Chiral Diol-Based Organocatalysts in Enantioselective Reactions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
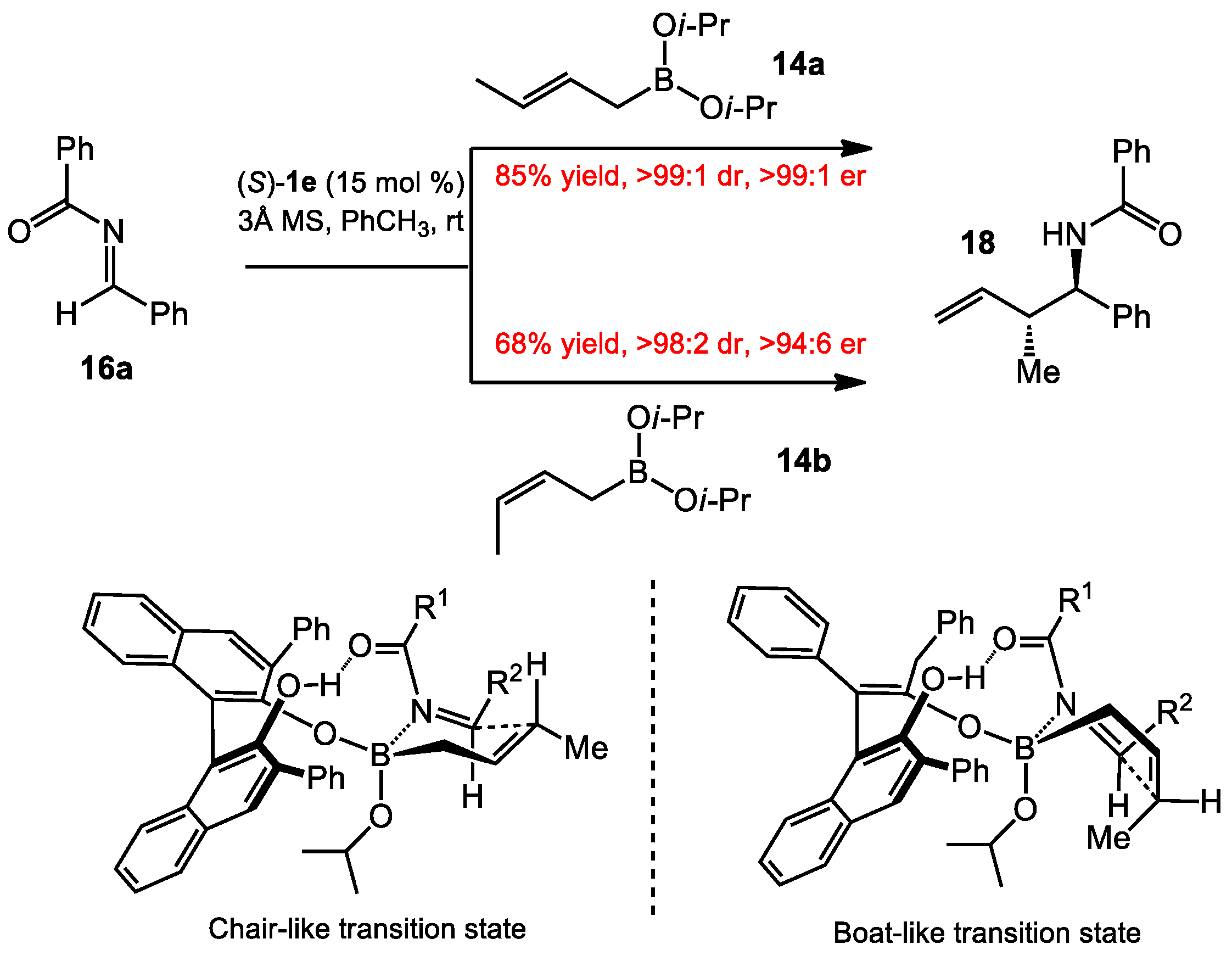{kind=link}
{kind=link}
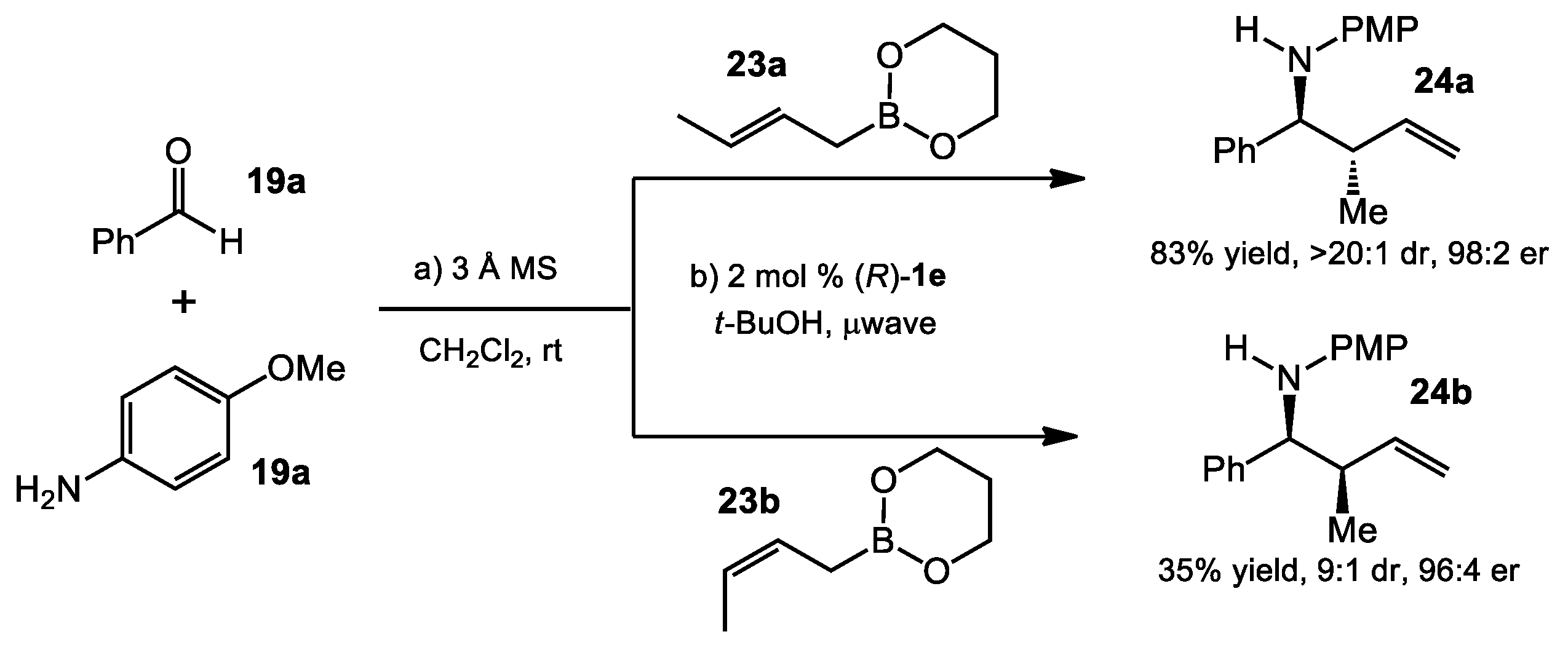{kind=link}
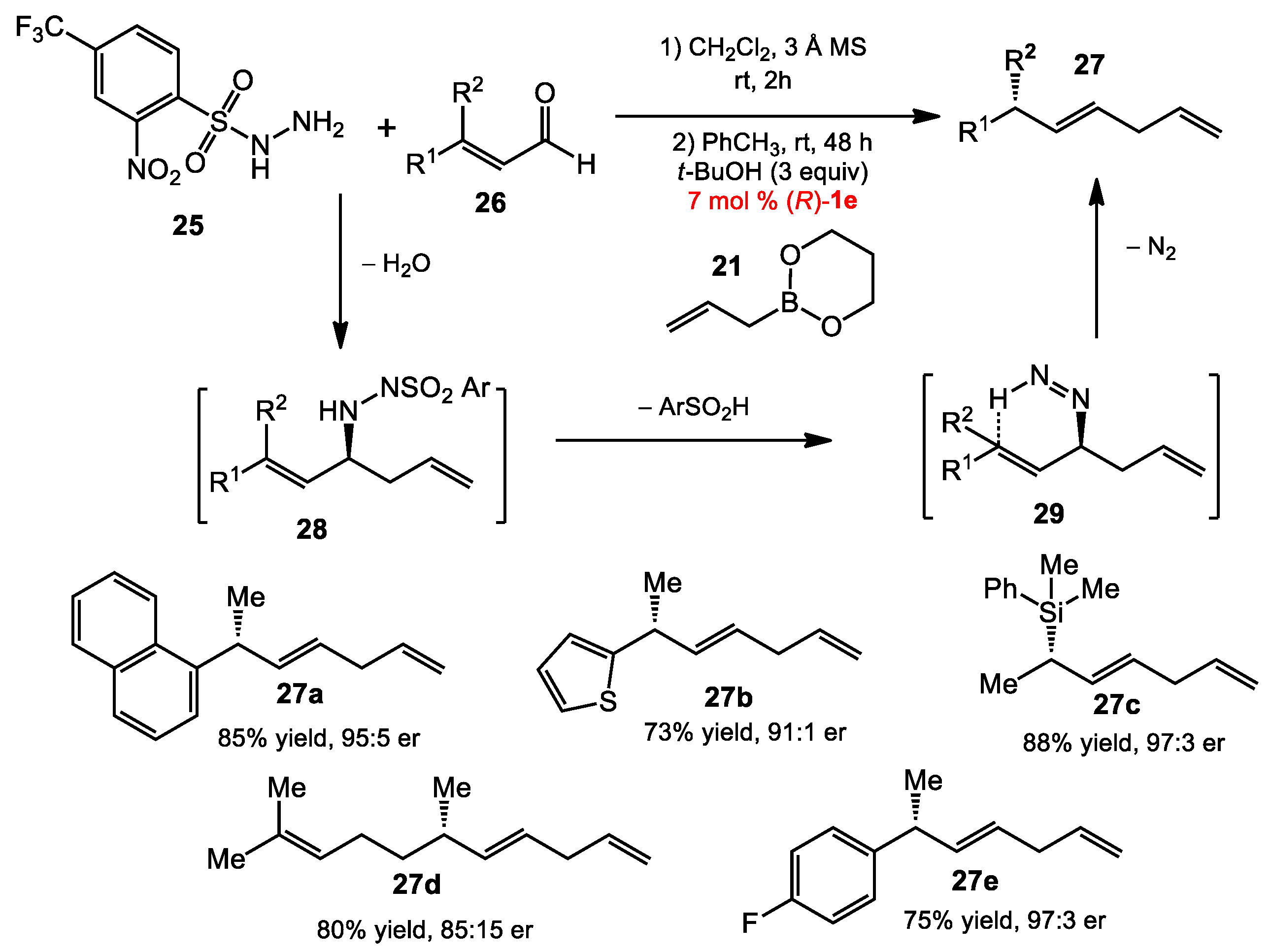{kind=link}
