.
(S)-4-(2-(5-(2-Chloro-6-fluorophenyl)-1H-pyrazole-3-carboxamido)-3-phenylpropanamido)benzoic acid (7i): white solid product (189 mg, 62.1% yield), m.p.: 203–205 °C, decomposition. 1H-NMR: δ 13.74–13.70 (m, 1H), 10.64 (s, 1H), 7.89–7.87 (d, J = 8.8 Hz, 2H), 7.71–7.69 (d, J = 8.4 Hz, 2H), 7.54–7.42 (m, 2H), 7.38–7.36 (m, 3H), 7.28–7.24 (m, 2H), 7.19–7.15 (m, 1H), 7.08–6.98 (m, 1H), 4.92–4.86 (m, 1H), 3.21–3.09 (m, 2H). 13C-NMR: δ 171.31, 168.49, 162.00, 143.11, 138.33, 134.48, 130.98, 129.94, 128.82, 127.69, 127.14, 126.53, 119.26, 115.63, 115.40, 108.35, 56.03, 37.88. ESI-MS (m/z) = 506.88 [M + H]+, HRMS (ESI) calcd. For C26H21ClFN4O4+: [M + H]+ m/z: 507.1230, found: 507.1213.
(S)-4-(2-(5-(2,6-Difluorophenyl)-1H-pyrazole-3-carboxamido)-3-phenylpropanamido)benzoic acid (7j): white solid product (153 mg, 50.6% yield), m.p.: 205–207 °C, decomposition. 1H-NMR: δ 13.66 (s, 1H), 12.79–12.70 (m, 1H), 10.54 (s, 1H), 7.91–7.89 (d, J = 8.4 Hz, 2H), 7.73–7.70 (d, J = 8.4 Hz, 2H), 7.50 (s, 1H), 7.35–7.03 (m, 6H), 7.03–6.99 (m, 2H), 4.91–4.89 (m, 1H), 3.16–3.15 (m, 2H). 13C-NMR: δ 170.65, 166.99, 160.63, 160.18, 158.08, 142.90, 137.62, 130.76, 130.46, 129.27, 128.20, 126.51, 125.53, 118.77, 112.31, 107.44, 55.16, 37.34. ESI-MS (m/z) = 490.98 [M + H]+, HRMS (ESI) calcd. For C26H21F2N4O4+: [M + H]+ m/z: 491.1525, found: 491.1500.
(S)-4-(2-(5-(3-Chloro-2,6-difluorophenyl)-1H-pyrazole-3-carboxamido)-3-phenylpropanamido)benzoicacid (7k): white solid product (220 mg, 70.2% yield), m.p.: 129–131 °C, decomposition. 1H-NMR: δ 13.80 (s, 1H), 12.74 (s, 1H), 10.53 (s, 1H), 7.91–7.89 (d, J = 8.4 Hz, 2H), 7.73–7.70 (m, 3H), 7.34–7.04 (m, 8H), 4.93–4.87 (m, 1H), 3.18–3.15 (m, 2H). 13C-NMR: δ 171.23, 167.60, 157.27, 143.45, 138.22, 131.08, 129.86, 128.81, 127.13, 126.20, 119.39, 117.07, 113.99, 113.77, 108.30, 55.82, 37.94. ESI-MS (m/z) = 525.00 [M + H]+, HRMS (ESI) calcd. For C26H20ClF2N4O4+: [M + H]+ m/z: 525.1136, found: 525.1116.
(S)-4-(2-(5-(4-Chloro-2,6-difluorophenyl)-1H-pyrazole-3-carboxamido)-3-phenylpropanamido)benzoic acid (7l): white solid product (220 mg, 70.2% yield), m.p.: 123–125 °C, decomposition. 1H-NMR: δ 13.93–13.77 (m, 1H), 12.78–12.72 (m, 1H), 10.59 (s, 1H), 9.04–9.02 (m, 1H), 7.91–7.89 (d, J = 8.8 Hz, 2 H), 7.73–7.71 (d, J = 8.8 Hz, 2H), 7.54 (s, 2H), 7.36–7.34 (m, 2H), 7.27–7.24 (m, 2H), 7.18–7.10 (m, 1H), 6.99–6.96 (m, 1H), 4.91–4.86 (m, 1H), 3.17–3.14 (m, 2H). 13C-NMR: δ 171.30, 167.68, 158.58, 143.52, 138.27, 131.07, 129.91, 128.80, 127.13, 126.19, 119.34, 114.11, 113.83, 108.14, 55.92, 37.86. ESI-MS (m/z) = 525.01 [M + H]+, HRMS (ESI) calcd. For C26H20ClF2N4O4+: [M + H]+ m/z: 525.1136, found: 525.1126.
(S)-4-(2-(5-(3-Chlorophenyl)-4-methyl-1H-pyrazole-3-carboxamido)-3-phenylpropanamido)benzoic acid (7m): white solid product (29 mg, 9.8% yield), m.p.: 164–166 °C, decomposition. 1H-NMR: δ 13.48 (s, 1H), 12.70 (s, 1H), 10.48 (s, 1H), 8.01–7.98 (m, 1H), 7.91–7.89 (m, 2H), 7.71–7.69 (m, 2H), 7.60 (s, 1H), 7.54–7.49 (m, 3H), 7.29–7.21 (m, 4H), 7.21–7.19 (m, 1H), 4.90–4.88 (m, 1H), 3.17–3.15 (m, 2H), 2.29 (s, 3H). ESI-MS (m/z) = 503.00 [M + H]+, HRMS (ESI) calcd. For C27H24ClN4O4+: [M + H]+ m/z: 503.1481, found: 503.1467.
(S)-4-(2-(5-(3-Chlorophenyl)-4-ethyl-1H-pyrazole-3-carboxamido)-3-phenylpropanamido)benzoic acid (7n): white solid product (227 mg, 73.5% yield), m.p.: 126–128 °C, decomposition. 1H-NMR: δ 13.46 (s, 1H), 12.74 (s, 1H), 10.54 (s, 1H), 8.01 (s, 1H), 7.91–7.89 (d, J = 8.4 Hz, 2H), 7.72–7.70 (d, J = 8.8 Hz, 2H), 7.54–7.45 (m, 4H), 7.31–7.24 (m, 4H), 7.20–7.16 (m, 1H), 4.93–4.88 (qd, J = 8.0 Hz, 1H), 3.21–3.10 (m, 2H), 2.75–2.69 (qd, J = 7.2 Hz, 2H), 1.04–1.01 (m, 3H). ESI-MS (m/z) = 516.91 [M + H]+, HRMS (ESI) calcd. For C28H26ClN4O4+: [M + H]+ m/z: 517.1637, found: 517.1624.
