1. Introduction
Proteases, one of the largest groups of enzymes in all organisms, are involved in diverse cellular processes by irreversibly cleaving peptide bonds. They represent potential drug targets for many human diseases [
1,
2,
3]. Based on which amino acid or atom is used to generate a nucleophile to attack the peptide carbonyl group, proteases are classified into six groups which have two general mechanisms of peptide hydrolysis. One is to use the hydroxyl or sulfhydryl group in a side chain as a nucleophile to make acyl-enzyme intermediates, as shown in serine, cysteine, and threonine proteases, and the other is to use an activated water molecule as a nucleophile to directly hydrolyze the peptide bonds, as presented in aspartic-, glutamic-, and metallo-proteases.
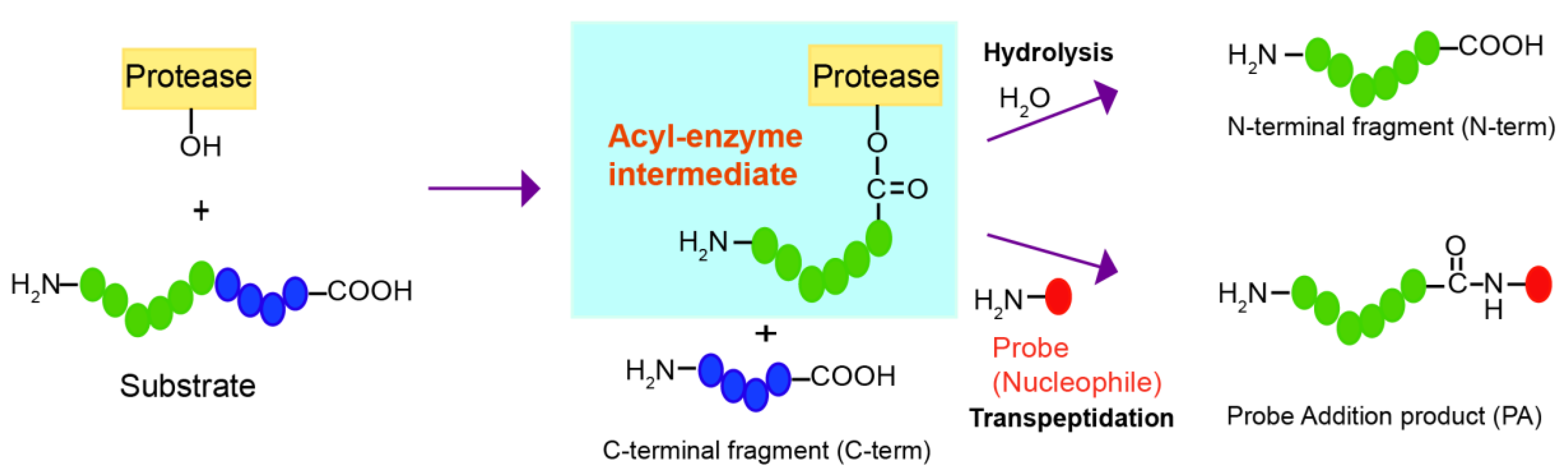
Although proteases have evolved to catalyze the efficient hydrolysis of peptide bonds, those making acyl-enzyme intermediates may also mediate transpeptidation or peptide ligation if amine nucleophiles react with the acyl-enzyme intermediate and induce aminolysis instead of hydrolysis (
Figure 1). Transpeptidation has been observed under normal conditions for protein digestion with several proteases, such as trypsin, V8 protease, Glu-C, and bleomycin hydrolase [
4,
5,
6,
7,
8]. By introducing several mutations, subtilisin was converted from a protease to a peptide ligase, subtiligase, which can mediate the selective biotin labeling of α-amines of peptides over ε-amines of lysines and has been applied to a proteomic approach to globally identify proteolytic cleavage sites [
9,
10,
11]. Notable cases of transpeptidation by a protease were reported with human proteasome, called peptide splicing, in which non-contiguous peptides from the proteasomal degradation were ligated and presented as major histocompatibility complex (MHC) class I antigens on the cell surface [
12,
13,
14,
15]. However, these examples are dependent on activated substrates or nucleophiles that are generated in situ, and they are generally very inefficient processes in which hydrolysis dominates. It is also unknown whether it is possible to find a good nucleophile that will efficiently react with acyl-enzyme intermediates under mild conditions.