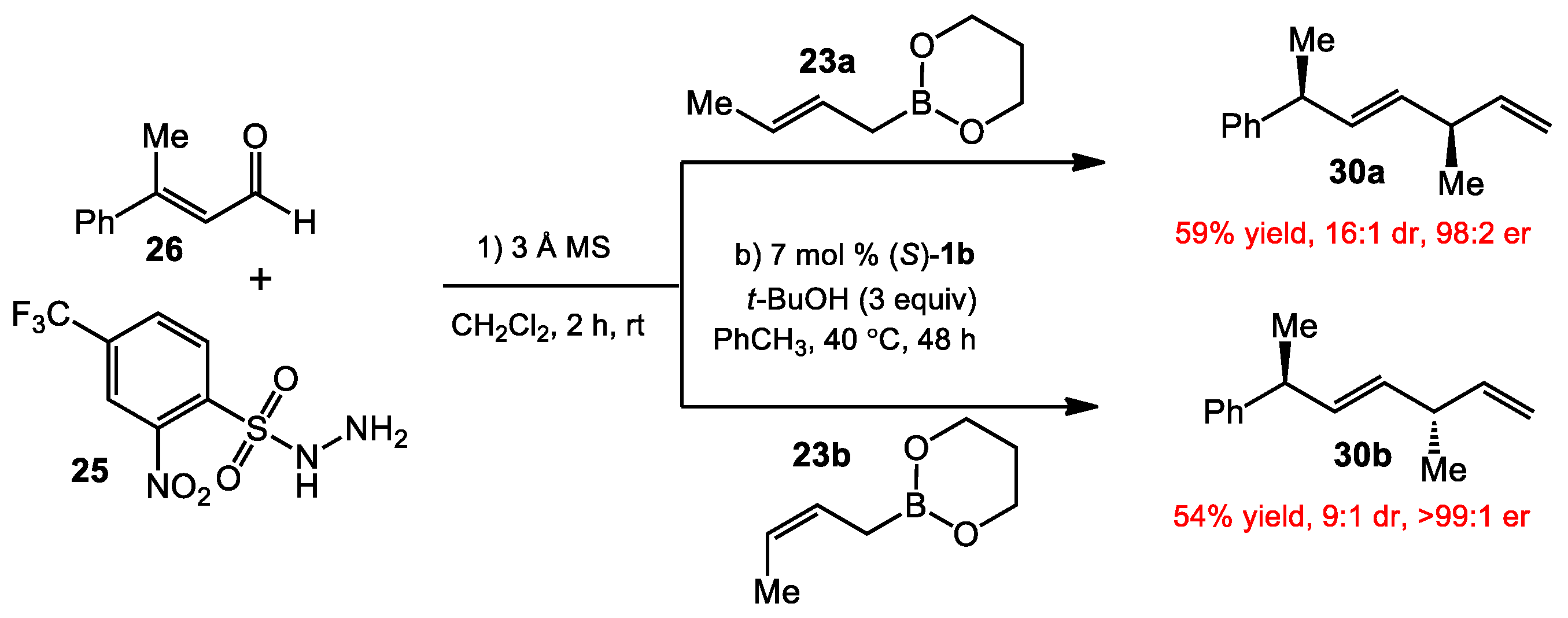{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
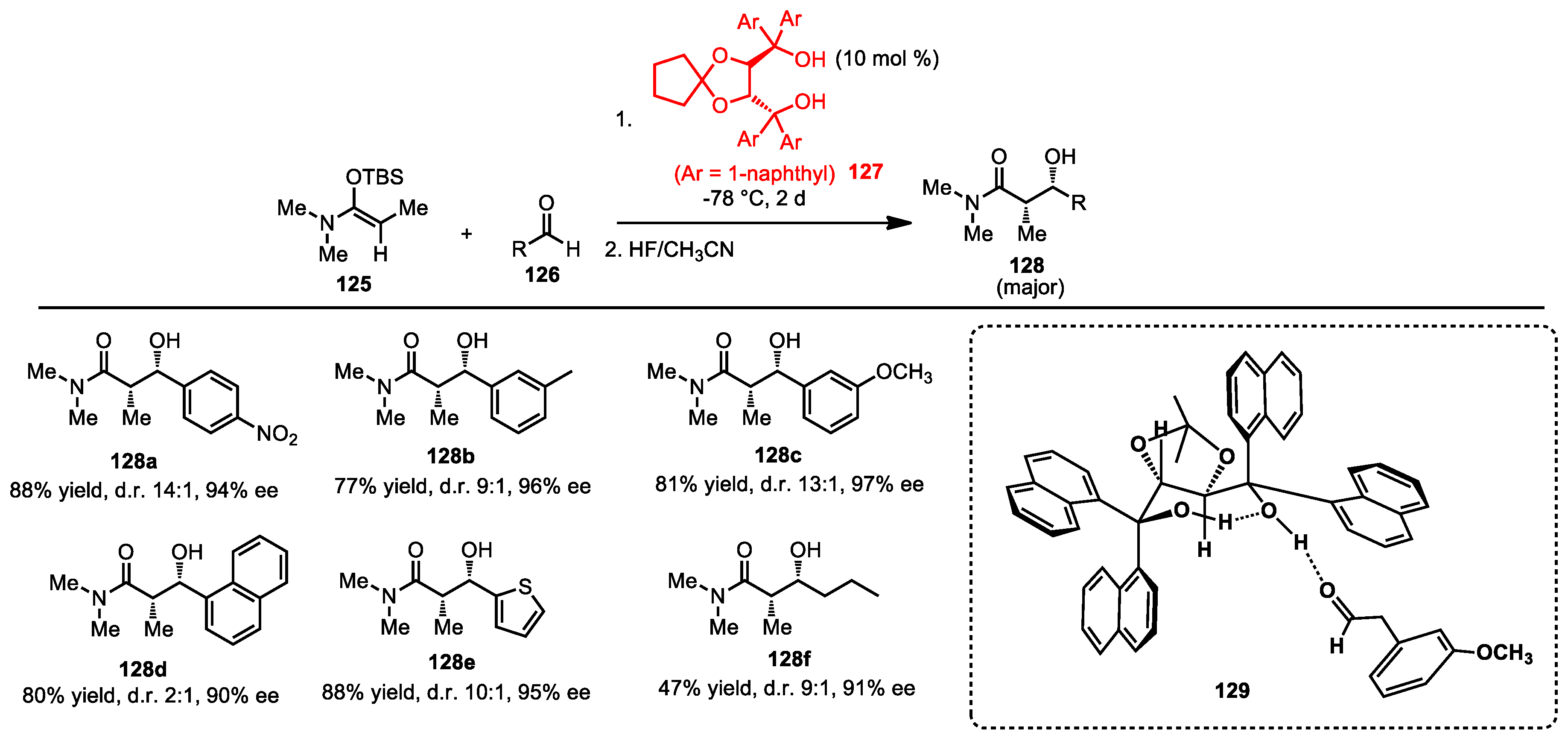{kind=link}
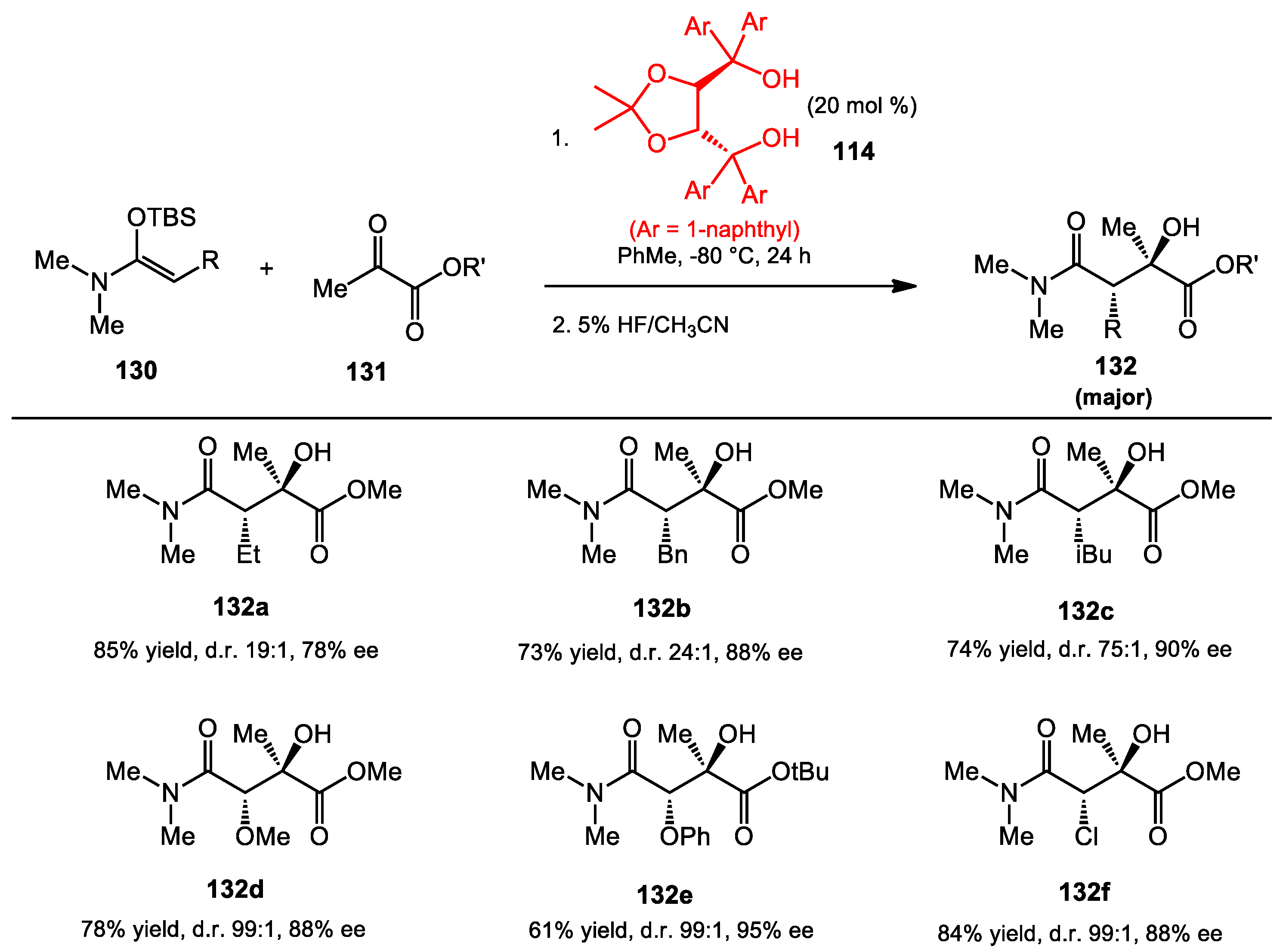{kind=link}
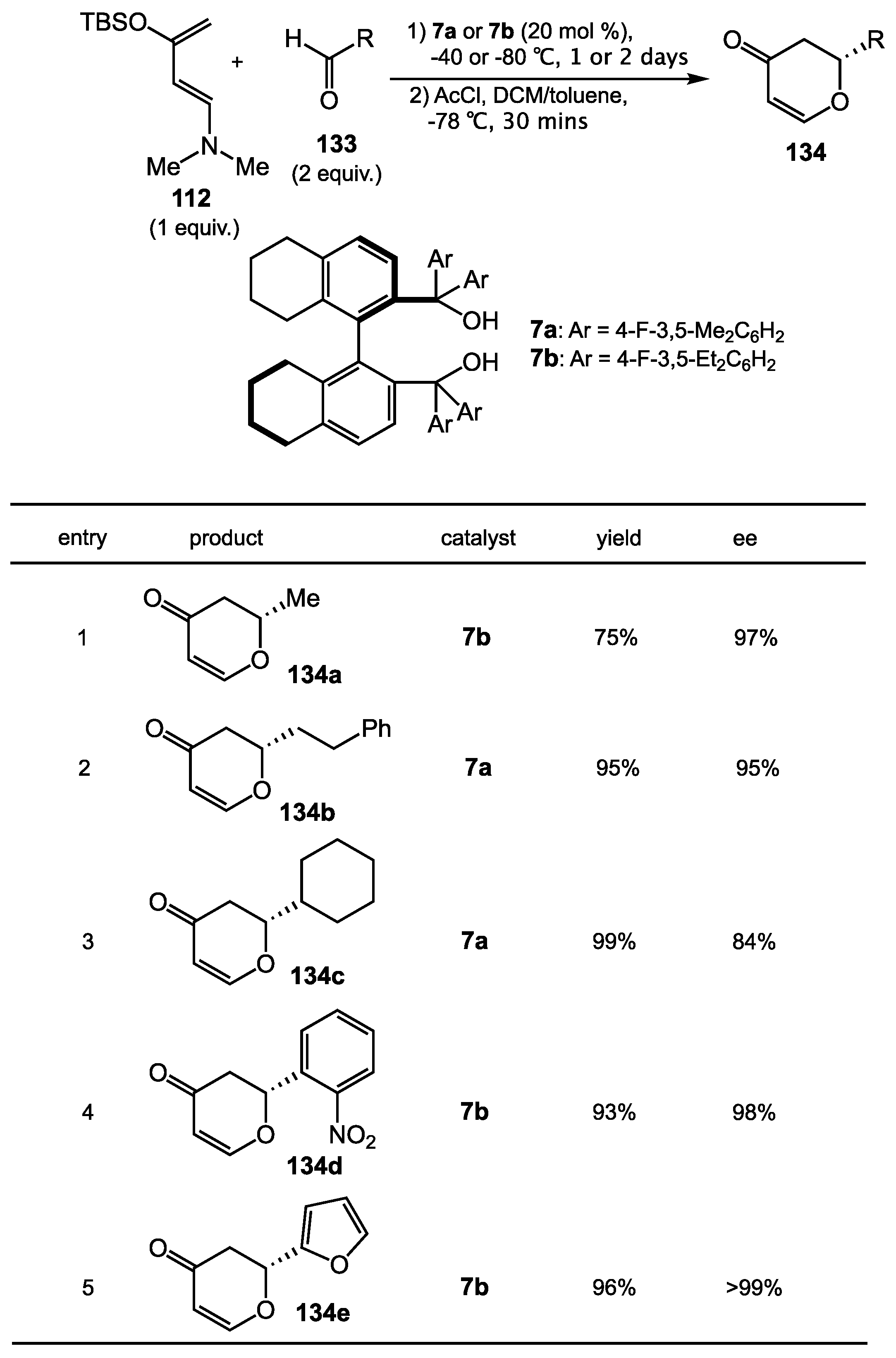{kind=link}
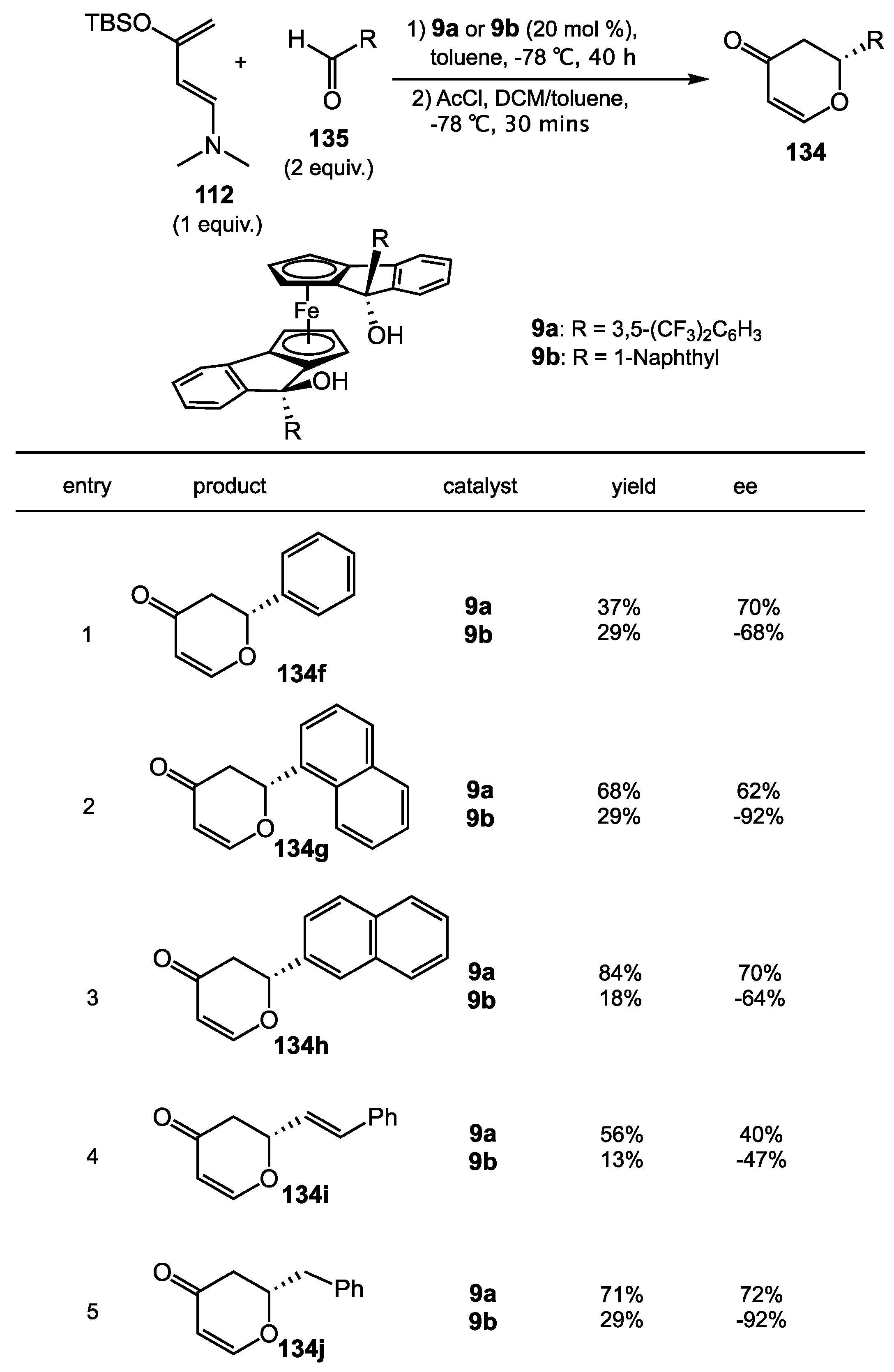{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