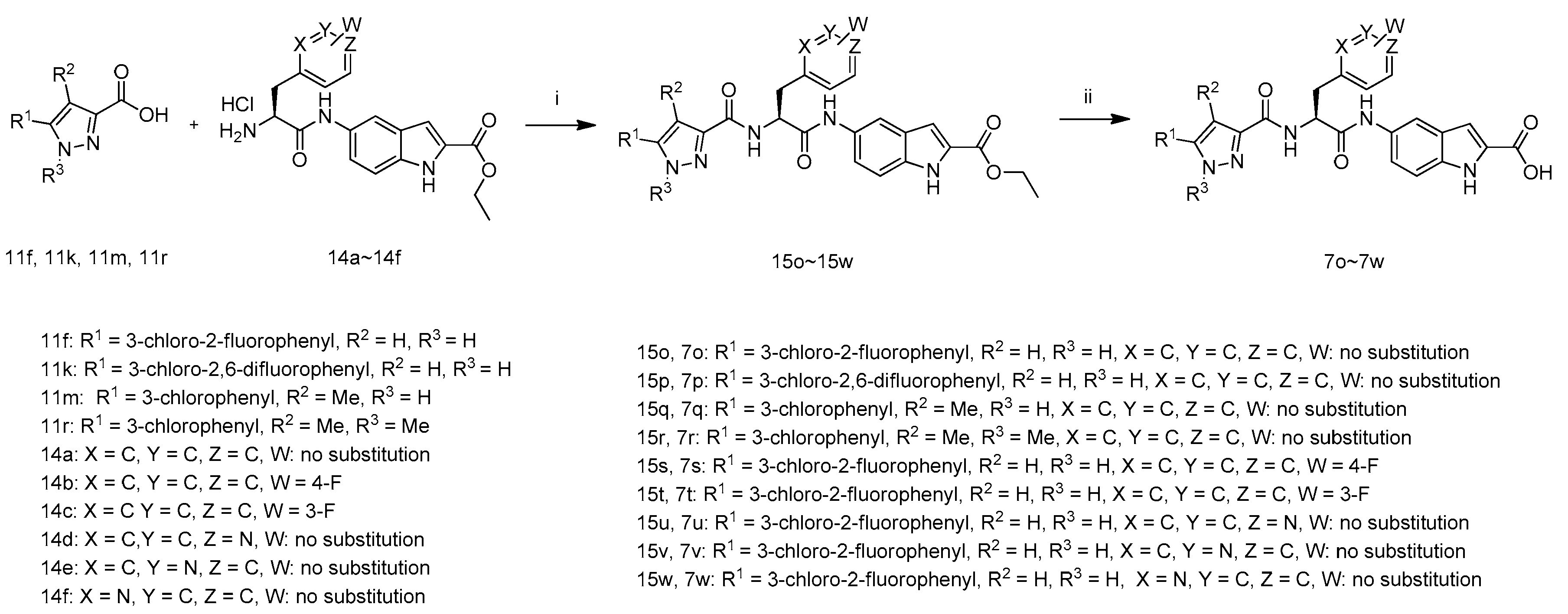
(S)-Ethyl 5-(2-(3-(3-chloro-2-fluorophenyl)-1H-pyrazole-5-carboxamido)-3-phenylpropanamido)-1H-indole-2-carboxylate (15o): To a mixture of 3-(3-chloro-2-fluorophenyl)-1H-pyrazole-5-carboxylic acid (11f, 136 mg, 0.57 mmol) in DMF (3 mL) was added 1-hydroxybenzotriazole (139 mg, 1.04 mmol), (S)-ethyl 5-(2-amino-3-phenylpropanamido)-1H-indole-2-carboxylate hydrochloride (14a, 200 mg, 0.52 mmol), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (198 mg, 1.04 mmol) and N,N-diisopropylethylamine (200 mg, 1.56 mmol), then the reaction mixture was stirred at room temperature overnight.TLC analysis indicated reaction was complete and H2O (30 mL) was added. The suspension was stirred for 0.5h and filtered. The filter cake was washed by H2O (5 mL) to get crude product 15o (130 mg, 44.2% yield) as a brown solid, which was used for next step without further purification.
(S)-5-(2-(3-(3-Chloro-2-fluorophenyl)-1H-pyrazole-5-carboxamido)-3-phenylpropanamido)-1H-indole-2-carboxylic acid (7o): To a suspension of compound 15o (130 mg, 0.23 mmol) in EtOH (3 mL) and H2O (1.5 mL) was added LiOH·H2O (38 mg, 0.92 mmol) and the reaction mixture was stirred at 40 °C for 6 h when TLC analysis indicated completion of reaction. The reaction mixture was evaporated on a rotary evaporator in vacuum and H2O (4 mL) was added. The solution was extracted by MTBE (2 mL), acidified by hydrochloric acid (1 M) to pH 3–4. After stirred for 0.5 h, the suspension was filtered, washed by H2O (5 mL) and the filter cake was dried at 50 °C for 2 h to afford 7o as a white solid (112 mg, 39.6% yield for 2 steps), m.p.: 172–174 °C, decomposition. 1H-NMR: δ 13.84 (s, 1H), 12.81 (s, 1H), 11.67 (s, 1H), 10.11 (s, 1H), 7.98 (s, 1H), 7.88 (s, 1H), 7.58 (s, 1H), 7.52–7.11 (m, 11H), 7.04 (s, 1H), 4.93–4.87 (m, 1H), 3.20–3.10 (m, 2H). HRMS (ESI) calcd. for C28H22ClFN5O4+: [M + H]+ m/z: 546.1339, found: 546.1325.
Compounds 7p~7w were synthesized according to the procedure described for the preparation of 7o.
(S)-5-(2-(3-(3-Chloro-2,6-difluorophenyl)-1H-pyrazole-5-carboxamido)-3-phenylpropanamido)-1H-indole-2-carboxylic acid (7p): white solid product (91 mg, 31.2% yield), m.p.: 164–166 °C, decomposition. 1H-NMR: δ 13.96 (s, 1H), 12.98 (s, 1H), 11.61 (s, 1H), 10.30 (s, 1H), 8.93 (s, 1H), 7.96 (s, 1H), 7.85 (s, 1H), 7.71–7.17 (m, 9H), 6.99 (s, 1H), 4.90–4.89 (m, 1H), 3.20–3.16 (m, 2H). ESI-MS (m/z) = 563.87 [M + H]+, HRMS (ESI) calcd. For C28H21ClF2N5O4+: [M+ H]+ m/z: 564.1245, found: 564.1234.