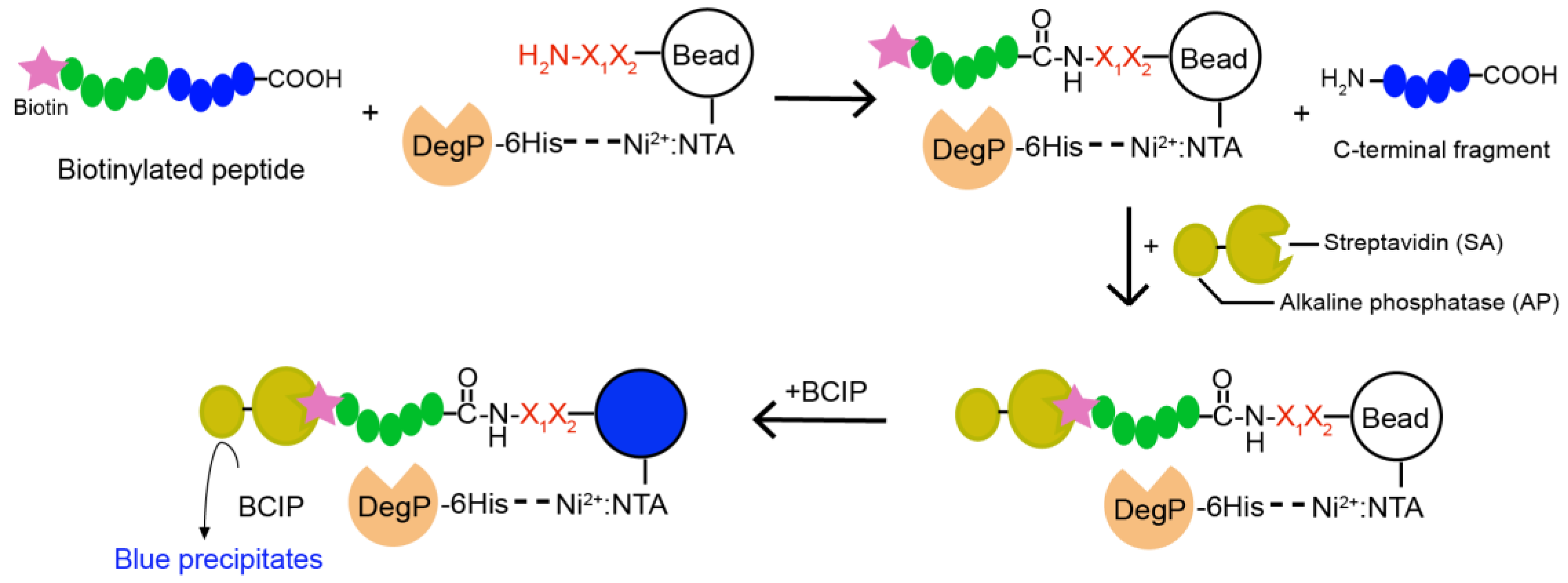
Here, we developed a strategy to find a good nucleophilic probe for protease-mediated transpeptidation using peptide libraries and on-bead screening. This method led to the identification of probes that are efficiently ligated to the C-termini of the peptides forming acyl-enzyme intermediates with the model DegP protease. The biotin tag in the probes allowed selective purification of the ligated peptides among various cleavage products.
3. Discussion
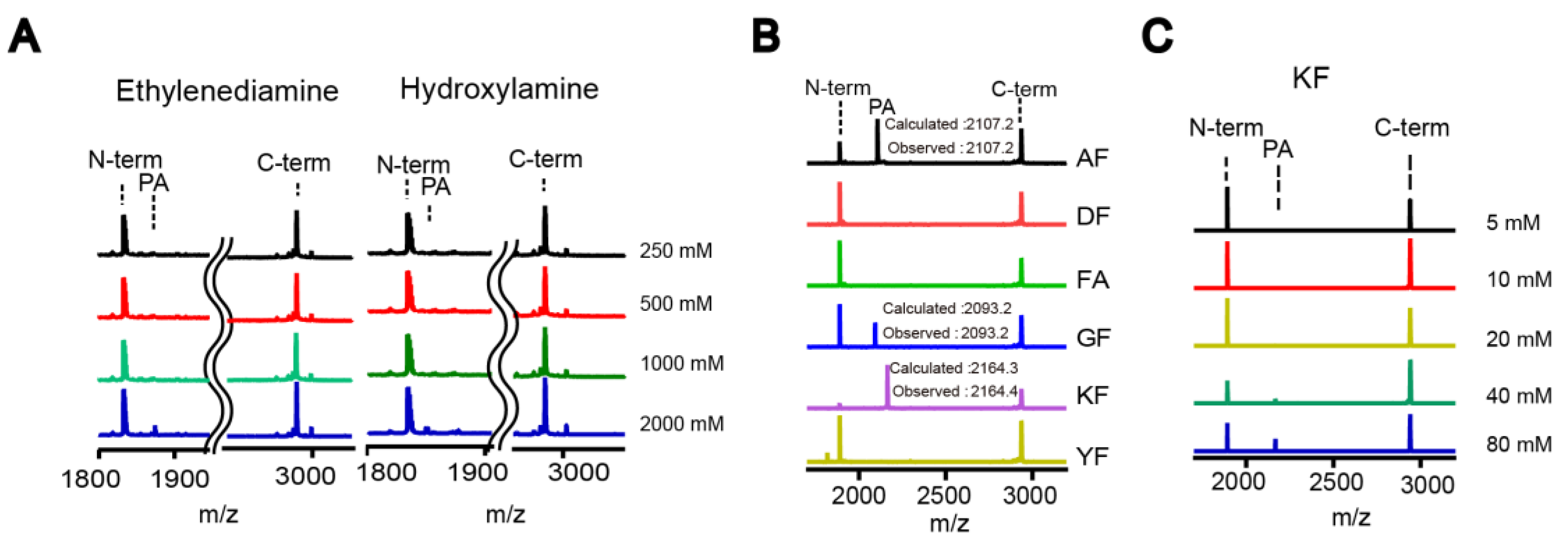
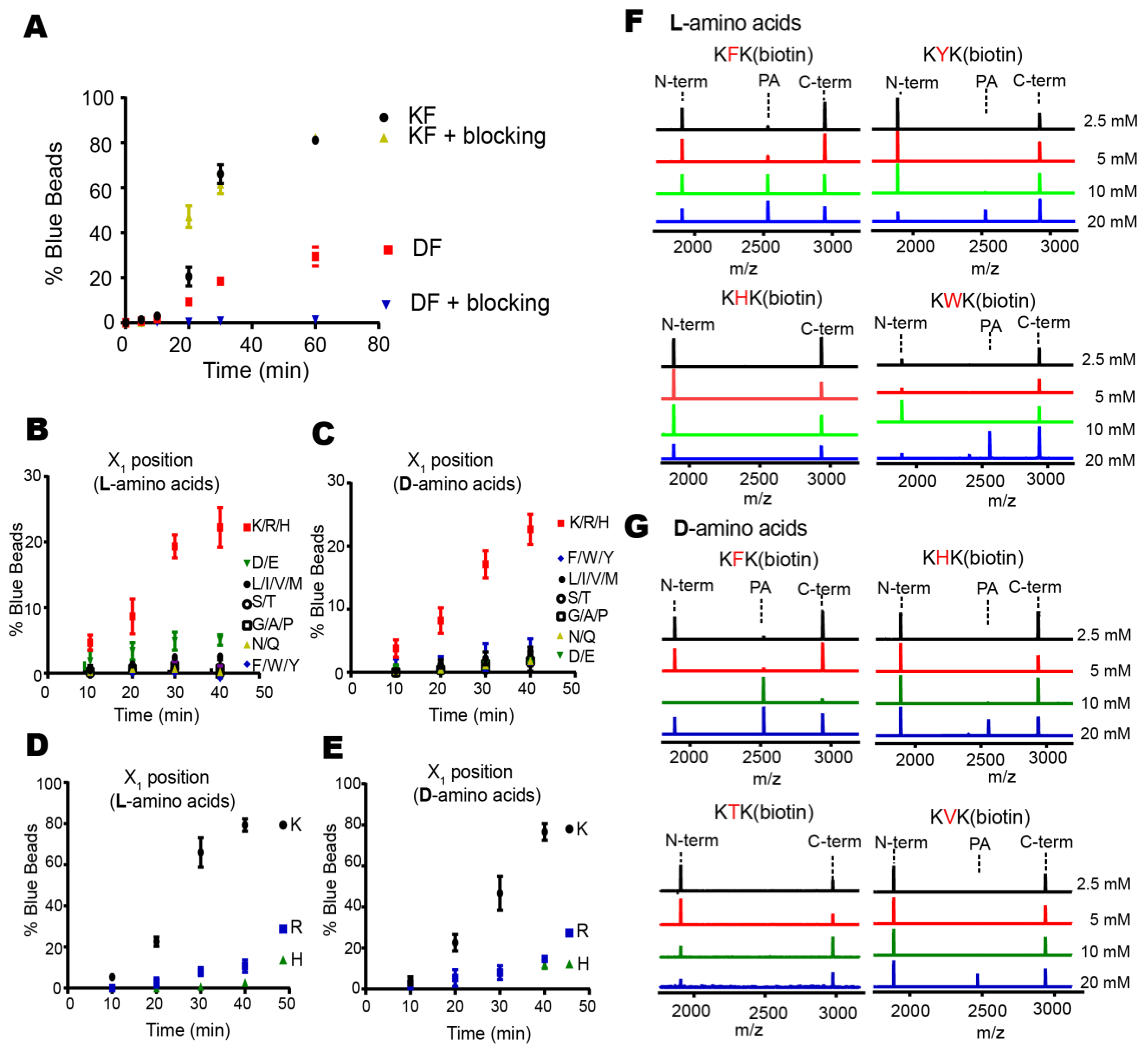
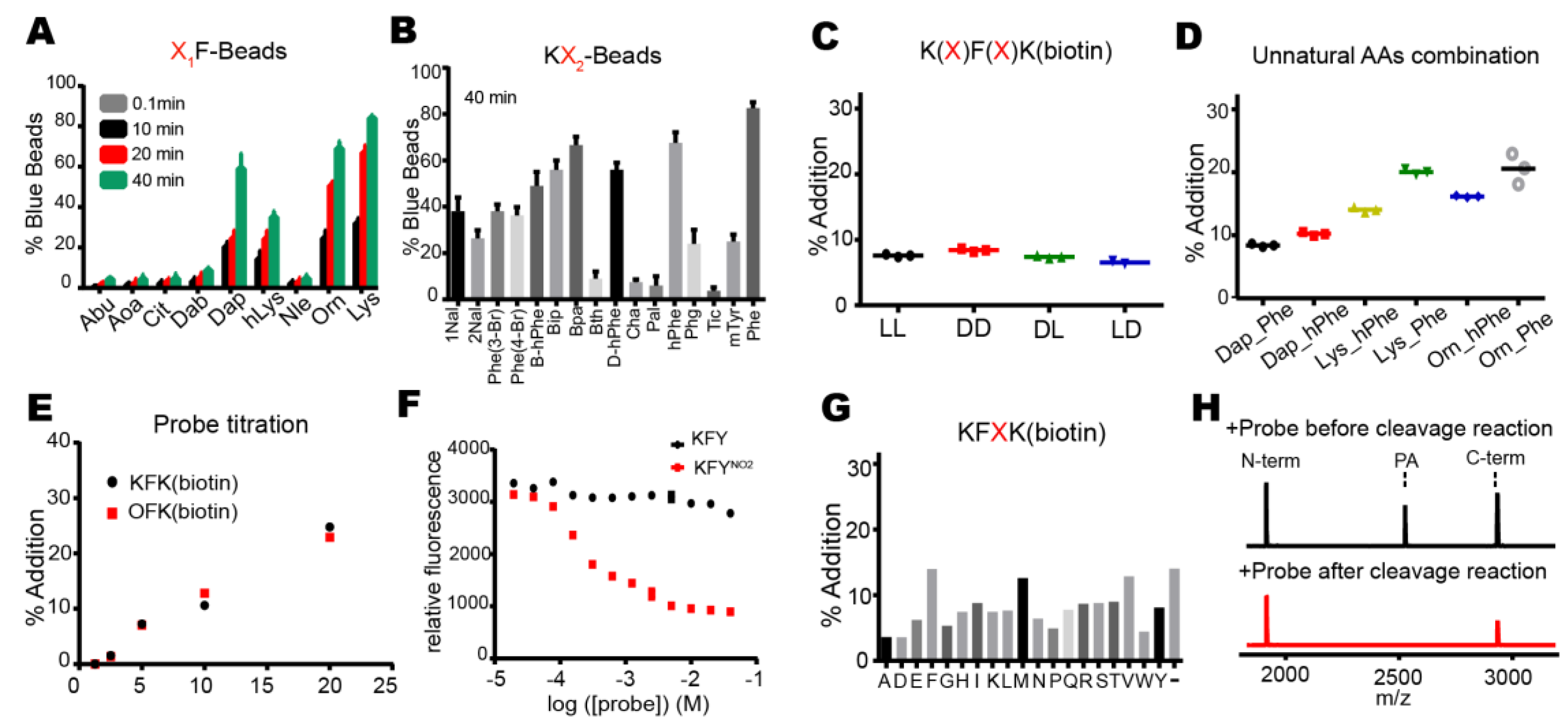
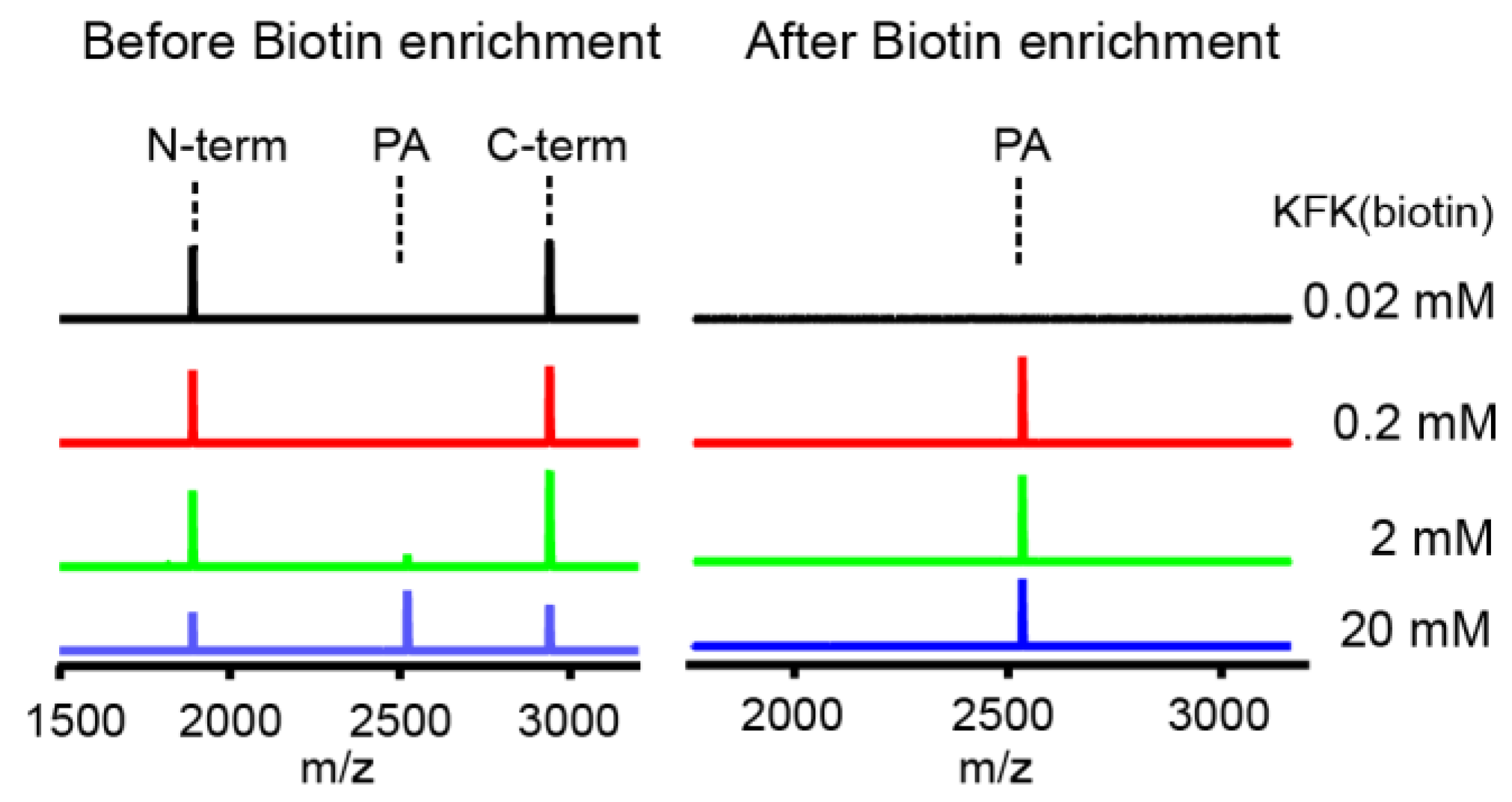
Proteases have evolved to mediate the hydrolysis of peptide bonds, which is an energetically downhill process. Therefore, transpeptidation by proteases normally occurs as a rare event or at a low efficiency. Here, we found nucleophilic probes that can readily react with acyl-enzyme intermediates and efficiently trap the cleavage products of a model protease, DegP, in a normal buffer condition. The OBOC-based on-bead screening approach was successfully applied to identify KFK(biotin) and OFK(biotin) as good probes which are covalently linked to the C-termini of the cleaved peptides and allow selective enrichment of the ligated peptides. This probe should react with the acyl-enzyme intermediates for