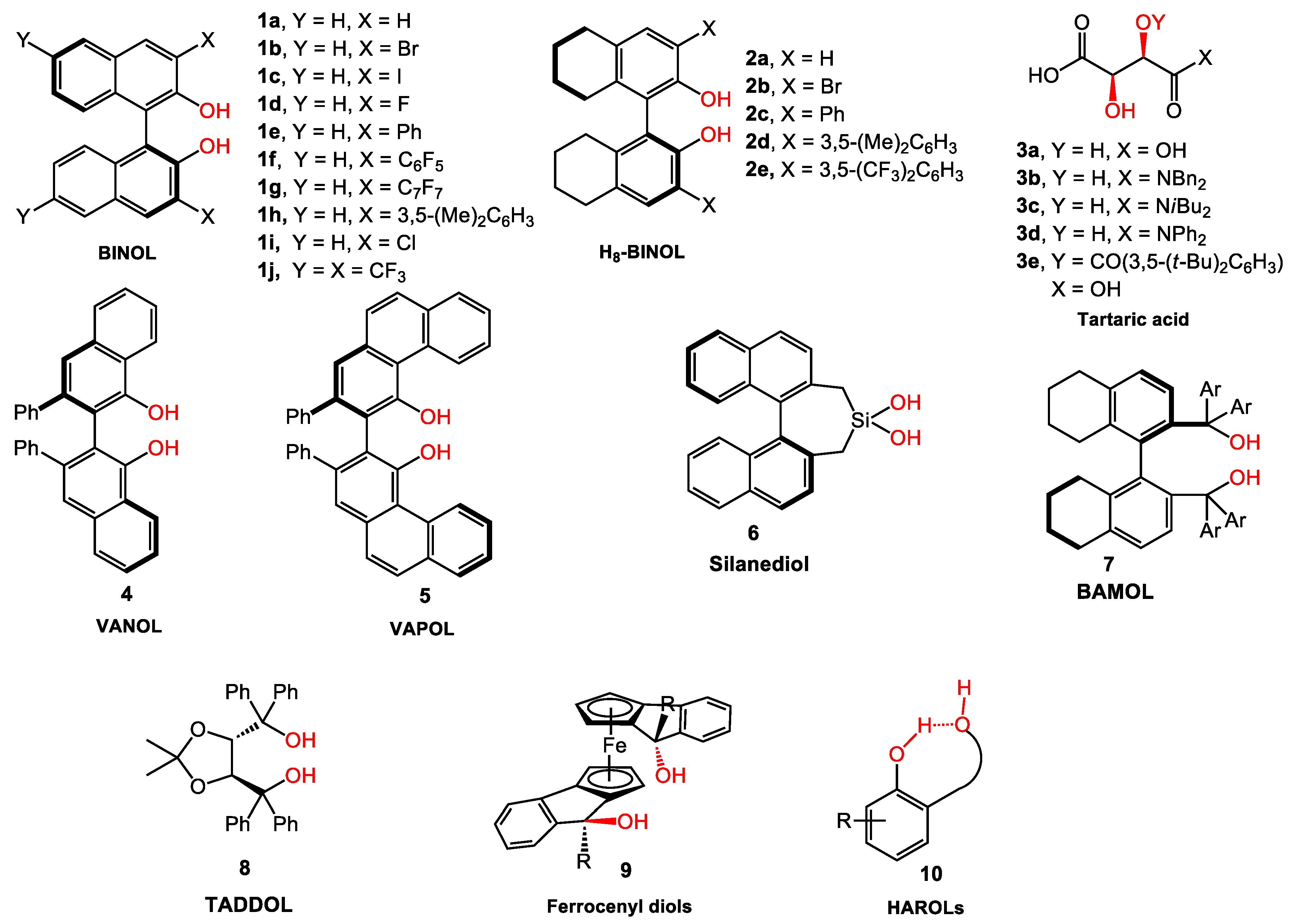
2. BINOL Derivatives
2.1. Allylboration
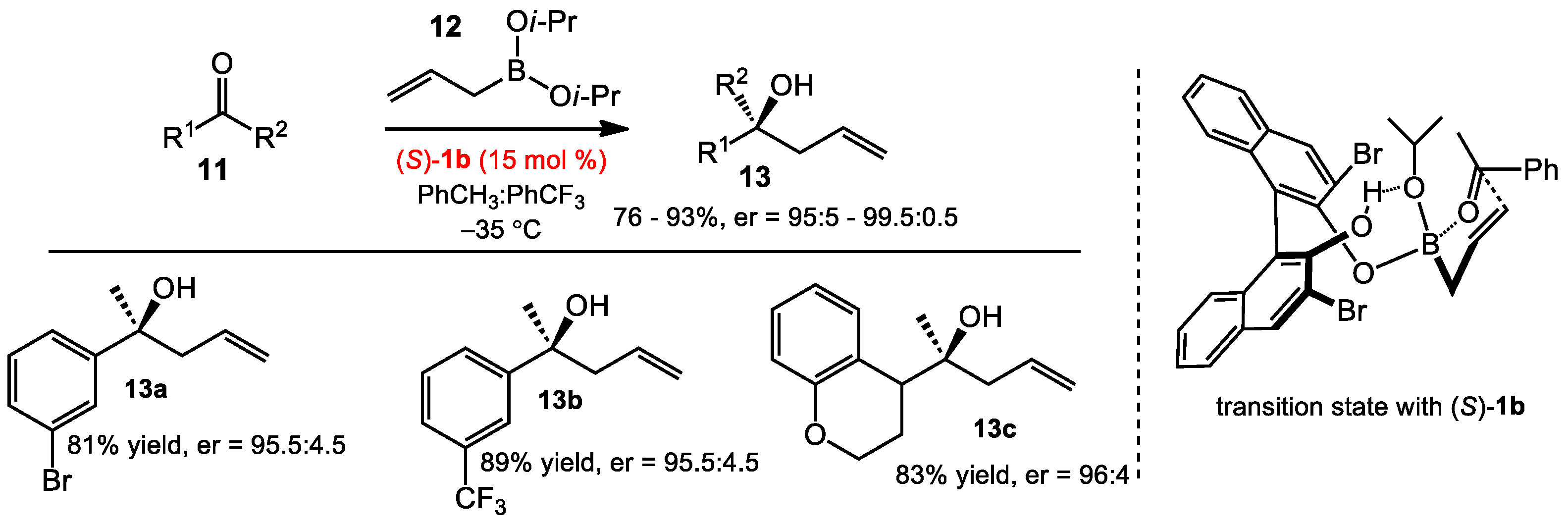
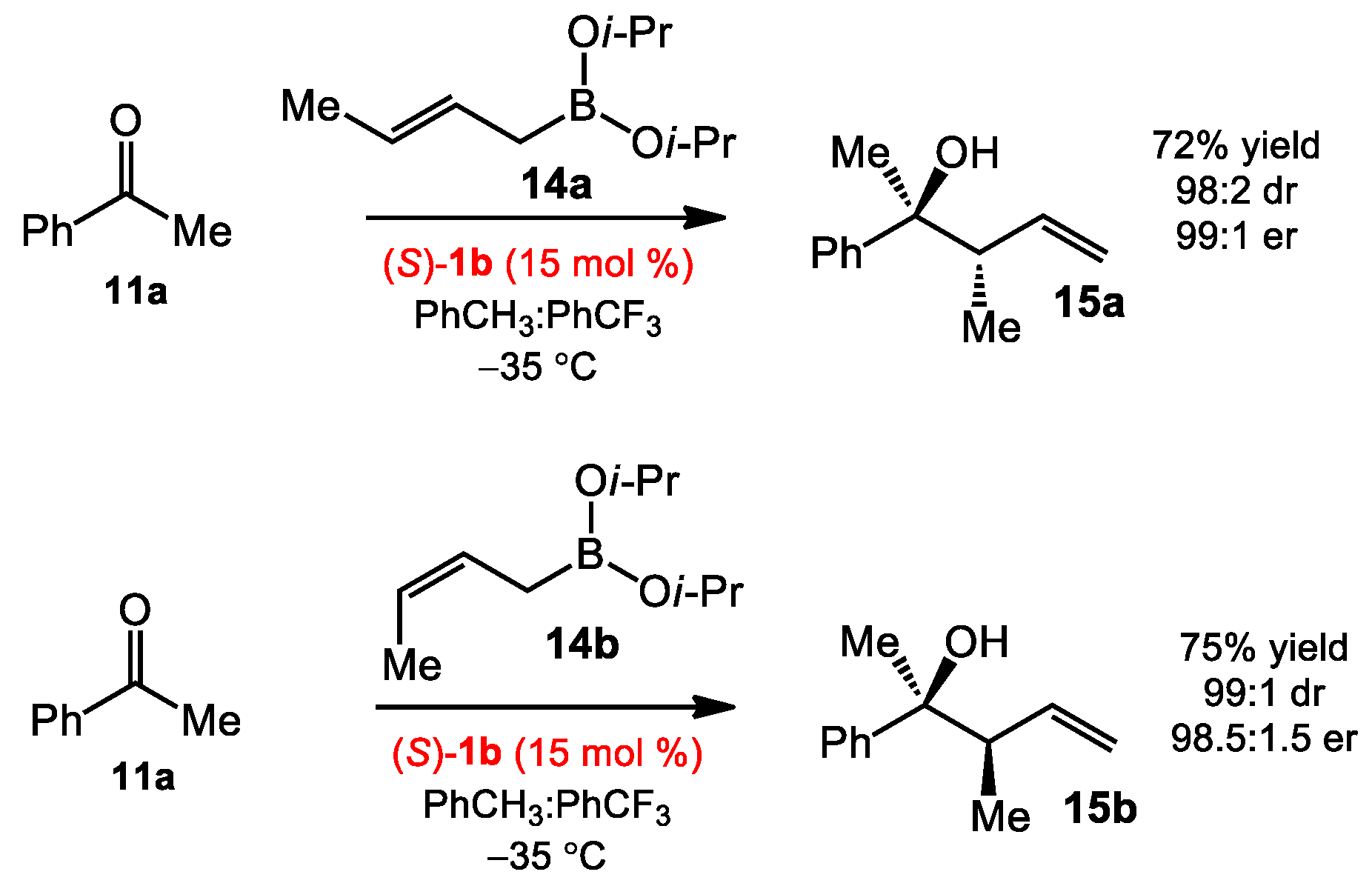
2.1.1. Allyboration/Crotylboration of Ketones
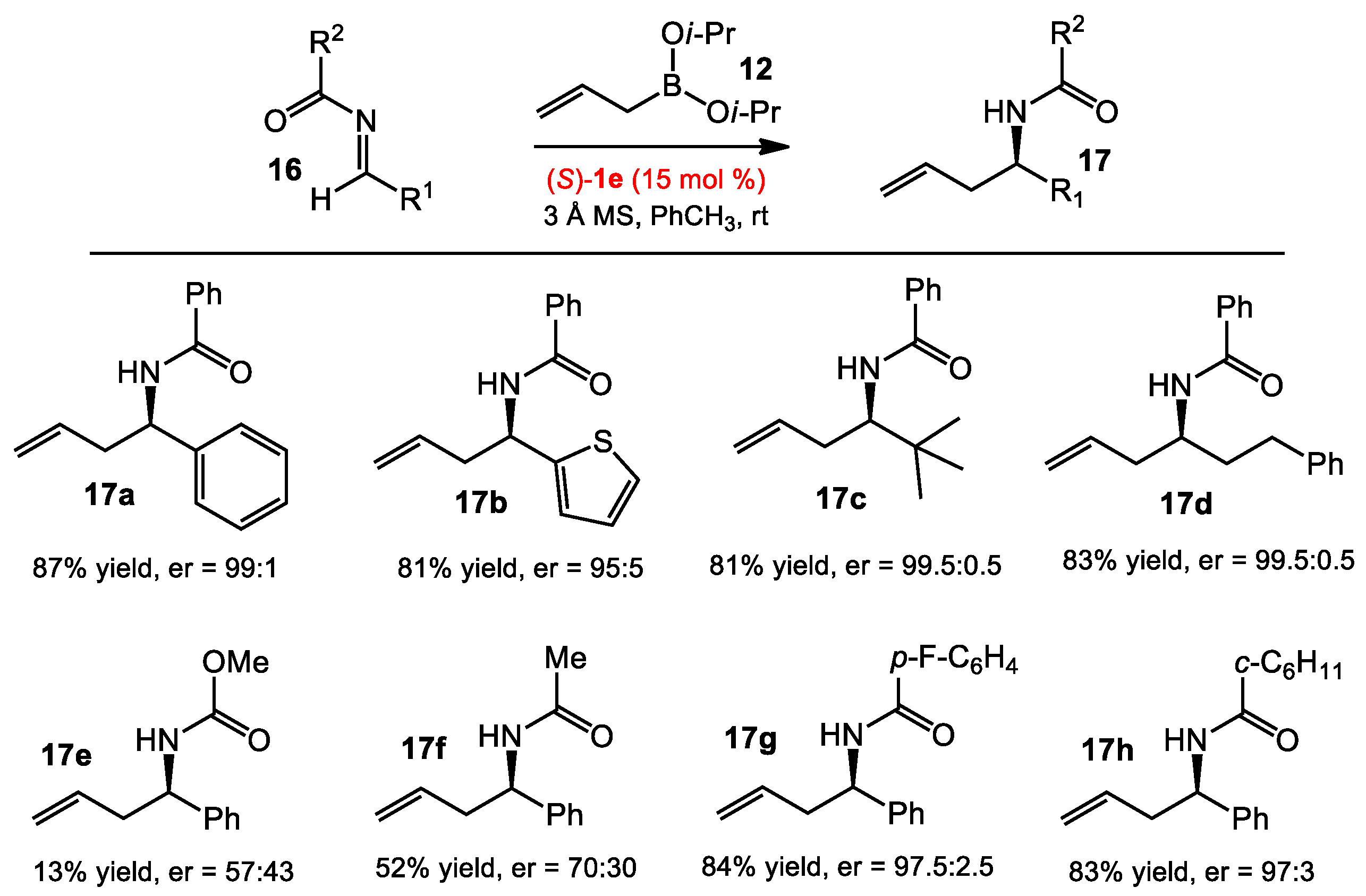
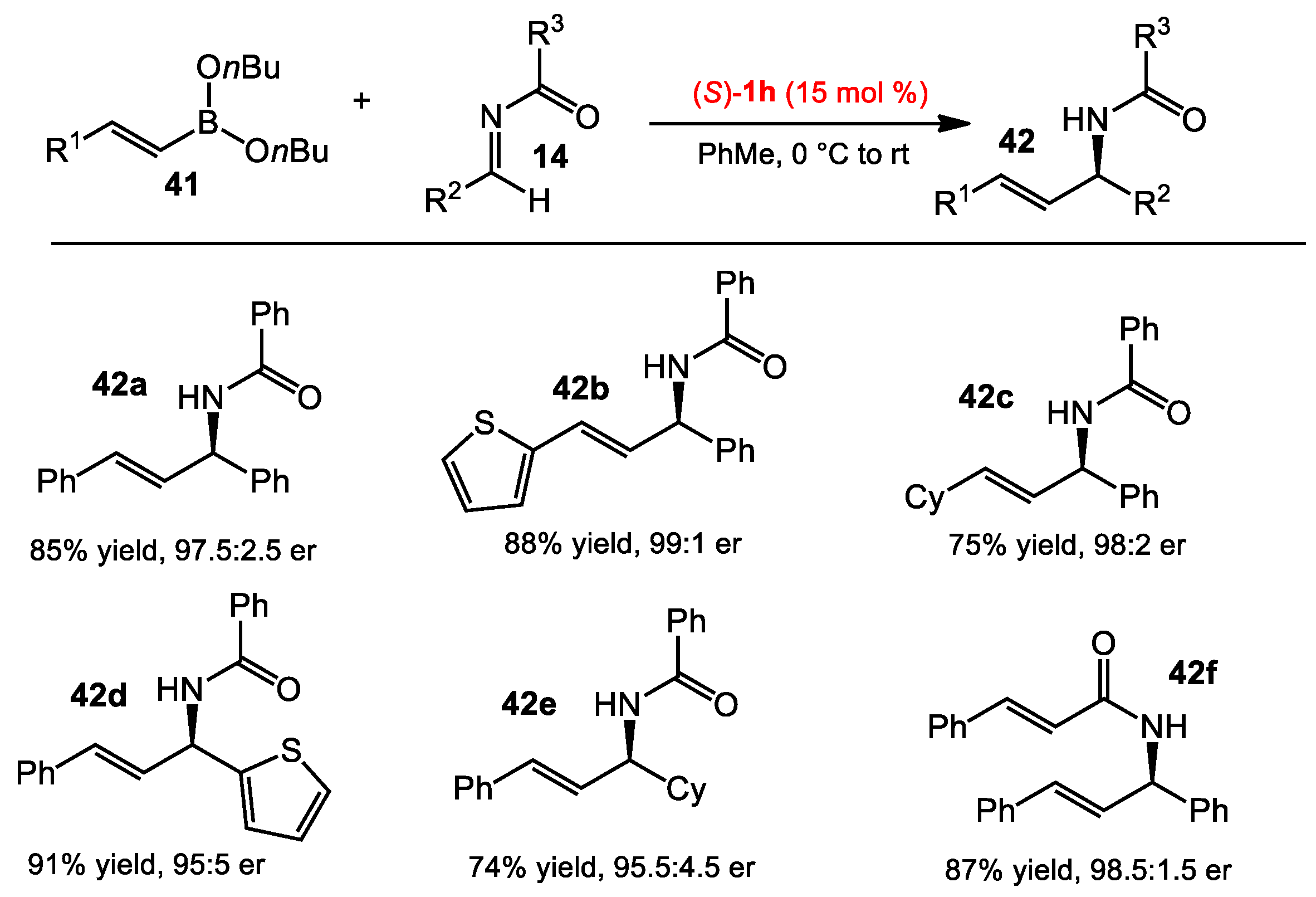
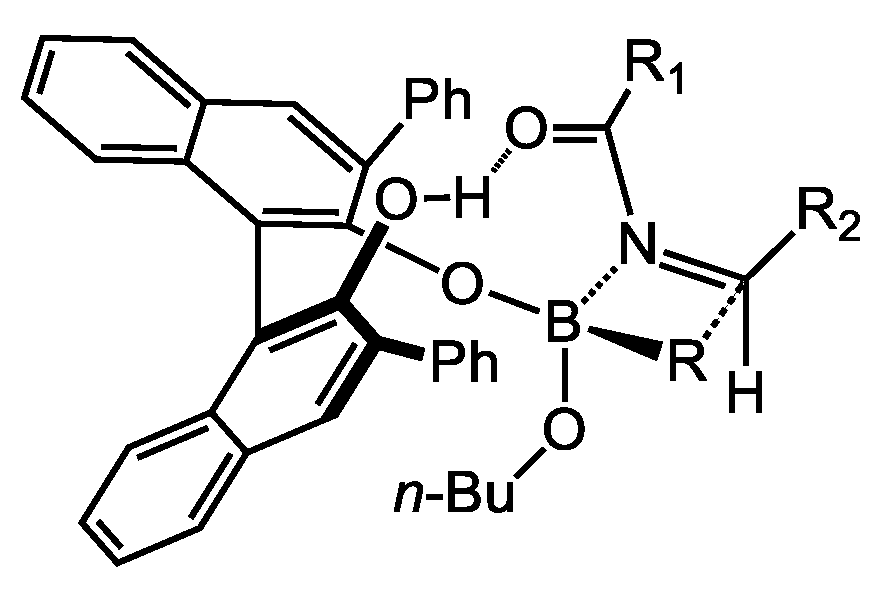