(S)-5-(2-(3-(3-Cchlorophenyl)-4-methyl-1H-pyrazole-5-carboxamido)-3-phenylpropanamido)-1H-indole-2-carboxylic acid (7q): white solid product (60 mg, 21.5% yield), m.p.: 154–156 °C, decomposition. 1H-NMR: δ 13.48 (s, 1H), 11.66 (s, 1H), 10.06 (s, 1H), 7.98 (s, 1H), 7.96–7.91 (m, 1H), 7.60 (s, 1H), 7.52 (s, 1H), 7.38–7.14 (m, 9H), 7.03 (s, 1H), 4.93–4.87 (m, 1H), 3.19–3.16 (m, 2H), 2.31 (s, 3H). ESI-MS (m/z) = 541.99 [M + H]+, HRMS (ESI) calcd. For C29H25ClN5O4+: [M + H]+ m/z: 542.1590, found: 542.1577.
(S)-5-(2-(3-(3-Chlorophenyl)-1,4-dimethyl-1H-pyrazole-5-carboxamido)-3-phenylpropanamido)-1H-indole-2-carboxylic acid (7r): white solid product (59 mg, 20.9% yield), m.p.: 148–150 °C, decomposition. 1H-NMR: δ 12.86 (s, 1H), 11.66 (s, 1H), 10.05 (s, 1H), 7.94 (s, 1H), 7.85–7.83 (d, J = 8.4 Hz, 1H), 7.56–7.53 (m, 3H), 7.40–7.24 (m, 6H), 7.20–7.18 (m, 1H), 7.03 (s, 1H), 4.94–4.89 (m, 1H), 3.75 (s, 3H), 3.18–3.10 (m, 2H), 2.09 (s, 3H). HRMS (ESI) calcd. For C30H27ClN5O4+: [M + H]+ m/z: 556.1746, found: 556.1735.
(S)-5-(2-(3-(3-Chloro-2-fluorophenyl)-1H-pyrazole-5-carboxamido)-3-(4-fluorophenyl)propanamido)-1H-indole-2-carboxylic acid (7s): white solid product (151 mg, 54.3% yield), m.p.: 186–188 °C, decomposition. 1H-NMR: δ 13.87 (s, 1H), 12.99–12.88 (m, 1H), 11.64 (s, 1H), 10.15 (s, 1H), 8.99–8.91 (m, 1H), 7.97 (s, 1H), 7.94 (s, 1H), 7.58 (s, 1H), 7.39–7.29 (m, 5H), 7.15–7.02 (m, 2H), 6.96 (s, 1H), 4.90–4.85 (m, 1H), 3.30–3.09 (m, 2H). 13C-NMR: δ 169.64, 163.18, 162.37, 159.96, 155.40, 152.91, 134.32, 134.16, 131.74, 131.32, 131.24, 130.08, 129.90, 126.99, 126.91, 125.79, 120.96, 118.76, 115.04, 114.83, 112.63, 112.26, 107.16, 106.21, 55.32, 36.93. ESI-MS (m/z) = 563.82 [M + H]+, HRMS (ESI) calcd. For C28H21ClF2N5O4+: [M + H]+ m/z: 564.1245, found: 564.1234.
(S)-5-(2-(3-(3-Chloro-2-fluorophenyl)-1H-pyrazole-5-carboxamido)-3-(3-fluorophenyl)propanamido)-1H-indole-2-carboxylic acid (7t): white solid product (154 mg, 55.7% yield), m.p.: 174–176 °C, decomposition. 1H-NMR: δ 13.92–13.75 (m, 1H), 13.03–12.81 (m, 1H), 11.66 (s, 1H), 10.12 (s, 1H), 7.97 (s, 1H), 7.87 (s, 1H), 7.59–7.56 (m, 1H), 7.54–7.27 (m, 5H), 7.21–6.93 (m, 4H), 4.94–4.88 (m, 1H), 3.29–3.13 (m, 2H). ESI-MS (m/z) = 563.84 [M + H]+, HRMS (ESI) calcd. For C28H21ClF2N5O4+: [M + H]+ m/z: 564.1245, found: 564.1234.
(S)-5-(2-(3-(3-Chloro-2-fluorophenyl)-1H-pyrazole-5-carboxamido)-3-(pyridin-4-yl)propanamido)-1H-indole-2-carboxylic acid (7u): grey solid product (92 mg, 32.7% yield), m.p.: 199–201 °C, decomposition.1H-NMR: δ 13.87 (s, 1H), 12.83 (s, 1H), 11.69 (s, 1H), 8.96 (s, 1H), 8.50 (s, 1H), 7.98(s, 1H), 7.93 (s, 1H), 7.69–7.39 (m, 6H), 7.05 (s, 1H), 5.00–4.95 (m, 1H), 3.27–3.17 (m, 2H). ESI-MS (m/z) = 547.01 [M + H]+, HRMS (ESI) calcd. For C27H21ClFN6O4+: [M + H]+ m/z: 547.1291, found: 547.1279.
(S)-5-(2-(3-(3-Chloro-2-fluorophenyl)-1H-pyrazole-5-carboxamido)-3-(pyridin-3-yl)propanamido)-1H-indole-2-carboxylic acid (7v): grey solid product (240 mg, 85.3% yield), m.p.: 175–177 °C, decomposition. 1H-NMR: δ 13.86 (s, 1H), 12.88 (s, 1H), 11.69 (s, 1H), 10.15 (s, 1H), 8.99 (s, 1H), 8.57 (s, 1H), 8.41–8.40 (d, J = 4.4 Hz, 1H), 7.98 (s, 1H), 7.88(s, 1H), 7.81–7.80 (d, J = 6.8 Hz, 1H), 7.52 (s, 1H), 7.39–7.10 (m, 4H), 7.04 (s, 1H), 4.95–4.89 (m, 1H), 3.25–3.14 (m, 2H). ESI-MS (m/z) = 547.05 [M + H]+, HRMS (ESI) calcd. For C27H21ClFN6O4+: [M + H]+ m/z: 547.1291, found: 547.1272.
(S)-5-(2-(3-(3-Chloro-2-fluorophenyl)-1H-pyrazole-5-carboxamido)-3-(pyridin-2-yl)propanamido)-1H-indole-2-carboxylic acid (7w): white solid product (109 mg, 38.7% yield), m.p.: 192–194 °C, decomposition. 1H-NMR: δ 11.68 (s, 1H), 10.19 (s, 1H), 8.71–8.70 (d, J = 4.4 Hz, 1H), 8.12 (s, 1H), 7.97 (s, 1H), 7.89–7.86 (m, 1H,), 7.71–7.70 (d, J = 6.0 Hz, 1H), 7.61–7.57 (t, J = 6.8 Hz, 2H), 7.36–7.30 (m, 4H), 7.03 (s, 1H), 5.13–5.11 (m, 1H), 3.56–3.39 (m, 2H). ESI-MS (m/z) = 547.04 [M + H]+, HRMS (ESI) calcd. For C27H21ClFN6O4+: [M + H]+ m/z: 547.1291, found: 547.1275.