transpeptidation before water molecules do for hydrolysis at a very high concentration (~55.5 M in aqueous solutions), but only 20 mM of the probe is required to obtain 20–25% of the addition product of the model substrate, indicating that this probe can compete with a water molecule at a 1000-fold lower concentration and generate a comparable amount of the transpeptidation product over the hydrolysis product. Also, we showed that the enrichment of the addition product allows its detection at a much lower probe concentration. We suggest that many other proteases acting via acyl-enzyme intermediates can also perform transpeptidation in the presence of a proper nucleophilic molecule and that these probes will help in the characterization of the substrates of proteases.
This study differs from the previous examples of protease-mediated transpeptidation or peptide ligation in the identification of efficient nucleophilic probes. Those with trypsin, V8 protease, Glu-C, bleomycin hydrolase, and human proteasome showed peptide ligation between cleaved peptides at a low efficiency [
4,
5,
6,
7,
8,
12,
13]. Subtiligase, a mutant variant of subtilisin BPN’, normally uses activated ester substrates for the selective labeling of proteins with free N-terminal α-amine, but our probe was applied to a wild-type protease for the selective C-terminal labeling of cleaved peptides [
9,
10,
11]. Many other examples of peptide ligation using proteases require activated ester substrates and harsh conditions, such as organic solvents, high pH, and high reactant concentrations, and are mainly applied to peptide synthesis in vitro [
24,
25]. Our study mainly concerned the selective ligation of the probe to the cleaved peptides under normal conditions, and therefore, this approach may have applications both in vitro and in vivo for substrate characterization.
It is unknown how exactly the probe triggers transpeptidation over hydrolysis, but it is likely that the probe transiently binds to the site near the active site and is positioned to readily attack acyl-enzyme intermediates [
26,
27]. In particular, two amino acids in the probe may function as the P1′ and P2′ residues which fit the S’-positions of the enzyme that are originally occupied by the C-terminal fragment of the substrate. This idea is consistent with our result whereby a specific residue, lysine for the DegP probe, is preferred at the N-terminal position of the probe for efficient transpeptidation. Therefore, the P1′ specificity of the protease may be a good starting point to find a good probe.
It has been suggested that peptide splicing in the human proteasome may generate a unique set of antigens with distinct immunological properties and may facilitate the development of vaccines and cancer immunotherapies [