2.1.2. Allyboration/Crotylboration of Preformed Acyl Imines
2.1.3. One-Pot Allyboration/Crotylboration of Acyl Imines Derived from Free Aldehydes
2.1.4. Traceless Petasis Borono–Mannich Allylation/Crotylation
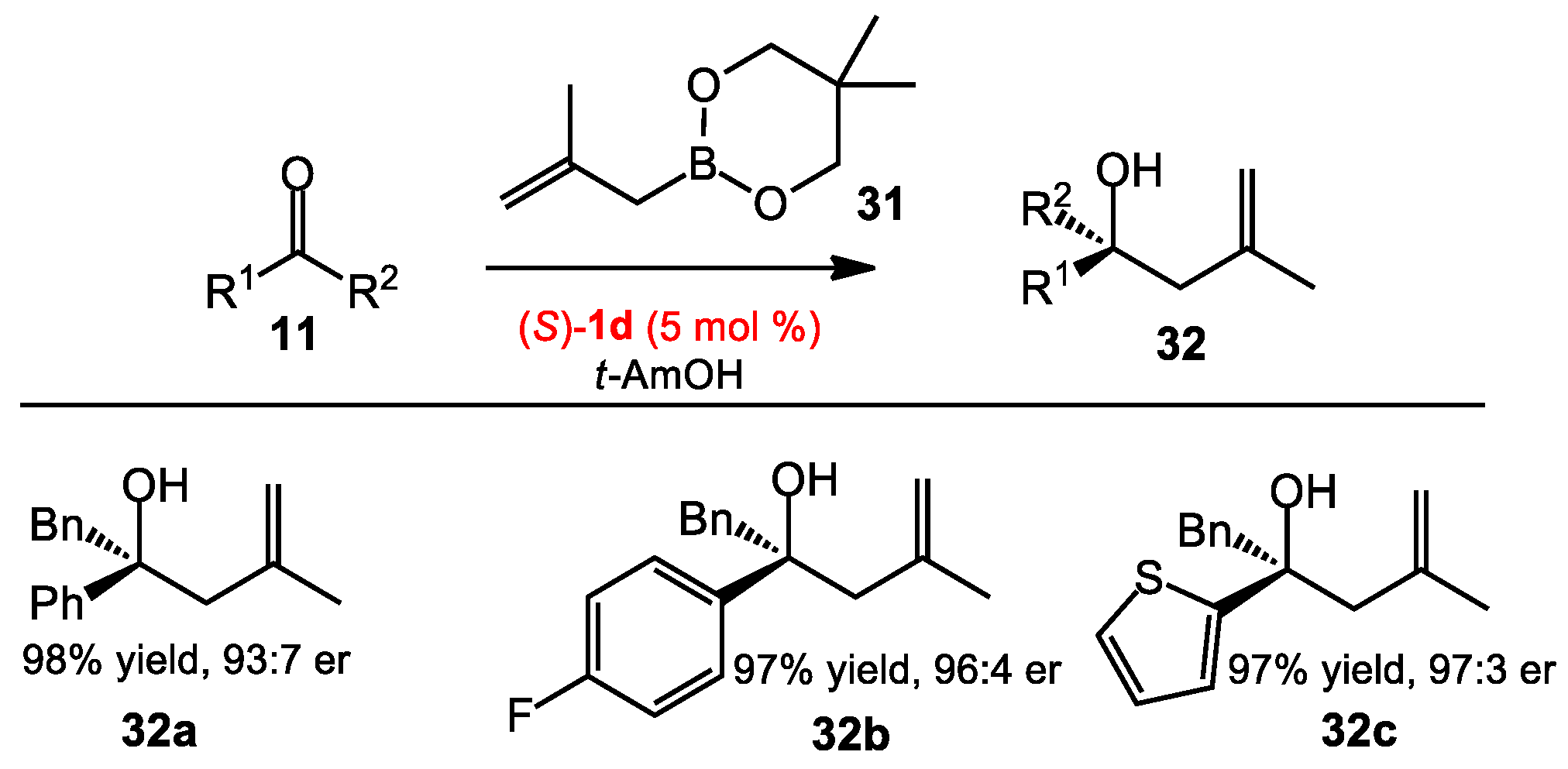
2.1.5. Methallylation
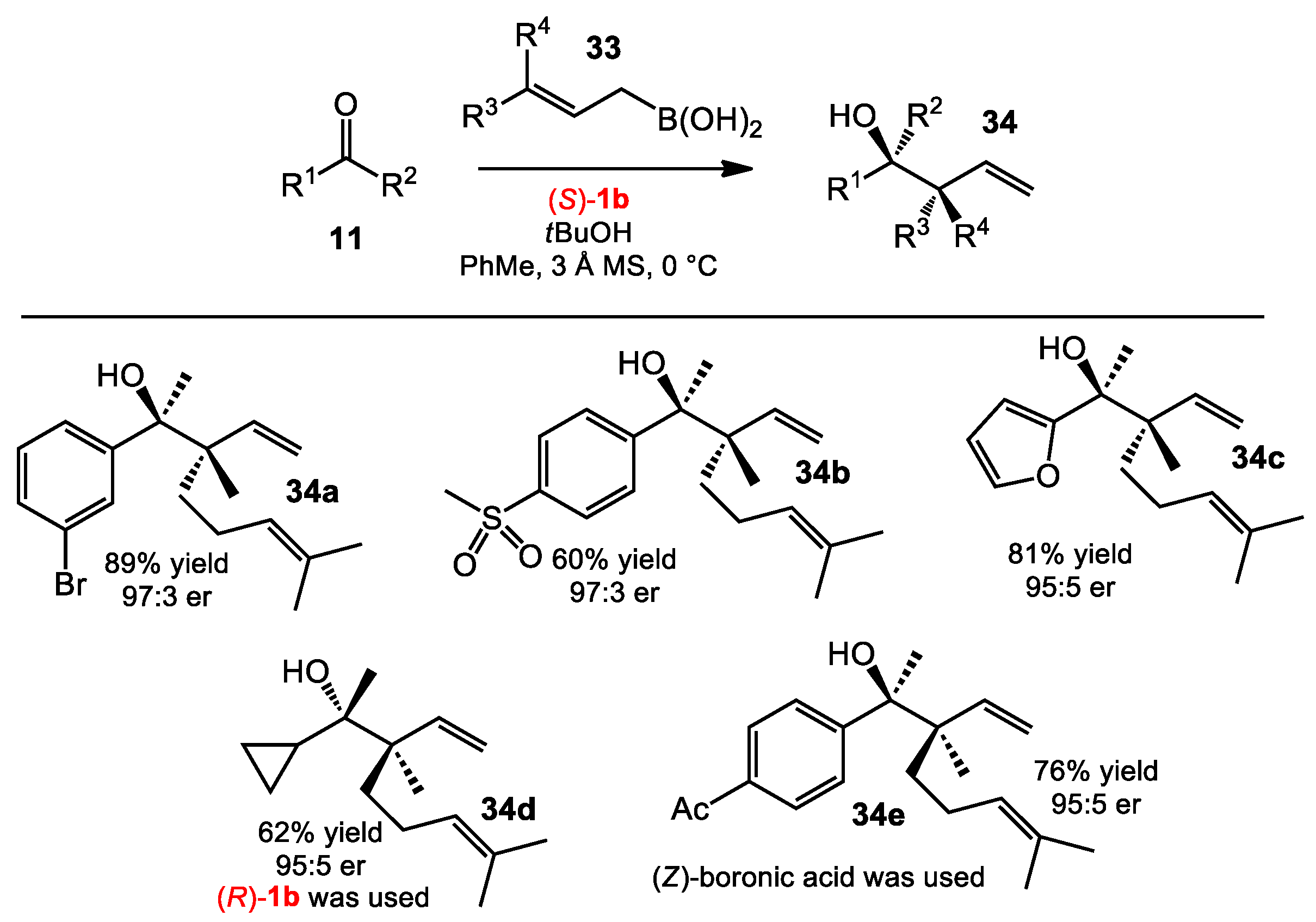