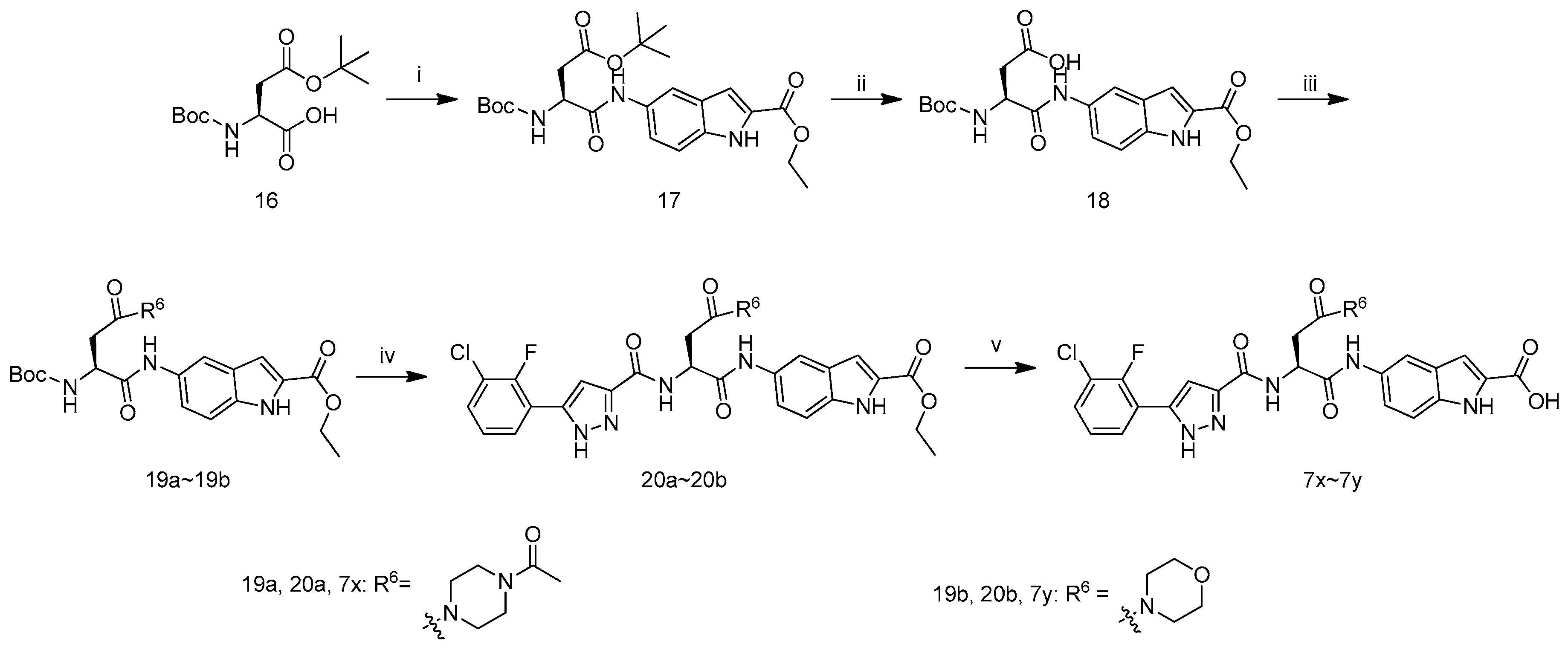
(S)-Ethyl 5-(4-(tert-butoxy)-2-((tert-butoxycarbonyl)amino)-4-oxobutanamido)-1H-indole-2-carboxylate (17): To a mixture of ethyl 5-amino-1H-indole-2-carboxylate (2.00 g, 9.8 mmol), pyridine (3.00 g, 19.6 mmol) and (S)-4-(tert-butoxy)-2-((tert-butoxycarbonyl)amino)-4-oxobutanoic acid (16, 2.83 g, 9.8 mmol) in CH2Cl2 (30 mL) was added POCl3 (1.16 g, 14.7 mmol) at −10 °C dropwise.After addition, the reaction mixture was stirred at −10 °C for 1 h when TLC analysis indicated completion of reaction. The mixture was washed by hydrochloric acid (1 M, 10 mL), dried by Na2SO4 and filtered. The filtrate was evaporated in vacuum and the residue was purified on column chromatography (n-hexane:ethyl acetate = 50:1 to 2:1) to get 17 as a white solid (1.77 g, 36.7% yield), m.p.: 119–121 °C, decomposition. 1H-NMR: δ 11.78 (s, 1H), 9.83 (s, 1H), 7.98 (s, 1H), 7.36 (s, 2H), 7.09–7.08 (m, 1H), 4.45–4.44 (m, 1H), 4.35–4.29 (m, 2H), 2.69–2.65 (m, 1H), 2.52–2.46 (m, 1H), 1.46–1.30 (m, 21H). ESI-MS (m/z) = 475.90 [M + H]+, HRMS (ESI) calcd. For C24H33KN3O7+: [M + K]+ m/z: 514.1950, found: 514.1947.
(S)-3-((tert-Butoxycarbonyl)amino)-4-((2-(ethoxycarbonyl)-1H-indol-5-yl)amino)-4-oxobutanoic acid (18): To a mixture of 17 (1.7 g, 3.6 mmol) in THF (5 mL) was added TFA (5 mL) at 0 °C. After addition, the mixture was stirred at room temperature for 6 h when TLC analysis indicated completion of reaction. The reaction mixture was evaporated in vacuum, then to the residue was added THF (20 mL), H2O (10 mL), Et3N (3.6 g, 36 mmol) and (Boc)2O (772 mg, 3.6 mmol) and the reaction was stirred at room temperature overnight.TLC analysis showed the reaction was complete and the reaction mixture was evaporated in vacuum. To the residue was added H2O (10 mL), extracted by MTBE (3 mL) and acidified by hydrochloric acid (1 M) to pH = 5. The aqueous layer was extracted by ethyl acetate for 3 times (10 mL × 3), and the combined organic layer was dried by Na2SO4, filtered and concentrated in vacuum to afford 18 as a grey solid (543 mg, 35.8% yield), m.p.: 123–125 °C, decomposition. 1H-NMR: δ 11.97 (s, 1H), 11.77 (s, 1H), 9.90 (s, 1H), 7.98 (s, 1H), 7.35 (s, 2H), 7.10–7.08 (m, 1H), 4.42–4.40 (m, 1H), 4.34–4.29 (m, 2H), 2.68–2.63 (m, 1H), 2.56–2.52 (m, 1H), 1.38–1.30 (m, 12H). ESI-MS (m/z) = 419.82 [M + H]+, HRMS (ESI) calcd. For C20H25KN3O7+: [M + K]+ m/z: 458.1324, found: 458.1322.