15]. The potential role of transpeptidation by other proteases is unknown, but one possibility may be to help proteolysis by removing relatively long-lived acyl-enzyme intermediates that may slow down the reaction. This idea suggests that the probe may be added more efficiently to a specific set of peptides with slow cleavage kinetics. In this case, the transpeptidiation efficiency may be dependent on the sequence of substrates as well as the probe. Further studies about the kinetics of hydrolysis and transpeptidation with various substrates will be required to support this idea.
4. Materials and Methods
4.1. Materials and Equipment
General chemicals and reagents were purchased from Fisher Acros (Hampton, NH, USA). Isothiocyanobenzyl-NTA was purchased from Biomass (Rockville, MD, USA). Tentagel S NH2 resin was purchased from RAPP polymer (Tuebingen, Germany). DynabeadsTM MyOneTM Streptavidin C1, Streptavidin-alkaline phosphatase (SAAP), and 5-bromo-4-chloro-3-indolyl-phosphate (BCIP) were obtained from Thermofisher (Hampton, NH, USA). Amino acids, coupling reagents, and biotin were purchased from GL-Biochem. BSA (Shanghai, China), lysozyme, and dipeptides were obtained from Sigma-Aldrich (St. Louis, MO, USA). Ni Sepharose 6 FastFlow beads were obtained from GE Healthcare (Chicago, IL, USA).