2.1.6. Allylboration of Ketones with γ-Disubstituted Allylboronic Acids
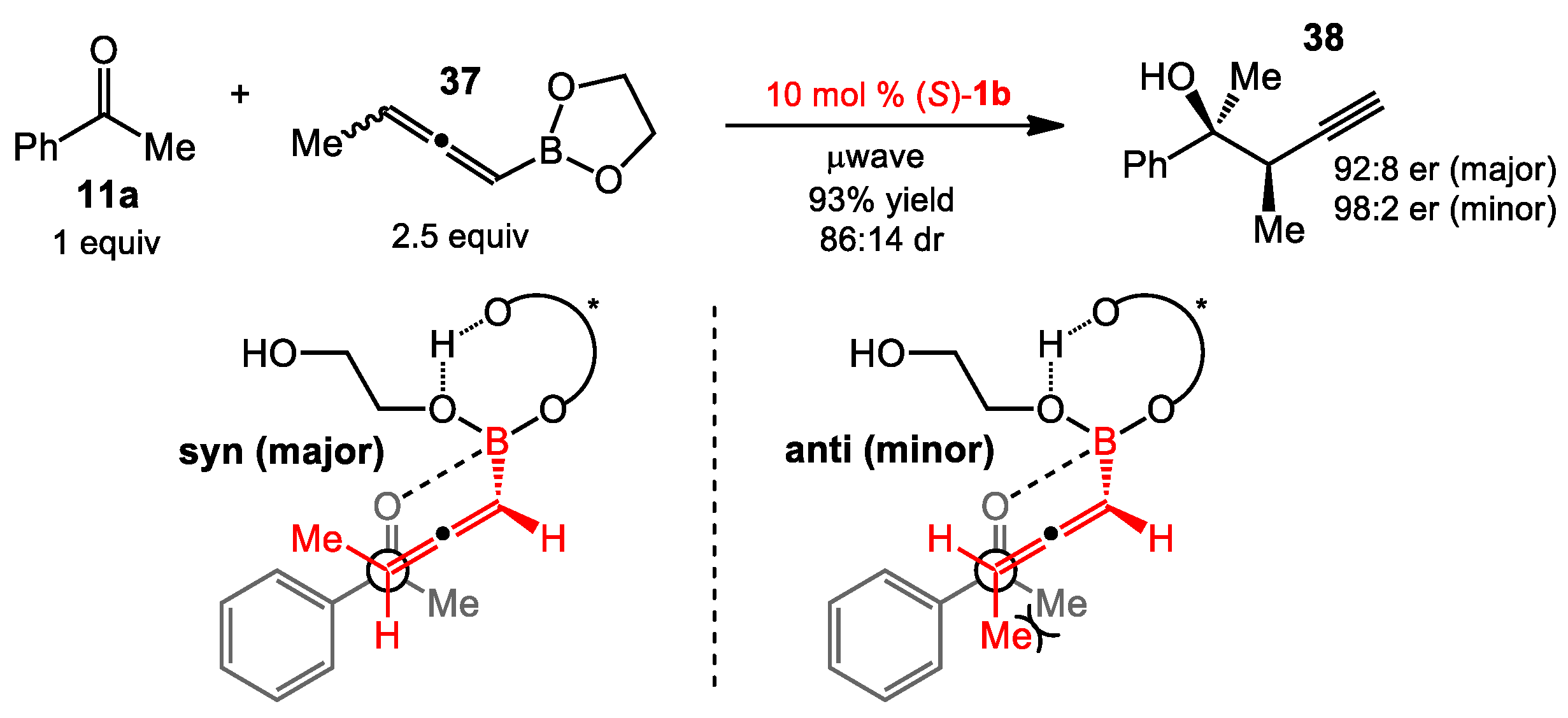
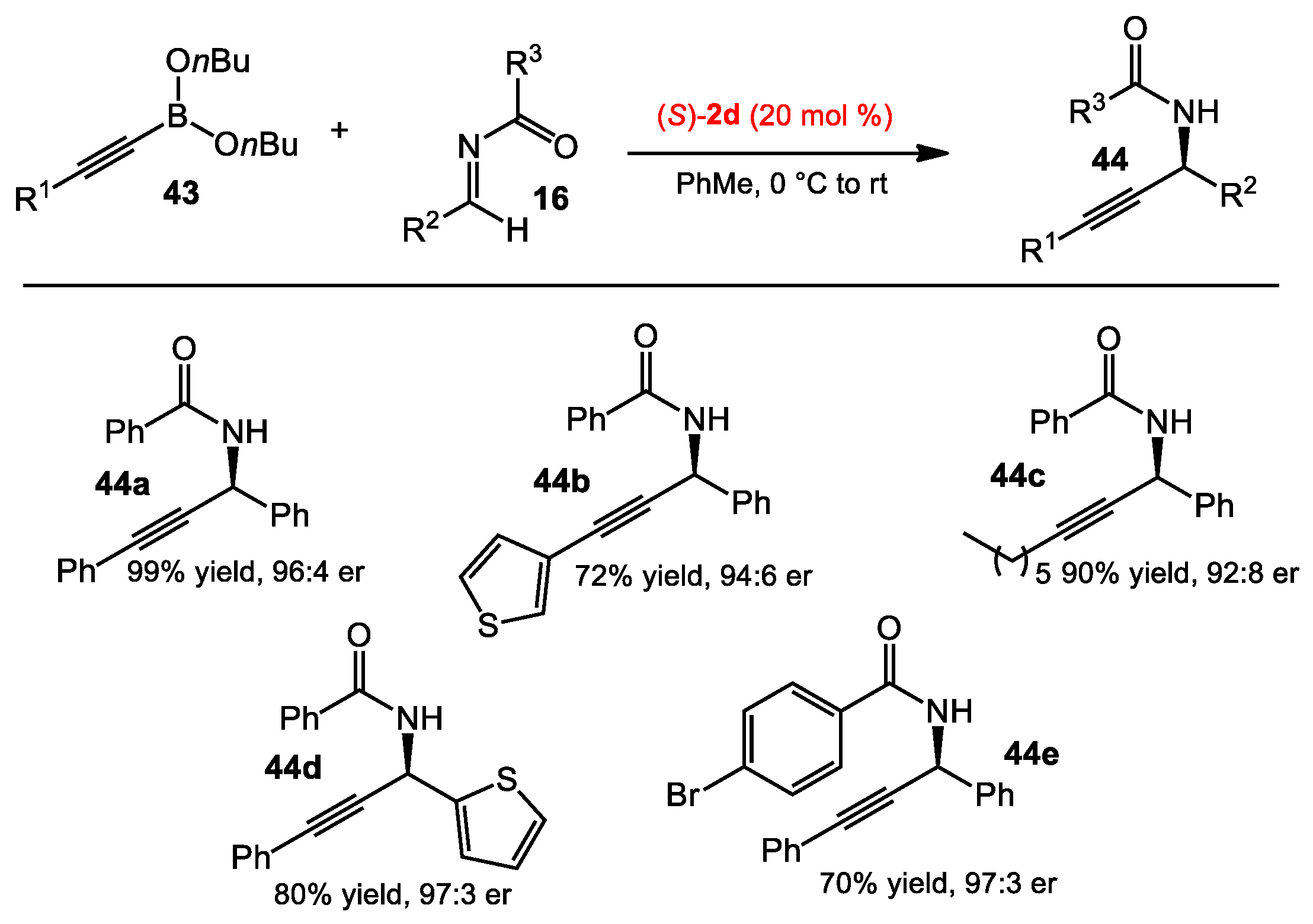
2.2. Propargylation
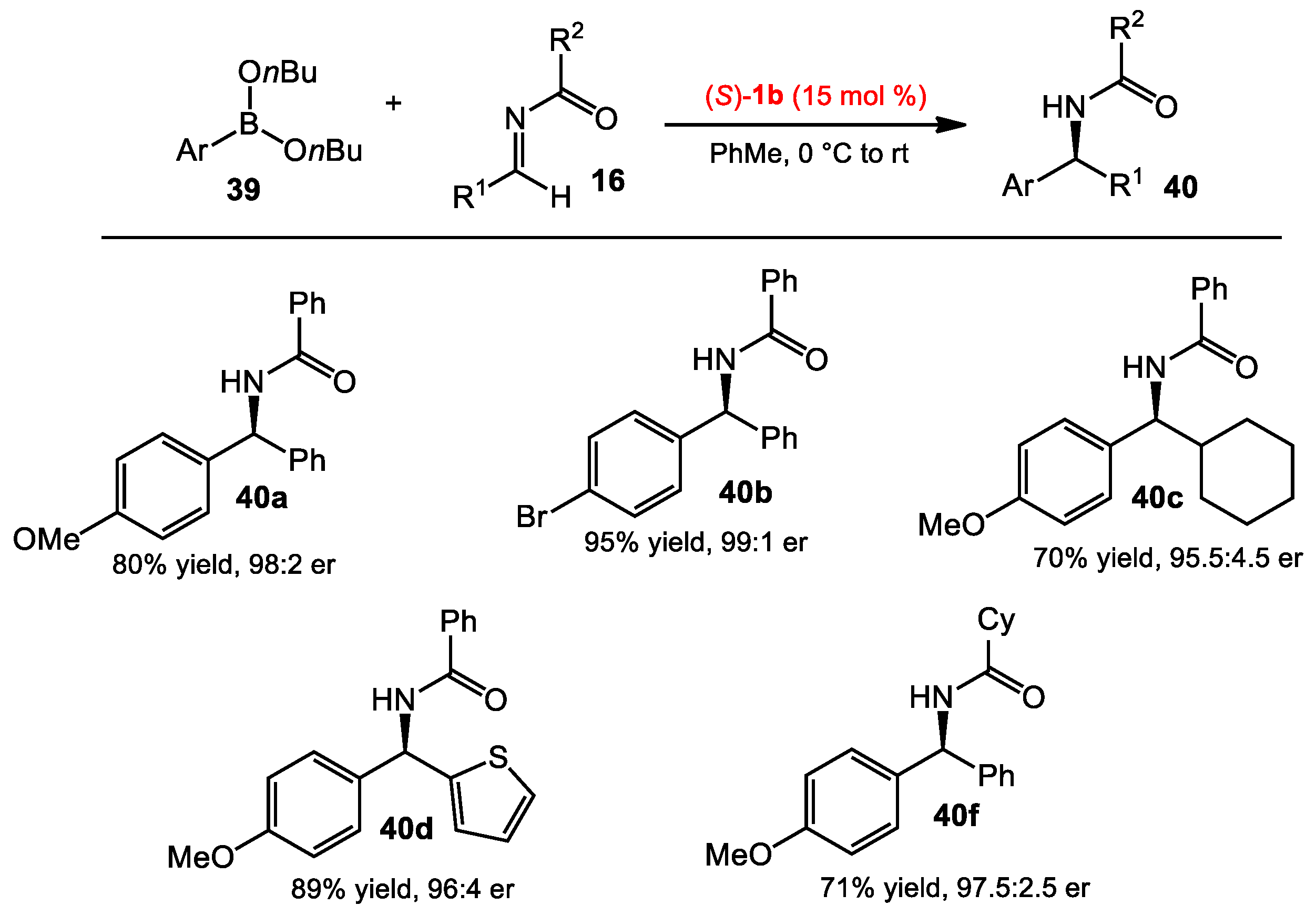
2.3. Addition of Organoboronates to Acyl Imines
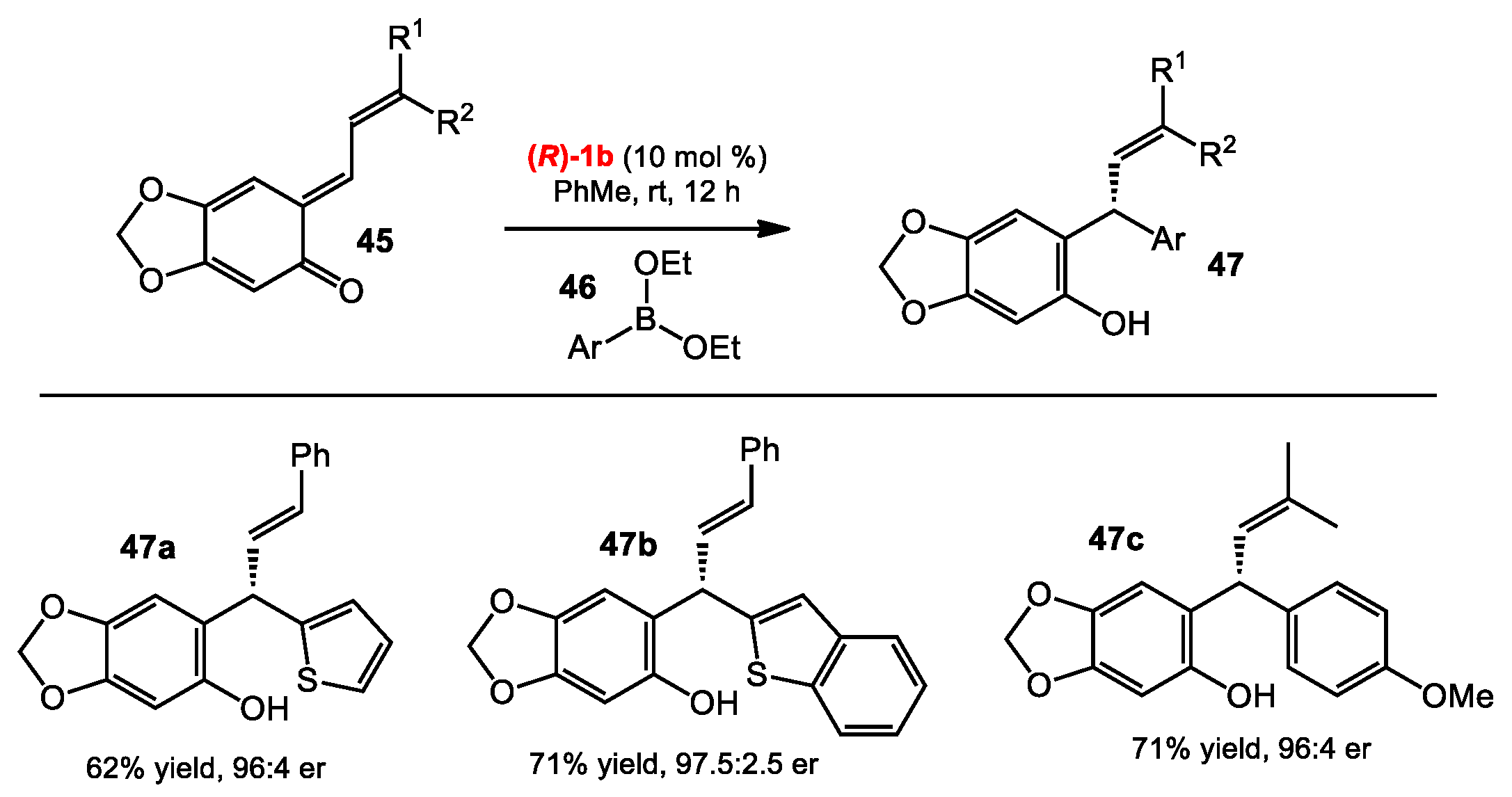