(S)-Ethyl 5-(4-(4-acetylpiperazin-1-yl)-2-((tert-butoxycarbonyl)amino)-4-oxobutanamido)-1H-indole-2-carboxylate (19a): To a mixture of 18 (200 mg, 0.48 mmol) in DMF (4 mL) was added 1-(piperazin-1-yl)ethanone (61 mg, 0.48 mmol), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (183 mg, 0.96 mmol), 1-hydroxybenzotriazole (129 mg, 0.96 mmol) and N,N-diisopropylethylamine (185 mg, 1.43 mmol). After addition, the reaction mixture was stirred at room temperature for 6 h when TLC analysis indicated completion of reaction. Then H2O (40 mL) was added and the mixture was extracted by ethyl acetate (15 mL × 3) for 3 times. The combined organic layer was dried, filtered and the filtrate was concentrated in vacuum to get 19a as a brown solid (176.3 mg, 69.8% yield). 1H-NMR: δ 11.76 (s, 1H), 9.81 (s, 1H), 7.94 (s, 1H), 7.36 (s, 1H), 7.09–7.07 (d, J = 2.0 Hz, 1H), 6.98–6.96 (d, J = 8.0 Hz, 1H), 4.50–4.48 (m, 1H), 4.34–4.29 (m, 2H), 3.45–3.39 (m, 8H), 2.75–2.72 (m, 2H), 1.99 (s, 3H), 1.37–1.30 (m, 12H). ESI-MS (m/z) = 529.95 [M + H]+.
(S)-Ethyl 5-(2-((tert-butoxycarbonyl)amino)-4-morpholino-4-oxobutanamido)-1H-indole-2-carboxylate (19b): Compounds 19b were synthesized from 18 and morpholine according to the procedure described for the preparation of 19a. Grey solid product (137.2 mg, 58.9% yield), m.p.: 144–146 °C, decomposition. 1H-NMR: δ 11.76 (s, 1H), 9.81 (s, 1H), 8.00 (s, 1H), 7.36 (s, 2H), 7.08 (s, 1H), 6.96–6.94 (d, J = 7.2 Hz, 1H), 4.49–4.48 (m, 1H), 4.34–4.29 (qd, J = 7.2, 2H), 3.54–3.41 (m, 8H), 2.72 (m, 2H), 1.38–1.30 (m, 12H). ESI-MS (m/z) = 488.97 [M + H]+, HRMS (ESI) calcd. For C24H32KN4O7+: [M + K]+ m/z: 527.1903, found: 527.1894.
(S)-Ethyl 5-(4-(4-acetylpiperazin-1-yl)-2-(5-(3-chloro-2-fluorophenyl)-1H-pyrazole-3-carboxamido)-4-oxobutanamido)-1H-indole-2-carboxylate (20a): To a mixture of 19a (170 mg, 0.32 mmol) in ethyl acetate (1 mL) was added hydrochloric/ethyl acetate (10 mL, saturated solution) and the reaction mixture was stirred at room temperature overnight. Then TLC analysis showed reaction was complete and the suspension was filtered to afford the intermediate. To the intermediate (150 mg, 0.32 mmo) in DMF (3 mL) was added 11f (78 mg, 0.32 mmo), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (124 mg, 0.64 mmol), 1-hydroxybenzotriazole (87 mg, 0.64 mmol), and N,N-diisopropylethylamine (124 mg, 0.96 mmol) and the reaction mixture was stirred at room temperature for 6 h when TLC analysis indicated completion of reaction. Then H2O (30 mL) was added and the suspension was stirred for 10 min, filtered, dried at 50 °C for 4 h to get 20a as a brown solid (91.2 mg, 43.3% yield), m.p.:156–158 °C, decomposition. 1H-NMR: δ 13.94 (m, 1H), 11.78 (s, 1H), 10.04–9.91 (m, 1H), 8.80–8.78 (m, 1H), 8.01 (s, 1H), 7.94–7.82 (m, 1H), 7.62–7.54 (m, 1H), 7.44–7.11 (m, 4H), 7.08 (s, 1H), 5.00 (s, 1H), 4.36–4.29 (m, 2H), 3.58–3.39 (m, 8H), 2.93–2.88 (m, 2H), 1.97 (s, 3H), 1.34–1.30 (m, 3H). ESI-MS (m/z) = 651.97 [M + H]+, HRMS (ESI) calcd. For C31H32ClFN7O6+: [M + H]+ m/z: 652.2081, found: 652.2070.
(S)-Ethyl 5-(2-(5-(3-chloro-2-fluorophenyl)-1H-pyrazole-3-carboxamido)-4-morpholino-4-oxobutanamido)-1H-indole-2-carboxylate (20b): Compounds 20b were synthesized from 19b and 11f according to the procedure described for the preparation of 20a. Grey solid product (90.3 mg, 52.2 % yield), m.p.:136–138 °C, decomposition. 1H-NMR: δ 13.90 (s, 1H), 11.78 (s, 1H), 9.98 (s, 1H), 8.01 (s, 1H), 7.88 (s, 1H), 7.60–7.53 (m, 2H), 7.41–7.30 (m, 4H), 7.08 (s, 1H), 5.03–4.97 (m, 1H), 4.38–4.29 (m, 2H), 3.63–3.38 (m, 8H), 2.93–2.80 (m, 2H), 1.38–1.30 (m, 3H). ESI-MS (m/z) = 610.95 [M + H]+, HRMS (ESI) calcd. For C29H29ClFN6O6+: [M + H]+ m/z: 611.1806, found: 611.1811.
(S)-5-(4-(4-Acetylpiperazin-1-yl)-2-(5-(3-chloro-2-fluorophenyl)-1H-pyrazole-3-carboxamido)-4-oxobutanamido)-1H-indole-2-carboxylic acid (7x): To a mixture of 20a (85 mg, 0.13 mmol) in EtOH (4 mL) and H2O (2 mL) was added LiOH·H2O (40 mg, 0.95) and the reaction mixture was stirred at room temperature for 5 h when TLC analysis indicated completion of reaction. The mixture was evaporated on a rotary evaporator in vacuum, then the residue was acidified by hydrochloric acid (1 M) to pH 3–4 and filtered. The filter cake was dried at 50 °C for 4 h to get 7x as a white solid (62 mg, 76.4% yield), m.p.: 101–103 °C, decomposition. 1H-NMR (400 MHz, DMSO-d6): δ 13.94 (s, 1H), 11.63 (s, 1H), 10.00 (s, 1H), 7.98 (s, 1H), 7.89 (s, 1H), 7.58 (s, 1H), 7.43–7.32 (m, 4H), 7.01 (s, 1H), 5.01–5.00 (m, 1H), 3.57–3.47 (m, 6H), 2.95–2.85 (m, 4H), 2.00 (s, 3H). ESI-MS (m/z) = 623.94 [M + H]+, HRMS (ESI) calcd. For C29H28ClFN7O6+: [M + H]+ m/z: 624.1768, found: 624.1755.