MALDI-TOF-MS and MALDI-TOF-MS/MS analysis were performed by a Bruker Micro-flex MALDI-TOF mass spectrometer (Billerica, MA, USA) and a Bruker Ultra-flextreme MALDI-TOF/TOF mass spectrometer. The HPLC analysis and HPLC purification were performed using Agilent 1260 Infinity (Agilent, Santa Clara, CA, USA). Positive bead (blue beads) screening was performed with a Nikon SMZ 745T microscope (Nikon, Tokyo, Japan).
4.2. Expression and Purification of Proteins and a Peptide
DegP
WT and the model substrate (18–58) were prepared as previously described [
19,
28]. The plasmid expressing 6His-MalE31 was constructed by mutating G32/I33 to D32/P33 in the MalE-expressing plasmid. Cells harboring the 6His-MalE31 plasmid were grown at 30 °C in 1 L LB medium with kanamycin (50 μg/mL). The 6His-MalE31 expression was induced at A
600 ~0.5 with isopropyl-1-thio-β-
d-galactopyranoside (IPTG, 0.1 mM) for 4–5 h. Cells were harvested, resuspended in 30 mL of 25 mM Tris buffer (pH 7.5), lysed by sonication, and centrifuged at 13,000 rpm for 30 min. The pellets were resuspended in 10 mL of the above buffer containing 2% Triton X-100. After centrifugation, inclusion bodies (pellets) were solubilized by urea Ni-column wash buffer (8 M urea, 300 mM NaCl, Tris 20 mM, pH 6.0). 6His-MalE31 was bound to Ni Sepharose 6 FastFlow beads, washed with wash buffer, and eluted with buffer B (8 M Urea, 300 mM NaCl, 20 mM Tris, pH 4.0). The eluted solution was concentrated with Amicon centrifugal filter (Millipore, Burlington, MA, USA).
4.3. Peptide Synthesis
OBOC peptide libraries were constructed using solid phase peptide synthesis. Tentagel S NH2 resin (0.7 g) was washed and soaked with 10 mL of 1:1 DMF/DCM for 20 min in a reaction vessel. Fmoc-Met-OH (5 equiv.) and HATU/DIEA (5/10 equiv.) dissolved in DMF were added to the reaction vessel and the reaction solution was incubated at room temperature for 30 min. Beads were washed three times with DMF and DCM. After the completion of coupling, monitored by ninhydrin and TNBS tests, beads were treated with capping solution (DMF/acetic anhydride/DIEA in a 9:1:0.05 ratio) for 8 min to block the unreacted amines. Beads were mixed with 20% (v/v) piperidine in DMF for 20 min to remove the Fmoc group and washed three times with DMF and DCM. The above coupling, capping, and deprotection steps were repeated to add four more amino acids, resulting in the AAARM peptide. Beads were washed six times with DMF and DCM, split into 19 equal portions in new reaction vessels. Beads were individually coupled to 19 amino acids (except cysteine) and subjected to the Fmoc deprotection and the DMF/DCM wash. All the beads were combined in one reaction vessel, split again into 19 equal portions, and subjected to the second coupling reactions with 19 amino acids. The N-terminal Fmoc group was removed with 20% (v/v) piperidine in DMF, and the side chain protection groups of the peptide library were removed with cleavage cocktail (95% TFA, 2.5% deionized water, 2.5% triisopropyl silane) for 4 h at room temperature. Finally, beads were washed with DMF and DCM and became ready for the on-bead screening experiment.
The X1X2K(biotin) peptides were synthesized using solid phase peptide synthesis. Fmoc-Lys(mtt)-OH (5 equiv.) and DMAP/DIC (0.1 and 4 equiv., respectively) were added to the washed and soaked Wang resin and mixed in DMF at room temperature for 30 h. After capping and washing beads, the 4-methyltrityl (MTT) protecting group was cleaved using 2% TFA in DCM for 30 min. The beads were washed with DMF and DCM, and mixed with the activated biotin solution, which was prepared by mixing 5 equiv. (d)-Biotin, 4 equiv. HATU, and 10 equiv. DIEA in 10 mL DMF/DMSO (1:1). After the beads were washed, two more rounds of the deprotection and amino acid coupling were performed. After the last coupling, a cleavage cocktail was applied to the resin for 2 h. The solution was then evaporated using nitrogen stream over the tube. For each 1 mL of the solution, 10 mL of ice-cold ether/hexane (1:1) was added for peptide precipitation. The precipitated peptides were centrifuged and washed with cold precipitation solution to remove the scavenger. Peptides were dissolved in DMSO, diluted with 0.05% TFA in water, and purified by HPLC and lyophilization.
The N-terminally biotinylated substrate of DegP (Biotin-SLGNWVSAAKFESNFNTQDYGILQI) and the p23 peptide (Abz-GNWVSAA KFEYNO2SKNTQDYGILQI; Abz, 2-aminobenzoic acid; YNO2, 3-nitrotyrosine) were synthesized using solid phase peptide synthesis, as described above.