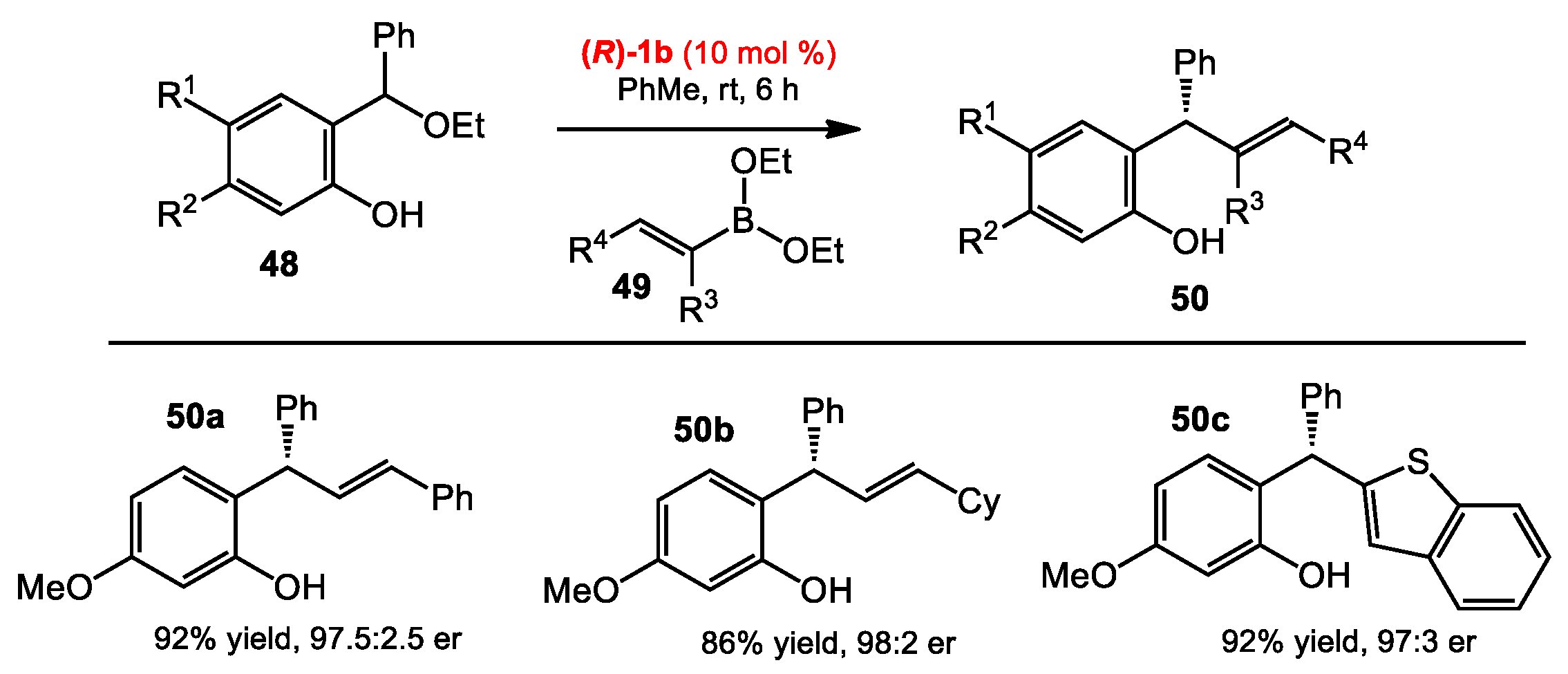
2.4. Addition of Boronates to o-Quinone Methides
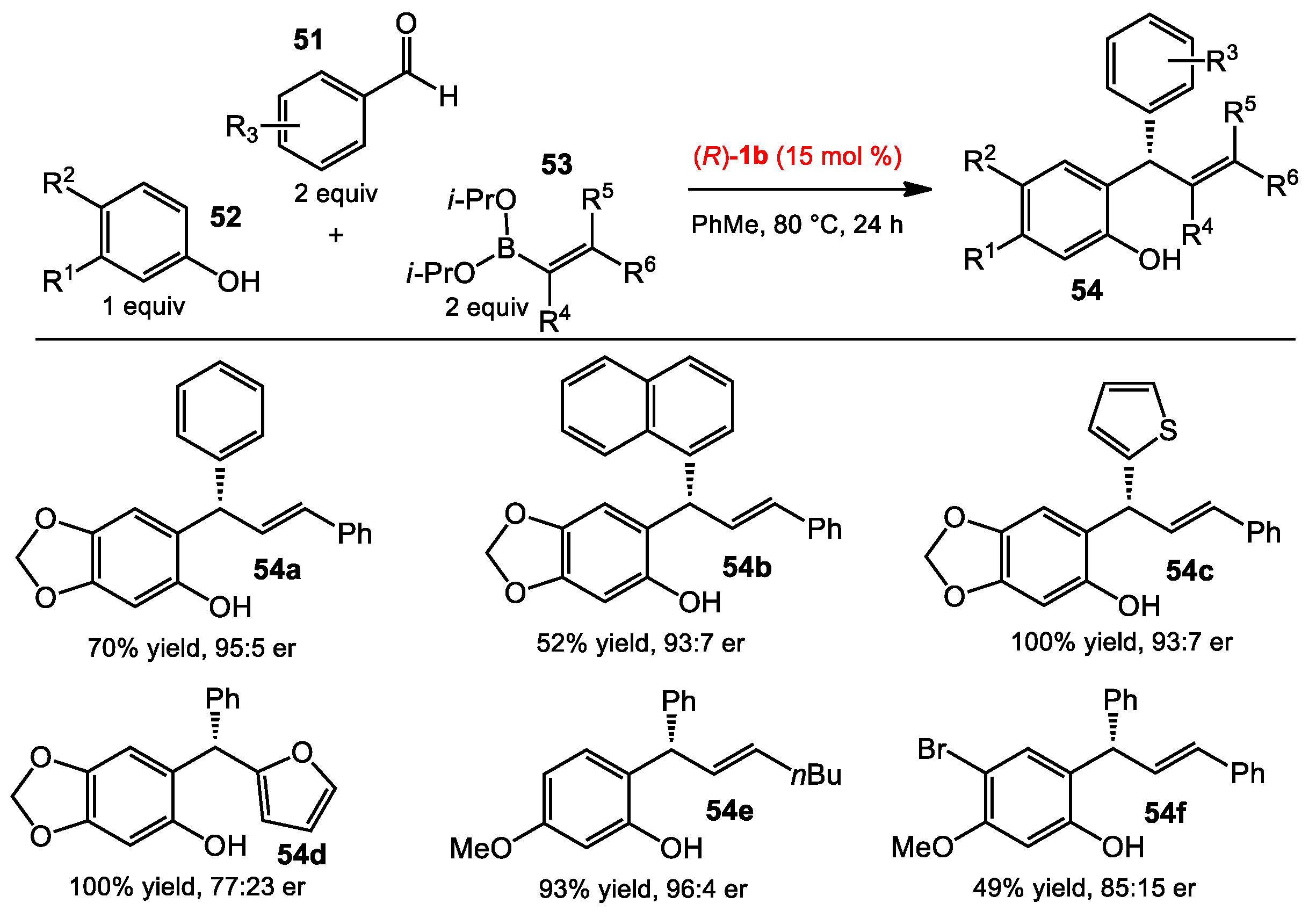
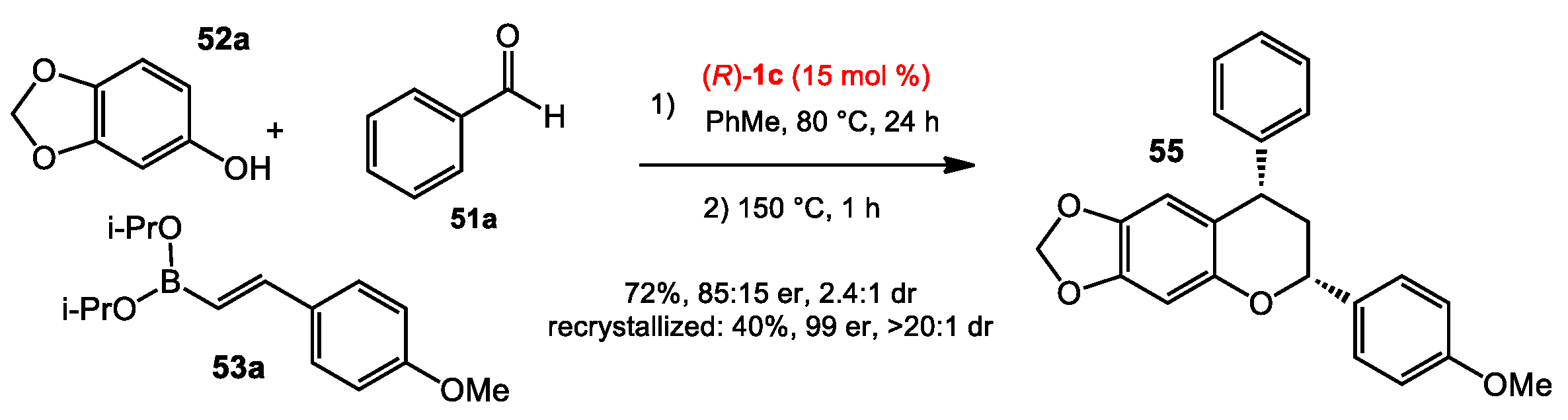
2.5. Multicomponent Quinone Methide Condensation
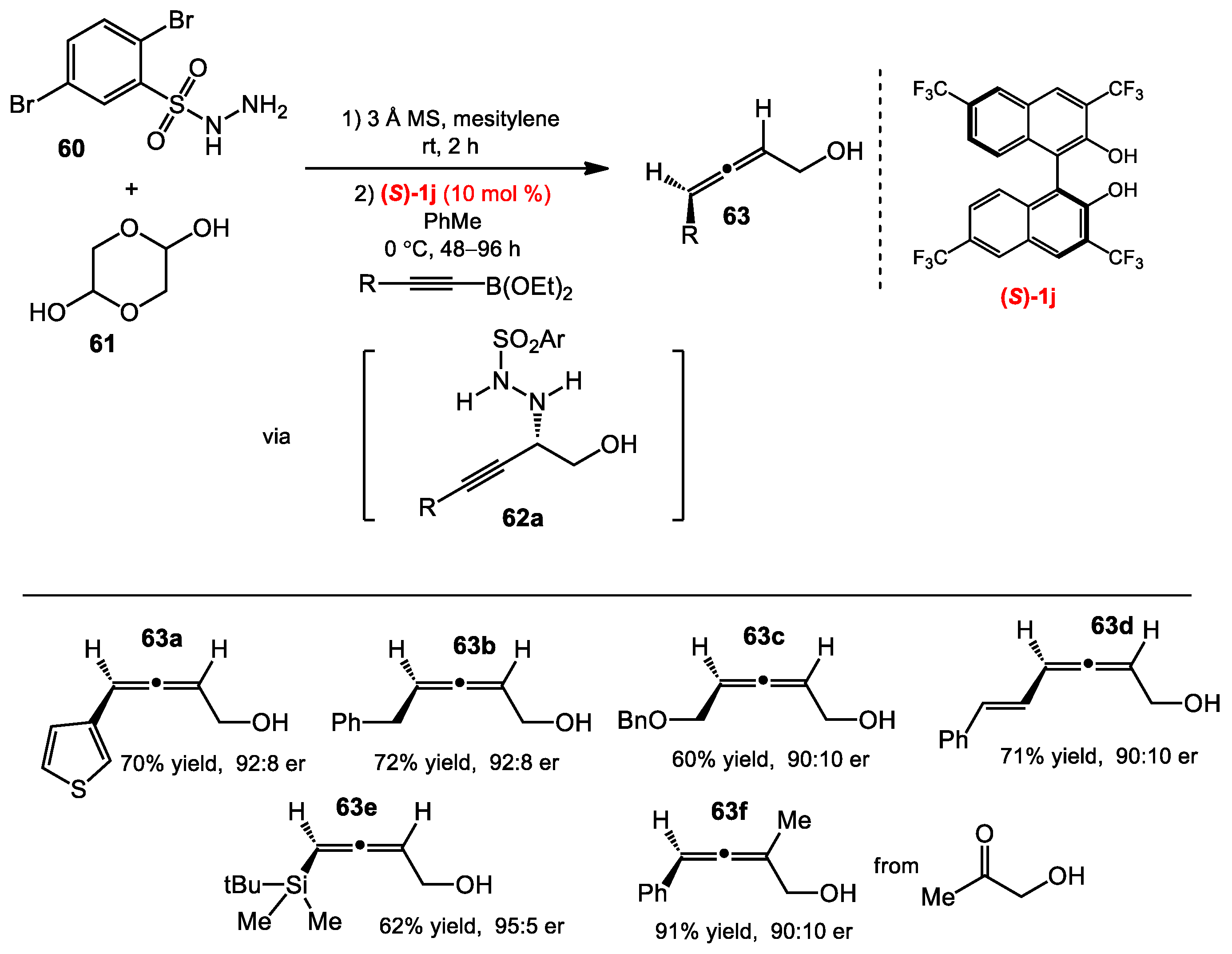
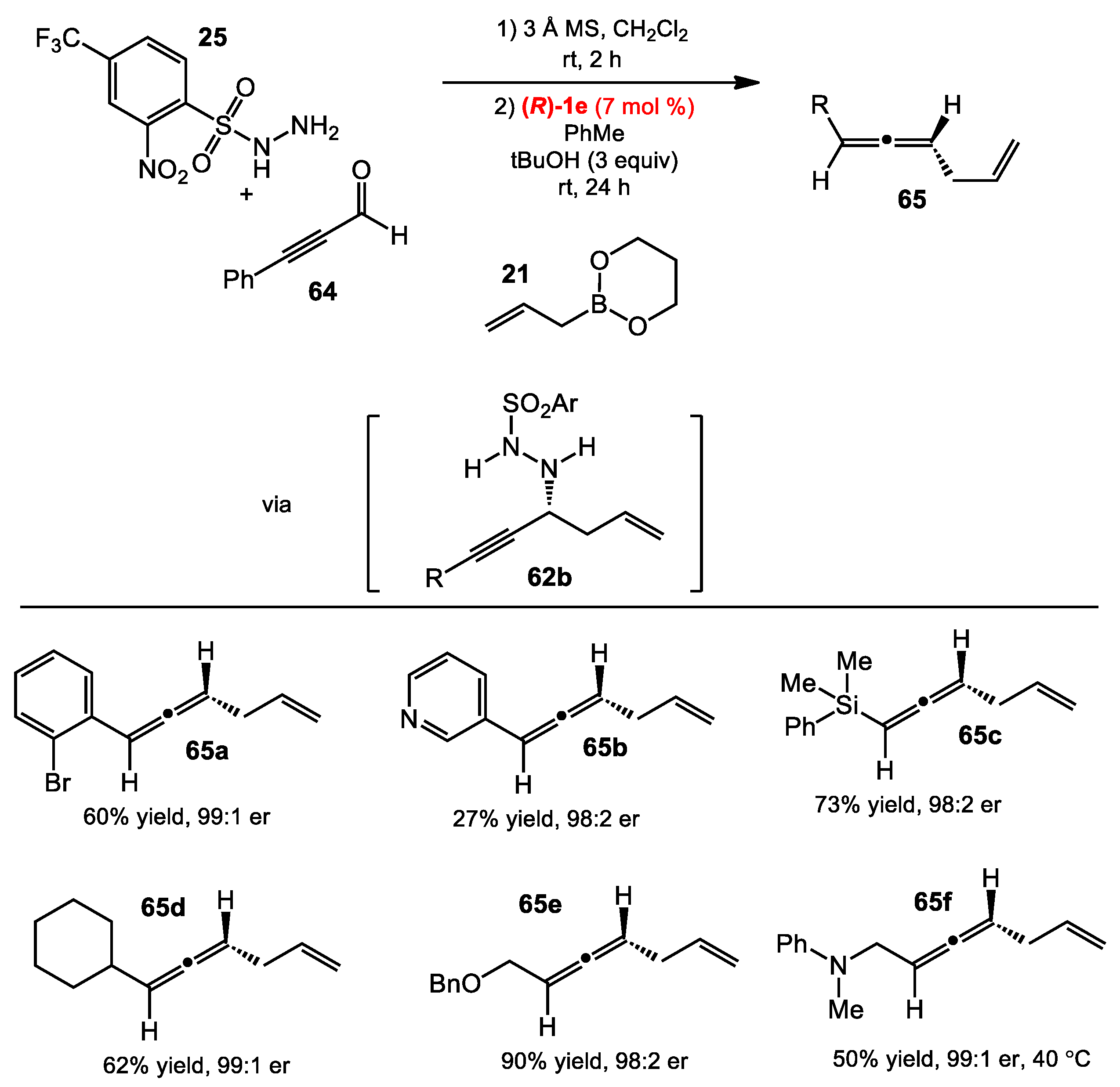