(S)-5-(2-(3-(3-Chloro-2-fluorophenyl)-1H-pyrazole-5-carboxamido)-4-morpholino-4-oxobutanamido)-1H-indole-2-carboxylic acid (7y): Compounds 7y were synthesized from 20b according to the procedure described for the preparation of 7x. White solid product (59 mg, 77.8% yield), m.p.: 175–177 °C, decomposition. 1H-NMR: δ 13.95–13.84 (m, 1H), 11.62–11.60 (m, 1H), 10.01–9.98 (m, 1H), 8.81 (s, 1H), 8.38–8.36 (m, 1H), 7.98 (s, 1H), 7.90–7.82 (m, 1H), 7.58 (s, 1H), 7.48–7.28 (m, 3H), 7.00 (s, 1H), 5.01–4.92 (m, 1H), 3.57–3.42 (m, 4H), 3.39–3.29 (m, 4H), 2.96–2.92 (m, 2H). ESI-MS (m/z) = 582.96 [M + H]+, HRMS (ESI) calcd. For C27H26ClFN6O6+: [M + H]+ m/z: 583.1503, found: 583.1485.
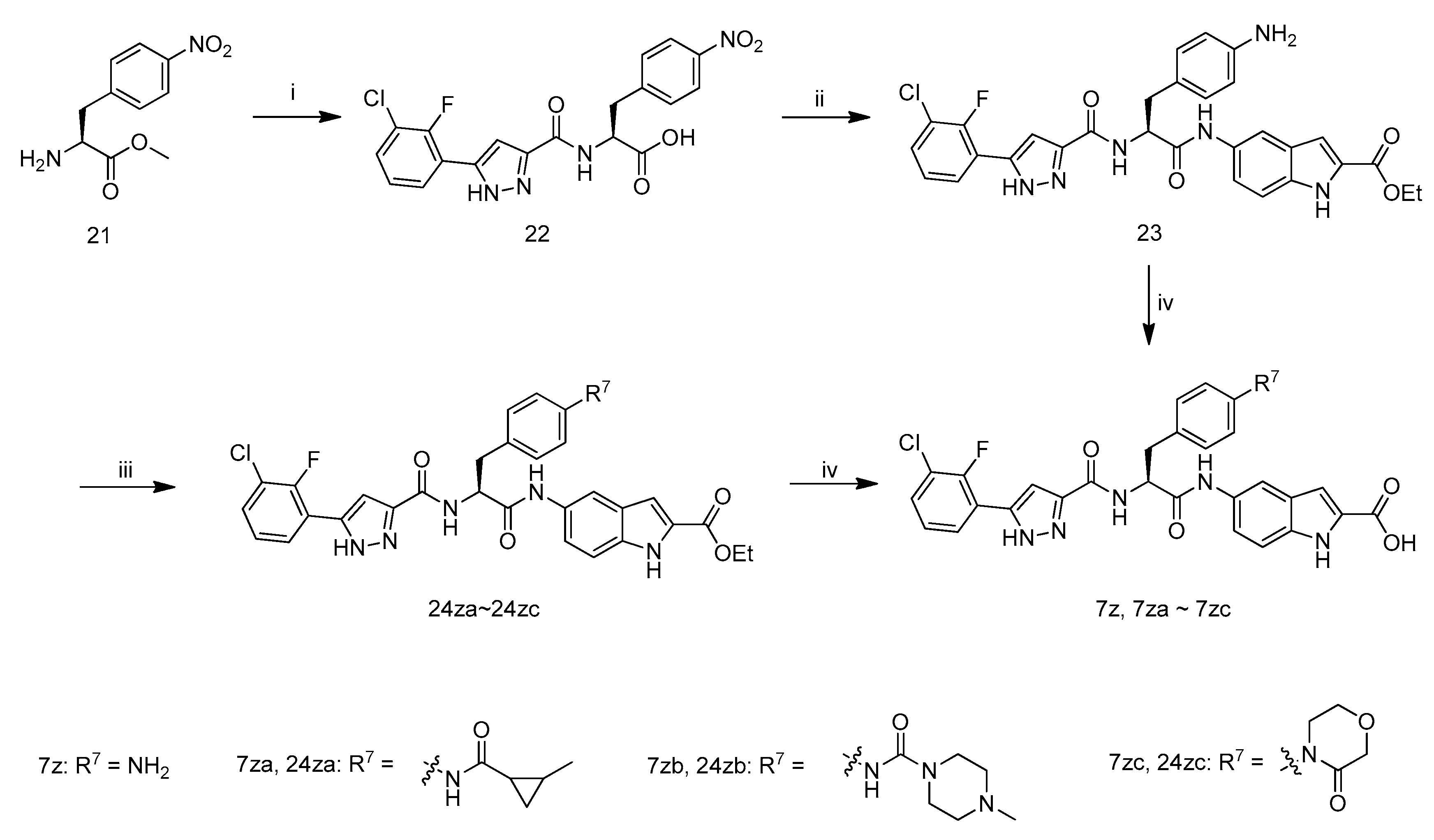
(S)-2-(5-(3-Chloro-2-fluorophenyl)-1H-pyrazole-3-carboxamido)-3-(4-nitrophenyl)propanoic acid (22): A mixture of 3-(3-chloro-2-fluorophenyl)-1H-pyrazole-5-carboxylic acid (11f, 2.2 g, 8.43 mmol), (S)-methyl 2-amino-3-(4-nitrophenyl)propanoate (21, 2.0 g, 8.43 mmol), 1-hydroxybenzotriazole (2.3 g, 17.04 mmol), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (3.2 g, 17.04 mmol) and N,N-diisopropylethylamine (3.2 g, 24.80 mmol)in DMF (20 mL) was stirred at room temperature overnight. Then TLC analysis showed reaction was complete and H2O (200 mL) was added, then the suspension was stirred for 0.5 h andfiltered. The filter cake was transferred to a round-bottomed flask, and MeOH (60 mL), H2O (30 mL) and LiOH·H2O (1.0 g, 23.8 mmol) was added. The reaction mixture was stirred at room temperature for 2 h when TLC analysis indicated completion of reaction, evaporated on a rotary evaporator in vacuum, then H2O (50 mL) was added, acidified by hydrochloric acid (1 M) to pH 3–4 and filtered. The filter cake was dried at 50 °C for 8 h to afford 22 as a yellow solid (3.25 g, 89.0% yield), m.p.: 167–169 °C, decomposition. 1H-NMR: δ 14.01–13.79 (m, 1H), 8.11–8.02 (m, 2H), 7.87–7.84 (m, 1H), 7.59–7.56 (m, 1H), 7.49–7.42 (m, 2H), 7.36–7.22 (m, 1H), 7.06 (s, 1H), 4.42–4.29 (m, 1H), 3.32–3.19 (m, 2H). ESI-MS (m/z) = 432.94 [M + H]+, HRMS (ESI) calcd. For C19H15ClFN4O5+: [M + H]+ m/z: 433.0710, found: 433.0708.