4.4. On-Bead Reaction and Screening
Two hundred and fifty nanomoles of Tentagel beads (64,790 beads) in 75 µL of NTA binding buffer (50 mM phosphate, 300 mM NaCl, pH 8.5) was mixed with 25 µL of Isothiocyanobenzyl-NTA (ITC-Bz-NTA; 100 μM) in DMSO and incubated at 37 °C for 1 h with shaking. Beads were washed five times with NTA binding buffer and mixed with 5 equivalent NiSO4 at room temperature for 30 min with shaking. Beads were washed with enzyme binding buffer (50 mM phosphate, 300 mM NaCl, 20 mM imidazole, pH 8.0) and mixed with 0.2 nM 6His-DegP at room temperature for 30 min in 100 μL of enzyme binding buffer. After washing with enzyme binding buffer, beads were incubated with 5 nM biotinylated peptide containing blocking buffer (50 mM phosphate, 100 mM NaCl, 4% BSA, pH 8.0) at 37 °C for 2 h with shaking. The reaction solution was decanted, and the beads were washed with the blocking buffer and SAAP buffer (30 mM Tris, 1 M NaCl, pH 7.4). Small amounts of beads (~200 beads each group) were transferred to a transparent 96-well plate with another 20 μL of SAAP buffer, and mixed with 1 μL of SAAP solution (1 mg/mL) for 10 min at 4 °C. After the solution was removed, the beads were washed with the SAAP buffer and BCIP staining buffer (30 mM Tris, 100 mM NaCl, 5 mM MgCl2, 2 μM ZnCl2, pH 8.4). One hundred microliters of BCIP staining buffer and 20 μL of 5 mg/mL BCIP were added to the beads, and the plate was incubated for about 30 min on a shaker. Some beads turned blue. The staining reaction was quenched by adding 100 µL of 1 M HCl. Beads were transferred to a transparent film for counting or picking up the blue beads using a microscope. For peptide cleavage, each blue bead was transferred into a microtube, washed with water, and treated with 20 μL of the CNBr solution (40 mg/mL in 70% (v/v) TFA and H2O) overnight. The solvent was evaporated with nitrogen stream, and 10 μL of the solution (10% Acetonitrile, 0.05% TFA) was added into each tube to dissolve the peptides. After desalting with C18 Ziptip, peptides were analyzed by MALDI-TOF-MS.
4.5. Reactions in Solution and HPLC Analysis
The small-scale reaction mixture (10–20 µL) contained DegP (2 μM), a peptide substrate (18–58; 20 μM), and various nucleophiles at the indicated concentrations in reaction buffer (50 mM phosphate, 100 mM NaCl, pH 8.0). The reaction tube was incubated at room temperature for 2 h. TFA was added at a final concentration of 0.1% (v/v) to quench the reaction. After desalting with C18 Ziptip, peptides were analyzed by MALDI-TOF-MS.
For the HPLC analysis, the peptide substrate (18–58; 100 µM) was added to the reaction solution (100 µL final) containing DegP (10 µM) and nucleophiles (indicated concentration) in the reaction buffer (50 mM phosphate, 100 mM NaCl, pH 8.0). After incubation at room temperature for 2 h, the reaction was quenched by adding TFA at a final concentration of 0.1% (v/v). The reaction solution was desalted with C18 Ziptip, diluted 10-fold with HPLC solvent A (0.05% TFA in water), and analyzed by HPLC. A C18 reverse phase HPLC column (ZORBAX SB-C18 4.6 × 250 mm, Santa Clara, CA, USA) was used to separate the peptides using a 10–40% acetonitrile gradient for 30 min at a flow rate of 1 mL/min. Each peak was analyzed by MALDI-TOF-MS. The probe addition efficiency was quantified by measuring the peak areas of the N-terminal fragment and the addition product.
4.6. FRET Assay
Reaction solutions (10 µL) containing DegP (1 μM), KFY or KFYNO2 (0–40 mM), and p23 (40 μM) in reaction buffer (50 mM phosphate, 100 mM NaCl, pH 8.0) were incubated at room temperature for 2 h. The probe addition was monitored after the reaction by measuring fluorescence (excitation at 320 nm; emission at 430 nm) using an Infinite F200Pro microplate reader (Tecan, Männedorf, Swiss).
4.7. Enrichment of the Addition Products
For enrichment of the addition product from the model peptide, the reaction solutions (100 µL) containing 18–58 (100 µM), DegP (10 µM), and OFK(biotin) (0.02, 0.2, 2, or 20 mM) were prepared in reaction buffer (50 mM phosphate, 100 mM NaCl, pH 8.0). After incubation at room temperature for 2 h, the solution was passed through a C18 Ziptip for peptide binding. The Ziptip was washed eight times with 200 μL of 10% acetonitrile/0.05% TFA (v/v) solution to remove the excessive probe, and peptides were eluted with 10 μL of 80% acetonitrile/0.05% TFA (v/v). The eluted solution was dried, and peptides were dissolved in binding buffer (50 mM phosphate, 300 mM NaCl, pH 7.4). Twenty microliters of streptavidin Dynabead C1 (Thermofisher) was washed three times with binding buffer and added to the peptide solution. After incubation at room temperature for one hour with shaking, the beads were washed five times with binding buffer. Biotinylated peptides were eluted by boiling at 95 °C for 10 min with 20 μL of elution solution (95% Formamide, 10 mM EDTA, pH 8.2). The eluted solution was transferred to a microtube, diluted with solvent A (200 μL H2O, 0.05% TFA (v/v)), and desalted with C18 Ziptip. Peptides were analyzed by MALDI-TOF-MS.