2.6. Allene Formation via Allylboration and Alkynylboration
2.7. Conjugate Addition
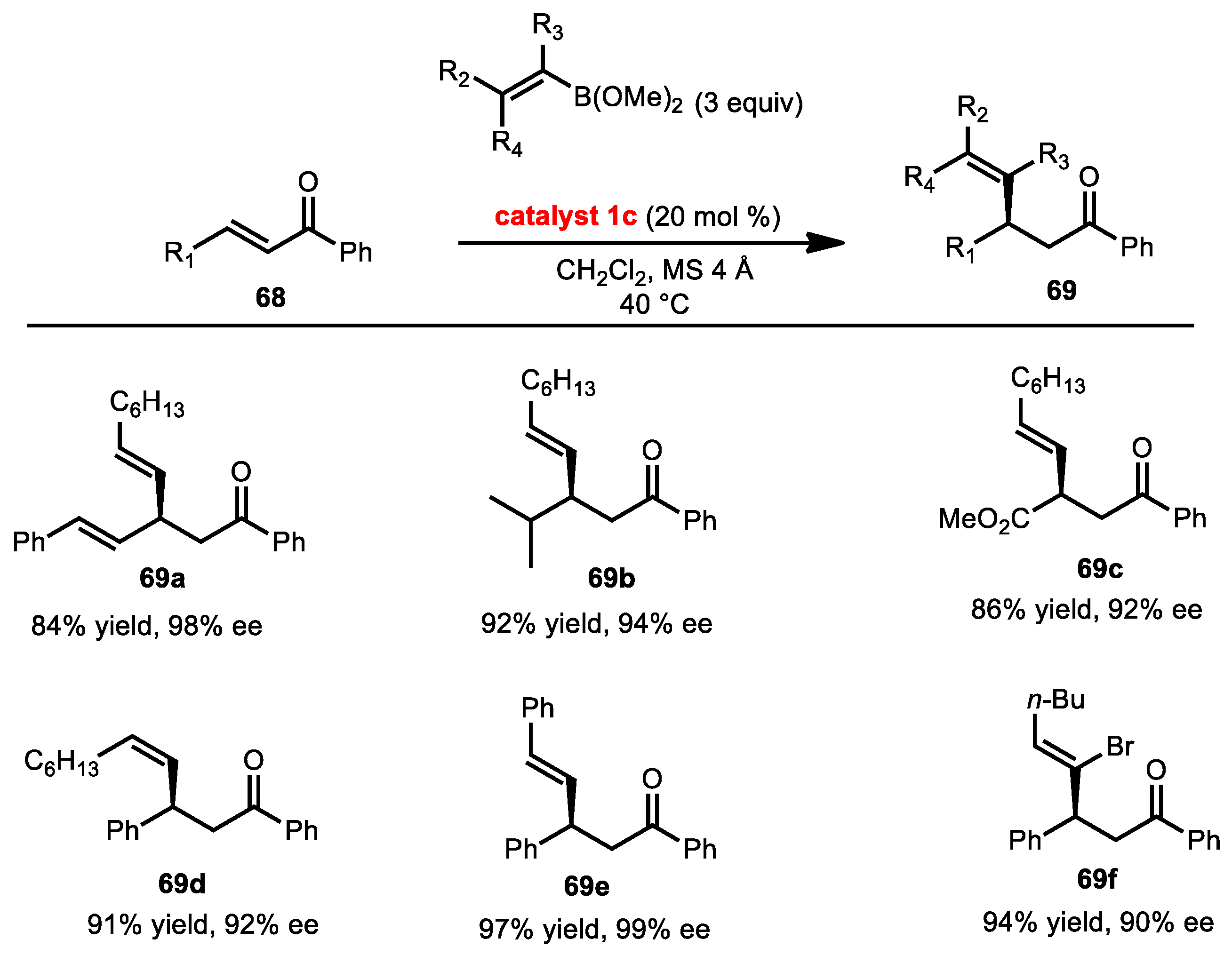
2.7.1. Conjugate Addition of Alkynyl Boronates
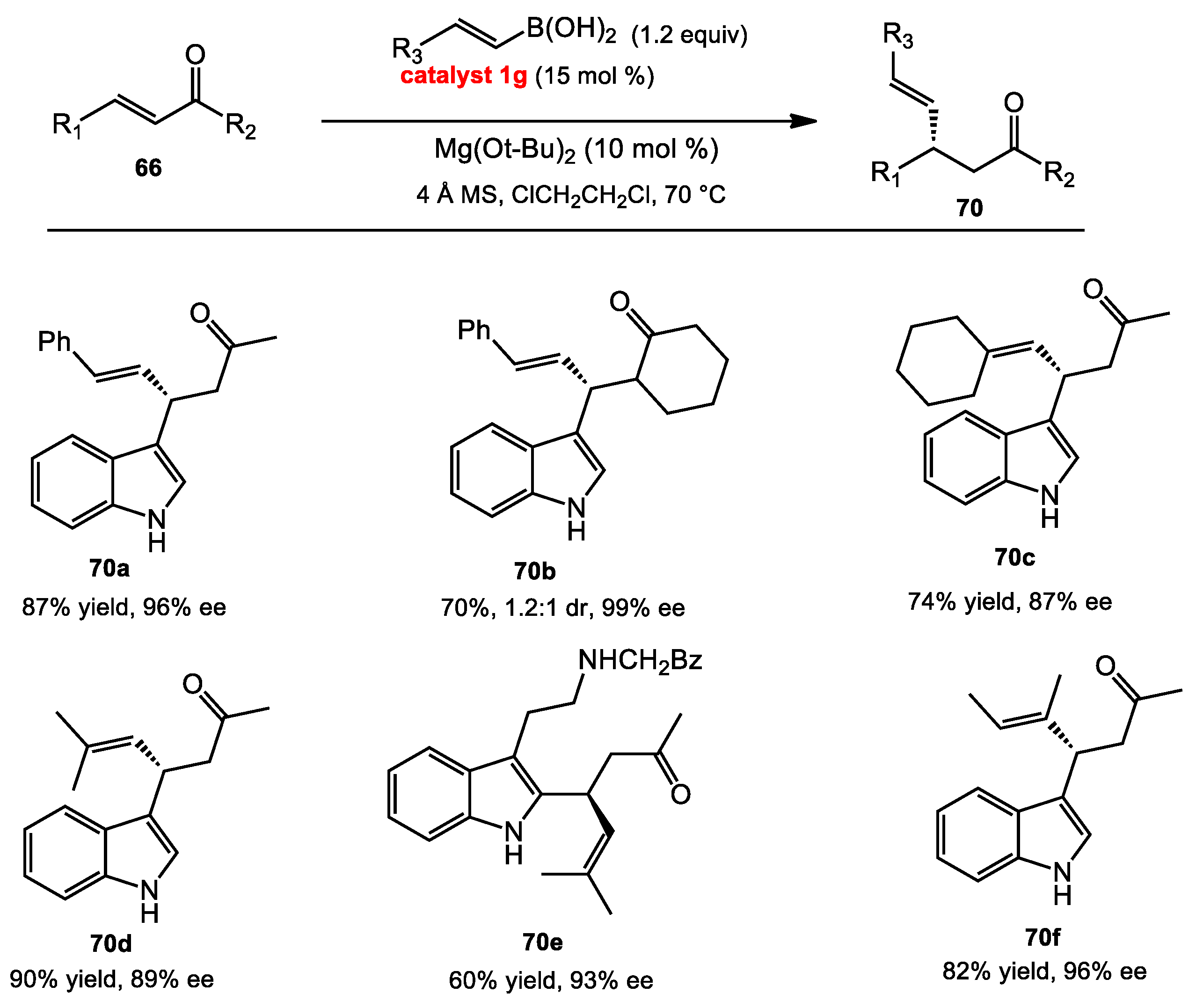
2.7.2. Conjugate Addition of Alkenyl Boronates
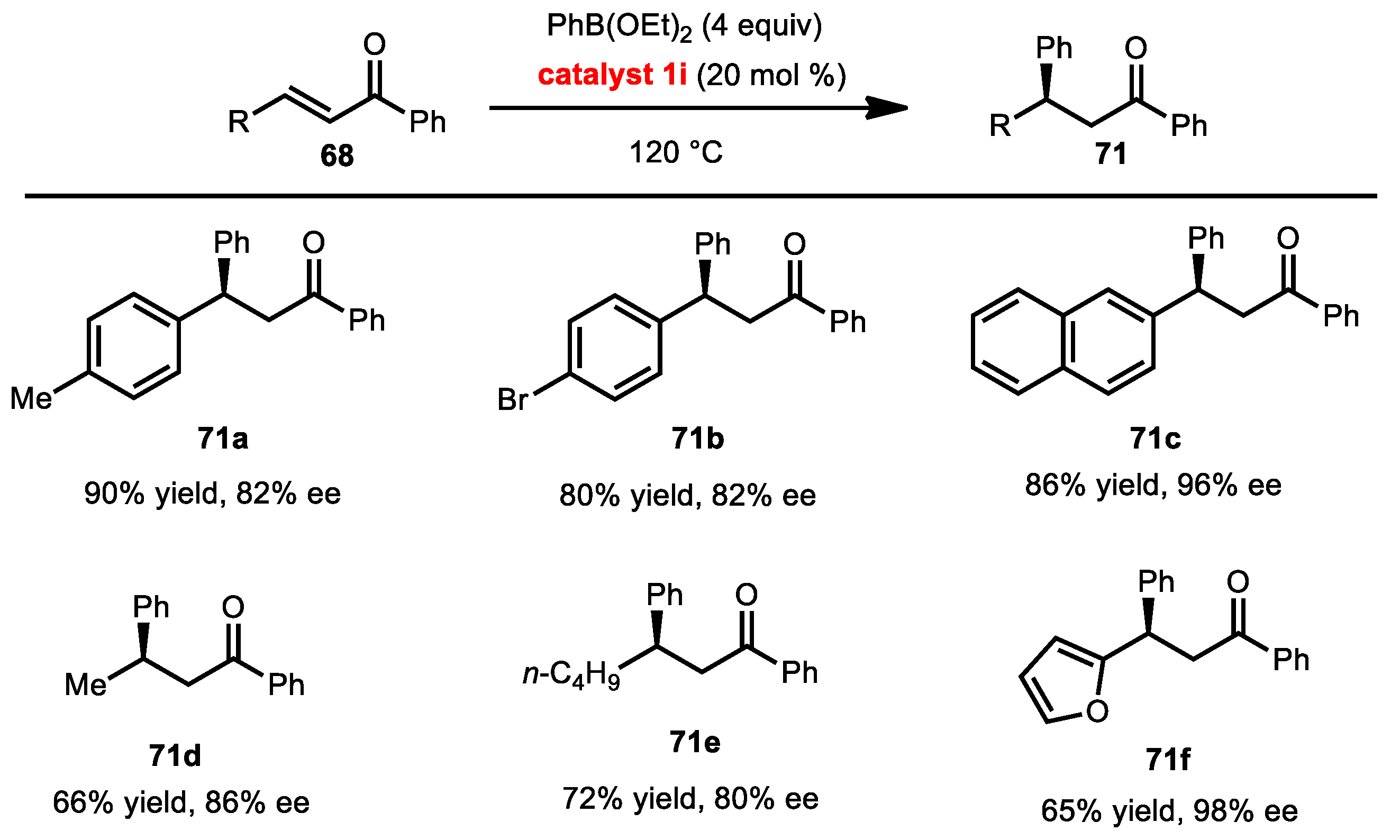
2.7.3. Conjugate Addition of Aryl Boronates