(S)-Ethyl 5-(3-(4-aminophenyl)-2-(5-(3-chloro-2-fluorophenyl)-1H-pyrazole-3-carboxamido)propanamido)-1H-indole-2-carboxylate (23): A mixture of compound 22 (3.00 g, 6.93 mmol), ethyl 5-amino-1H-indole-2-carboxylate (1.42 g), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (2.66 g, 13.9 mmol), 1-hydroxybenzotriazole (1.87 g, 13.9 mmol) and N,N-diisopropylethylamine (1.79 g, 13.9 mmol) in DMF(20 mL) was stirred at room temperature overnight when TLC analysis indicated completion of reaction, then H2O (200 mL) was added. The suspension was stirred for 10 min and filtered. The filter cake was dried at room temperature overnight and resolve in MeOH (200 mL) and ethyl acetate (100 mL). To the solution was added Pd/C (10%, 0.30 g) and the reaction mixture was stirred at the atmosphere of H2overnight when TLC analysis indicated completion of reaction. The suspension was filtered and the filtrate was concentrated in vacuum to afford intermediate 23 as a grey solid (3.1 g, 75.6% yield), m.p.: 120–122 °C, decomposition. 1H-NMR: δ 13.77 (s, 1H), 11.80 (s, 1H), 10.20 (s, 1H), 8.99–8.69 (m, 2H), 7.99 (s, 1H), 7.91 (s, 1H), 7.38–6.98 (m, 7H), 6.91 (s, 1H), 6.81–6.80 (d, J = 2.4 Hz, 2H), 4.88–4.86 (m, 1H), 4.34–4.29 (qd, J = 6.8 Hz, 2H), 3.17–3.10 (m, 2H), 1.34–1.31 (m, 3H). 13C-NMR: δ 170.51, 161.92, 147.33, 135.04, 132.57, 130.40, 128.58, 127.48, 127.26, 126.30, 125.51, 119.84, 114.71, 113.25, 112.79, 108.33, 106.65, 61.05, 54.03, 40.76, 14.94. ESI-MS (m/z) = 588.98 [M + H]+, HRMS (ESI) calcd. For C30H27ClFN6O4+: [M + H]+ m/z: 589.1688, found: 589.1755.
(S)-5-(3-(4-Aminophenyl)-2-(5-(3-chloro-2-fluorophenyl)-1H-pyrazole-3-carboxamido)propanamido)-1H-indole-2-carboxylic acid (7z): To a mixture of compound 23 (140 mg, 0.24 mmol) in MeOH (4 mL) and H2O (2 mL) was added LiOH·H2O (52 mg, 1.20 mmol) and the mixture was stirred at 40 °C for 4 h when TLC analysis indicated completion of reaction. The mixture was concentrated and H2O (2 mL) was added. The solution was acidified with hydrochloric acid (1 M) to pH = 5, filtered and the filter cake was washed by H2O (2 mL), dried at 50 °C for 3 h to afford 7z as a brown solid (98.0 mg, 72.8% yield), m.p.: 209–211 °C, decomposition. 1H-NMR: δ 13.88–13.86 (m, 1H), 11.64 (s, 1H), 10.10–10.04 (m, 1H), 8.82 (s, 1H), 7.96 (s, 1H), 7.89 (s, 1H), 7.58 (s, 1H), 7.57–7.29 (m, 6H), 7.22–6.98 (m, 3H), 6.45–6.43 (d, J = 8.0 Hz, 2H), 4.80–4.74 (m, 1H), 3.02–2.92 (m, 2H). ESI-MS (m/z) = 560.92 [M + H]+, HRMS (ESI) calcd. For C28H23ClFN6O4+: [M + H]+ m/z: 561.1448, found: 561.1439.
Ethyl 5-((2S)-2-(5-(3-chloro-2-fluorophenyl)-1H-pyrazole-3-carboxamido)-3-(4-(2-methylcyclopropane-carboxamido)phenyl)propanamido)-1H-indole-2-carboxylate (24za): To a mixture of compound 23 (200 mg, 0.34 mmol), 2-methylcyclopropanecarboxylic acid (34 mg, 0.34 mmol) and pyridine (54 mg, 0.68 mmol) in CH2Cl2 (5 mL) was added POCl3(78 mg, 0.51 mmol) at −10 °C dropwise. After addition, the reaction mixture was stirred at −10 °C for 1 h when TLC analysis indicated completion of reaction. The mixture was added H2O (0.5 mL), concentrated in vacuum, and the residue was purified by Prep-TLC to afford desired product 24za as a brown solid (76 mg, 33.3% yield), m.p.: 227–229 °C, decomposition. 1H-NMR: δ 13.85 (s, 1H), 11.80 (s, 1H), 10.10 (s, 1H), 9.99 (s, 1H), 7.98 (s, 1H), 7.91 (s, 1H), 7.57 (s, 1H), 7.48–7.10 (m, 9H), 7.05 (s, 1H), 4.86–4.85 (m, 1H), 4.35–4.29 (qd, J = 6.8 Hz, 2H), 3.12–3.02 (m, 2H), 1.46–1.45 (m, 1H), 1.34–1.31 (m, 3H), 1.22–1.16 (m, 1H), 1.08–1.05 (m, 3H), 0.96–0.94(m, 1H), 0.58(s, 1H). ESI-MS (m/z) = 670.92 [M + H]+, HRMS (ESI) calcd. For C35H33ClFN6O5+: [M + H]+ m/z: 671.2180, found: 671.2178.