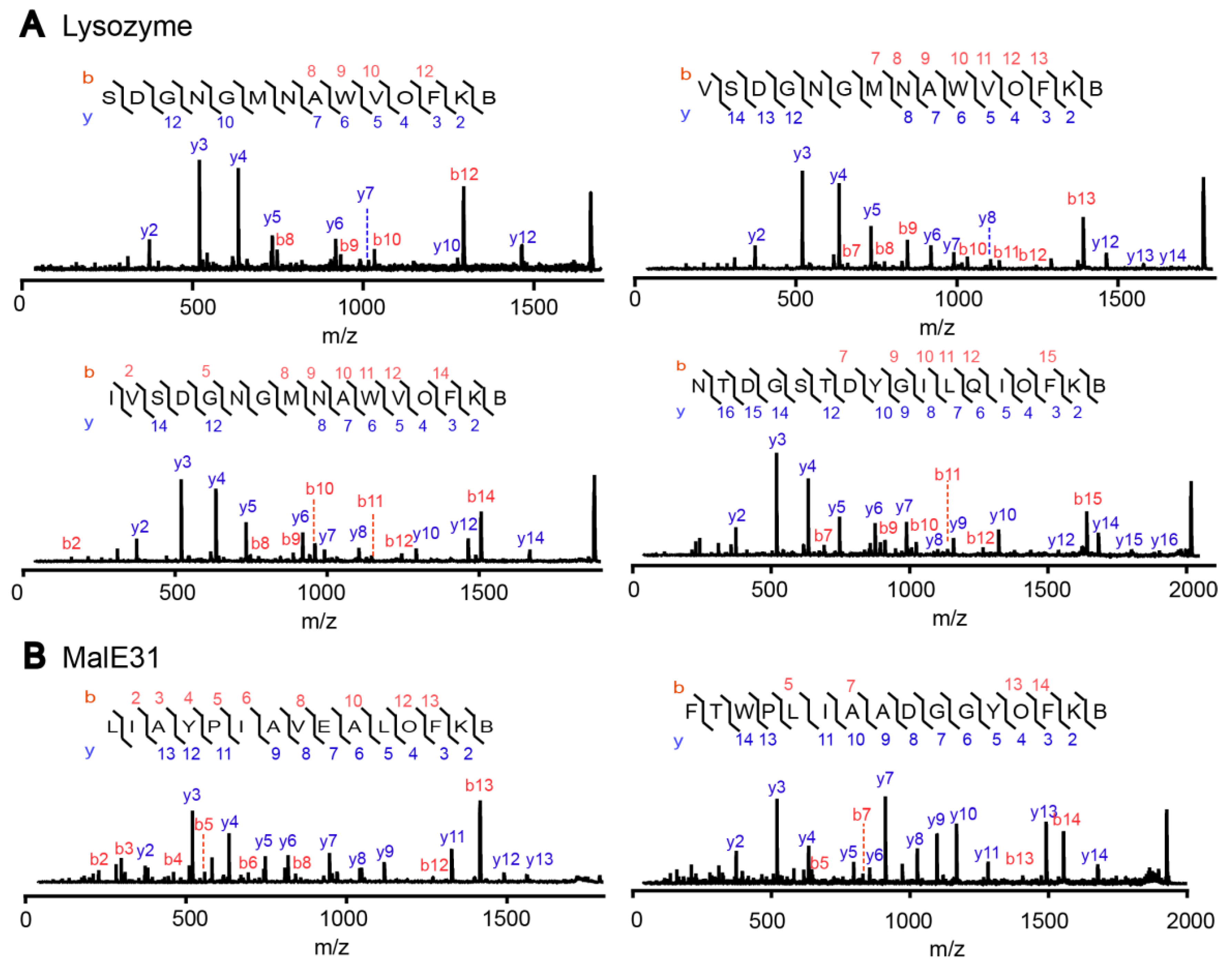
For the enrichment of the addition product from the model proteins, lysozyme (80 µM) and MalE31 (80 µM final, diluted from 2 mM solution in 8 M urea) were individually degraded by DegP (10 µM) in the presence of OFK(biotin) (10 mM) and DTT (5 mM) in reaction buffer (50 mM phosphate, 100 mM NaCl, pH 8.0; 100 µL final volume). The reaction solutions were incubated at 42 °C (lysozyme) or at room temperature (MalE31) for 4 h. Protein degradation was confirmed by SDS-PAGE. The reaction solutions were diluted 20-fold with the reaction buffer (50 mM phosphate, 100 mM NaCl, pH 8.0), treated with trypsin in a final protein:protease ratio of 20:1 (w/w), and incubated at room temperature overnight. The trypsin digestion was stopped by adding 0.1% TFA. The solutions were loaded onto a Ziptip streptavidin Dynabead C1, and then to a second Ziptip for the enrichment of biotinylated peptides, as described above. Peptides were analyzed by MALDI-TOF-MS/MS (Bruker Daltonics) in which the tandem mass spectra were used to identify the probe-addition products.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