5-((2S)-2-(5-(3-Chloro-2-fluorophenyl)-1H-pyrazole-3-carboxamido)-3-(4-(2-methylcyclopropane-carboxamido)phenyl)propanamido)-1H-indole-2-carboxylic acid (7za): Compounds 7za was synthesized from compound 24za according to the procedure described for the preparation of 7z. Grey solid product (45 mg, 63.7% yield), m.p.: 212–214 °C, decomposition. 1H-NMR: δ 13.85 (s, 1H), 12.83 (s, 1H), 11.67 (s, 1H), 10.12 (s, 1H), 8.12 (s, 1H), 7.98 (s, 1H), 7.60–7.57 (m, 1H), 7.52–7.04 (m, 6H), 7.03 (s, 1H), 4.88–4.70 (m, 1H), 3.14–3.01 (m, 2H), 1.48–1.44 (m, 1H), 1.21–1.17(m, 1H), 1.16–1.08 (m, 3H), 1.06–1.04 (m, 1H), 0.56–0.59 (m, 1H). 13C-NMR: δ 171.23, 169.68, 162.96, 155.35, 152.85, 138.12, 137.95, 134.30, 132.17, 131.71, 130.05, 129.56, 126.92, 125.71, 120.90, 120.73, 118.80, 112.57, 112.21, 107.24, 106.11, 55.26, 37.26, 23.07, 17.61, 15.21, 11.90. ESI-MS (m/z) = 642.92 [M + H]+, HRMS (ESI) calcd. For C33H29ClFN6O5+: [M + H]+ m/z: 643.1867, found: 643.1848.
(S)-Ethyl 5-(2-(5-(3-chloro-2-fluorophenyl)-1H-pyrazole-3-carboxamido)-3-(4-(4-methylpiperazine-1-carboxamido)phenyl)propanamido)-1H-indole-2-carboxylate (24zb): A mixture of compound 23 (300 mg, 0.51 mmol), pyridine (403 mg, 5.1 mmol), 4-methylpiperazine-1-carbonyl chloride hydrochloride (166 mg, 1.02 mmol), and 4-dimethylaminopyridine (13 mg, 0.10 mmol) in DMF (5 mL) was stirred at room temperature overnight when TLC analysis indicated completion of reaction. The mixture was added H2O (40 mL) and extracted by ethyl acetate for 3 times (15 mL × 3). The combined organic layer was dried and concentrated in vacuum to get intermediate 24zb (223 mg, 60.8% yield), m.p.: 174–176 °C, decomposition. 1H-NMR: δ 13.86 (s, 1H), 11.80 (s, 1H), 10.14 (s, 1H), 8.44 (s, 1H), 7.99–7.89 (m, 2H), 7.58 (s, 1H), 7.49–7.34 (m, 6H), 7.32–7.21 (m, 2H), 7.09 (s, 1H), 4.86–4.84 (m, 1H), 4.34–4.29 (qd, J = 7.2 Hz, 2H), 3.60–3.04 (m, 10H), 2.29 (s, 3H), 1.34–1.31 (m, 3H,). ESI-MS (m/z) = 715.22 [M + H]+, HRMS (ESI) calcd. For C36H37ClFN8O5+: [M + H]+ m/z: 715.2554, found: 715.2543.
(S)-5-(2-(5-(3-Chloro-2-fluorophenyl)-1H-pyrazole-3-carboxamido)-3-(4-(4-methylpiperazine-1-carboxamido)phenyl)propanamido)-1H-indole-2-carboxylic acid (7zb): sCompound 7zb was synthesized from compound 24zb according to the procedure described for the preparation of 7z. Grey solid product (153 mg, 73.4% yield), m.p.: 203–205 °C, decomposition. 1H-NMR: δ 13.88 (s, 1H), 11.66 (s, 1H), 10.10 (s, 1H), 8.44 (s, 1H), 7.97 (s, 1H), 7.88 (s, 1H), 7.58 (s, 1H), 7.38–7.20 (m, 9H), 7.03 (s, 1H), 4.86–4.85 (m, 1H), 3.48–3.44 (m, 6H), 3.13–3.02 (m, 4H), 2.30 (3H, s). ESI-MS (m/z) = 687.11 [M + H]+, HRMS (ESI) calcd. For C34H33ClFN8O5+: [M + H]+ m/z: 687.2241, found: 687.2222.
(S)-Ethyl 5-(2-(5-(3-chloro-2-fluorophenyl)-1H-pyrazole-3-carboxamido)-3-(4-(3-oxomorpholino)phenyl)-propanamido)-1H-indole-2-carboxylate (24zc): A mixture of 2-(2-chloroethoxy)acetic acid (48 mg, 0.34 mmol), compound 23 (200 mg, 0.34 mmol) and N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline (100 mg, 0.41 mmol) in THF (4 mL) was stirred at 60 °C for 4 h when TLC analysis indicated completion of reaction. The reaction mixture was concentrated in vacuum and THF (5 mL) was added to the residue. To the stirred mixture was added NaH (40 mg, 1 mmol) in portions at room temperature. After addition, the mixture was stirred at room temperature for 5 h and TLC showed reaction was complete. Then NH4Cl solution (0.2 mL, concentrated solution) was added and the mixture was concentrated in vacuum. The residue was recrystallized fromn-hexane and ethyl acetate to give 24zc as a brown solid (174 mg, 76.0% yield). 1H-NMR: δ 11.84 (s, 1H), 10.45–10.39 (m, 1H), 8.47 (s, 3H), 8.02 (s, 2H), 7.41–7.39 (m, 3H), 7.28–7.26 (m, 3H), 7.23–7.19 (m, 2H), 4.90 (m, 1H), 4.34–4.28 (qd, J = 7.2 Hz, 2H), 3.92–3.89 (m, 2H), 3.68–3.59 (m, 2H), 3.40–3.38 (m, 2H), 3.17–3.07 (m, 2H), 1.33–1.30 (t, J = 7.2 Hz, 3H). ESI-MS (m/z) = 695.17 [M + Na]+.
(S)-5-(2-(5-(3-Chloro-2-fluorophenyl)-1H-pyrazole-3-carboxamido)-3-(4-(3-oxomorpholino)phenyl)-propanamido)-1H-indole-2-carboxylic acid (7zc): Compounds 7zc was synthesized from compound 24zc according to the procedure described for the preparation of 7z. Grey solid product (78.0 mg, 50.4% yield), m.p.: 200–202 °C, decomposition. 1H-NMR: δ 13.89 (s, 1H), 11.18 (s, 1H), 10.14 (s, 1H), 7.92–7.88 (m, 2H), 7.56–7.41 (m, 2H), 7.39–7.25 (m, 6H), 4.90–4.89 (m, 1H), 4.15 (s, 2H), 3.93–3.90 (m, 2H), 3.68–3.66 (m, 2H), 3.57–3.55 (m, 2H). ESI-MS (m/z) = 644.90 [M + H]+, HRMS (ESI) calcd. For C32H27ClFN6O6+: [M + H]+ m/z: 645.1659, found: 645.1637.


 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