3. Tartaric Acid Derivatives
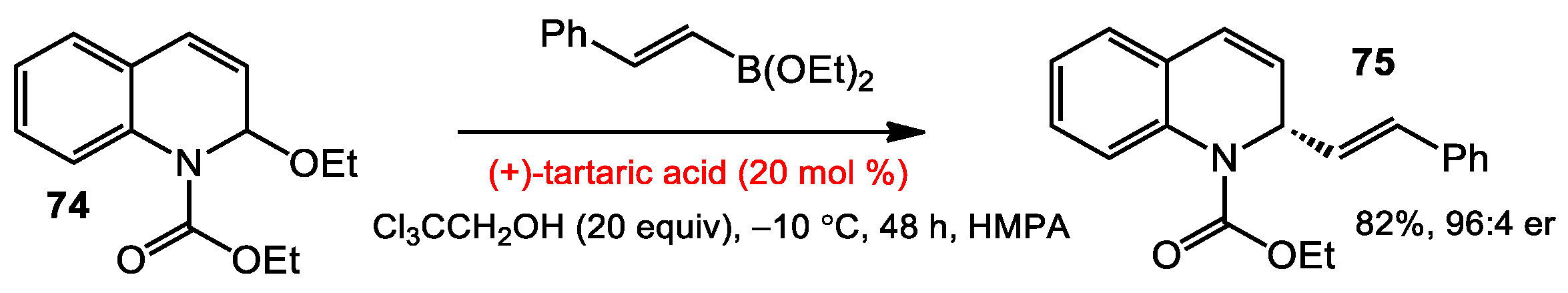
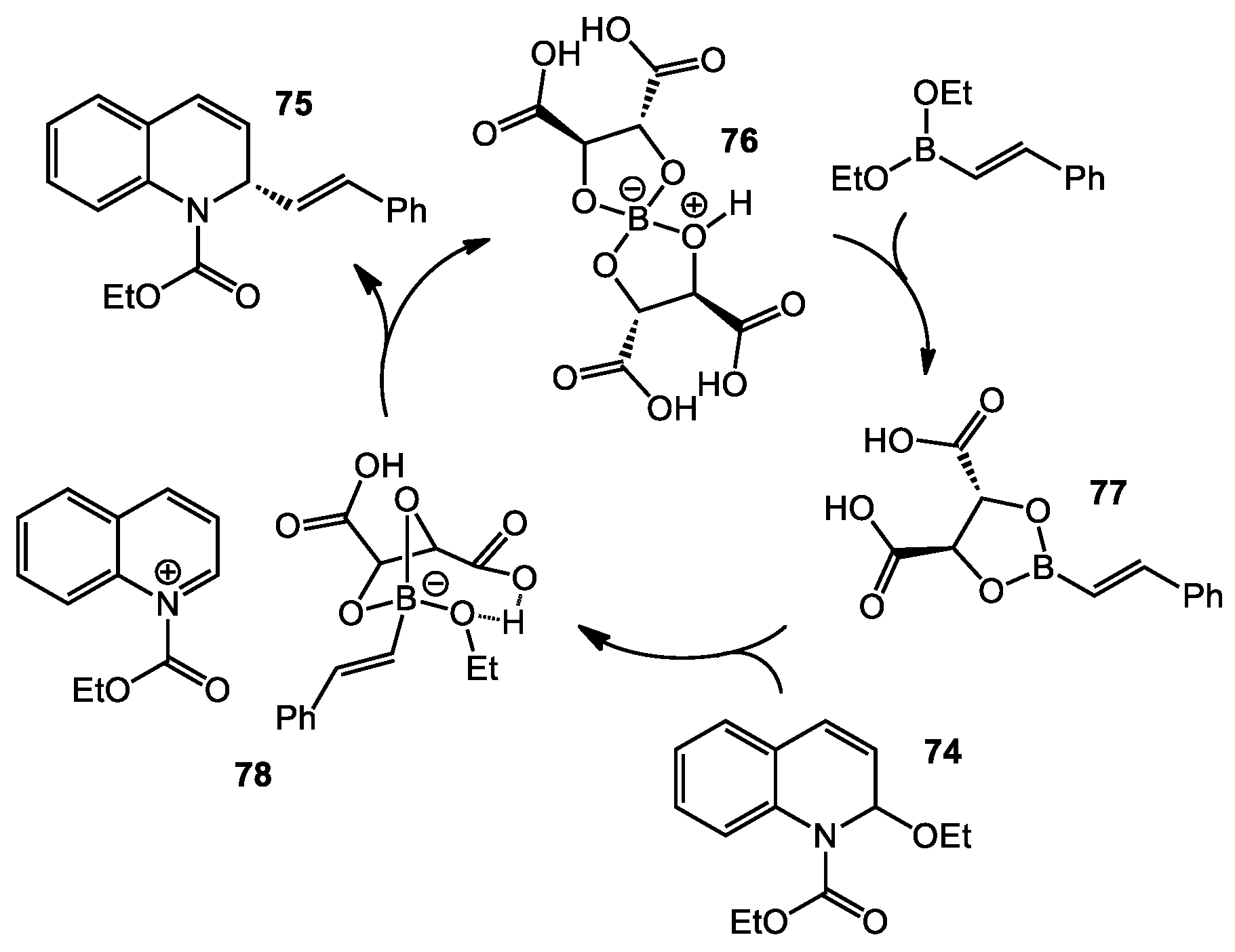
3.1 Asymmetric Addition of Alkenylborates to N-acyl Quinoliniums
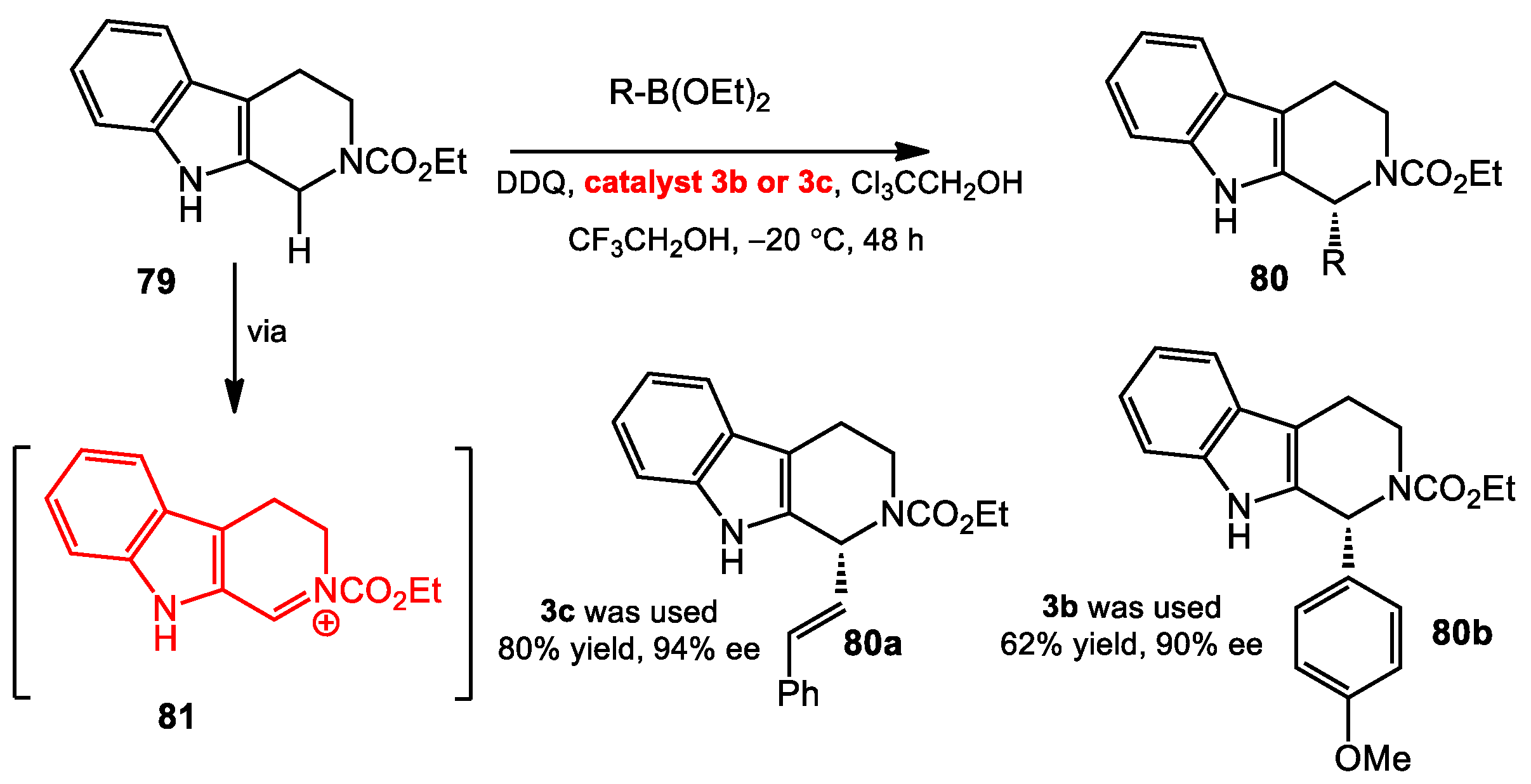
3.2. Enantioselective Oxidative C–H Alkenylation and Arylation
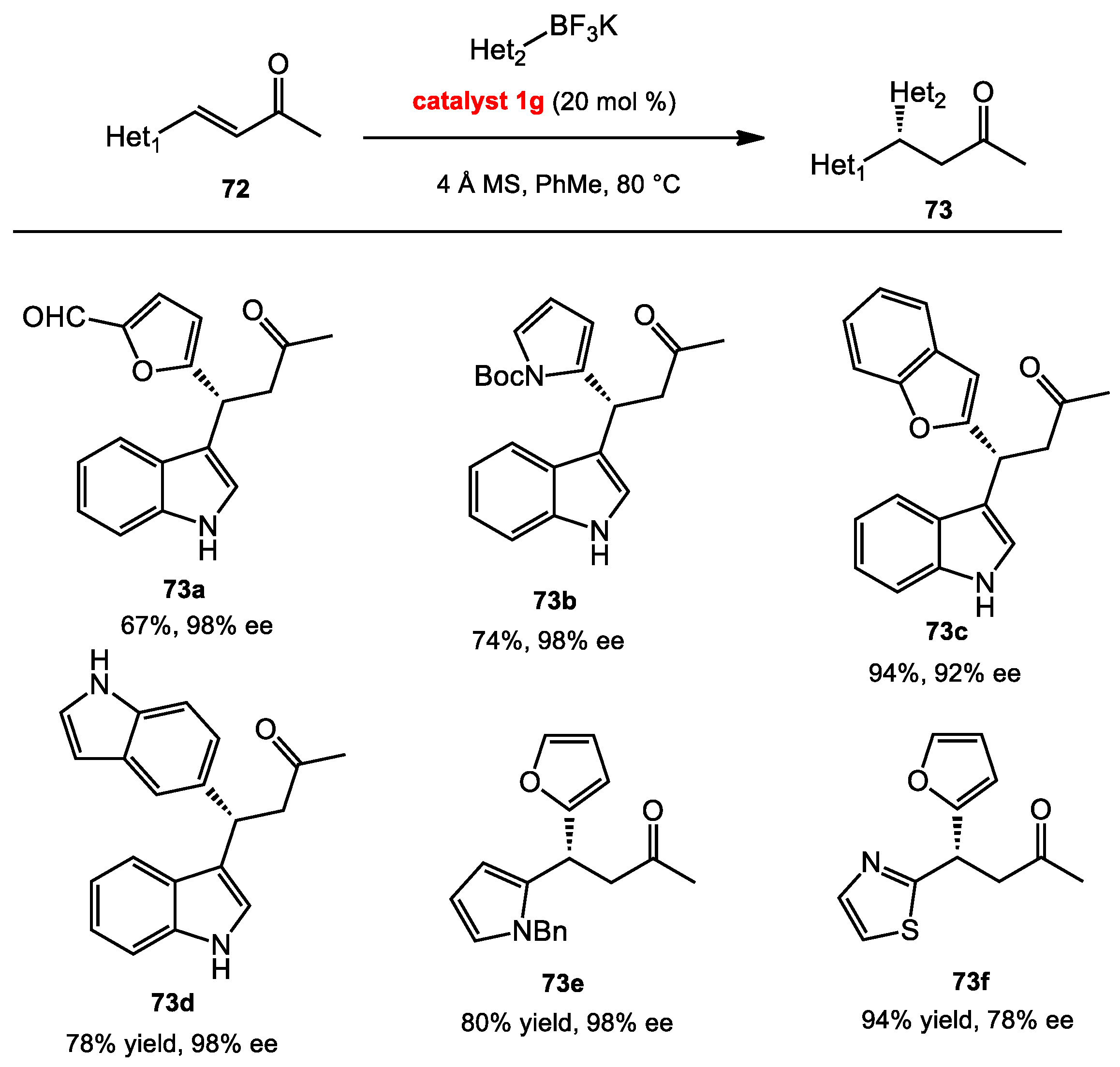
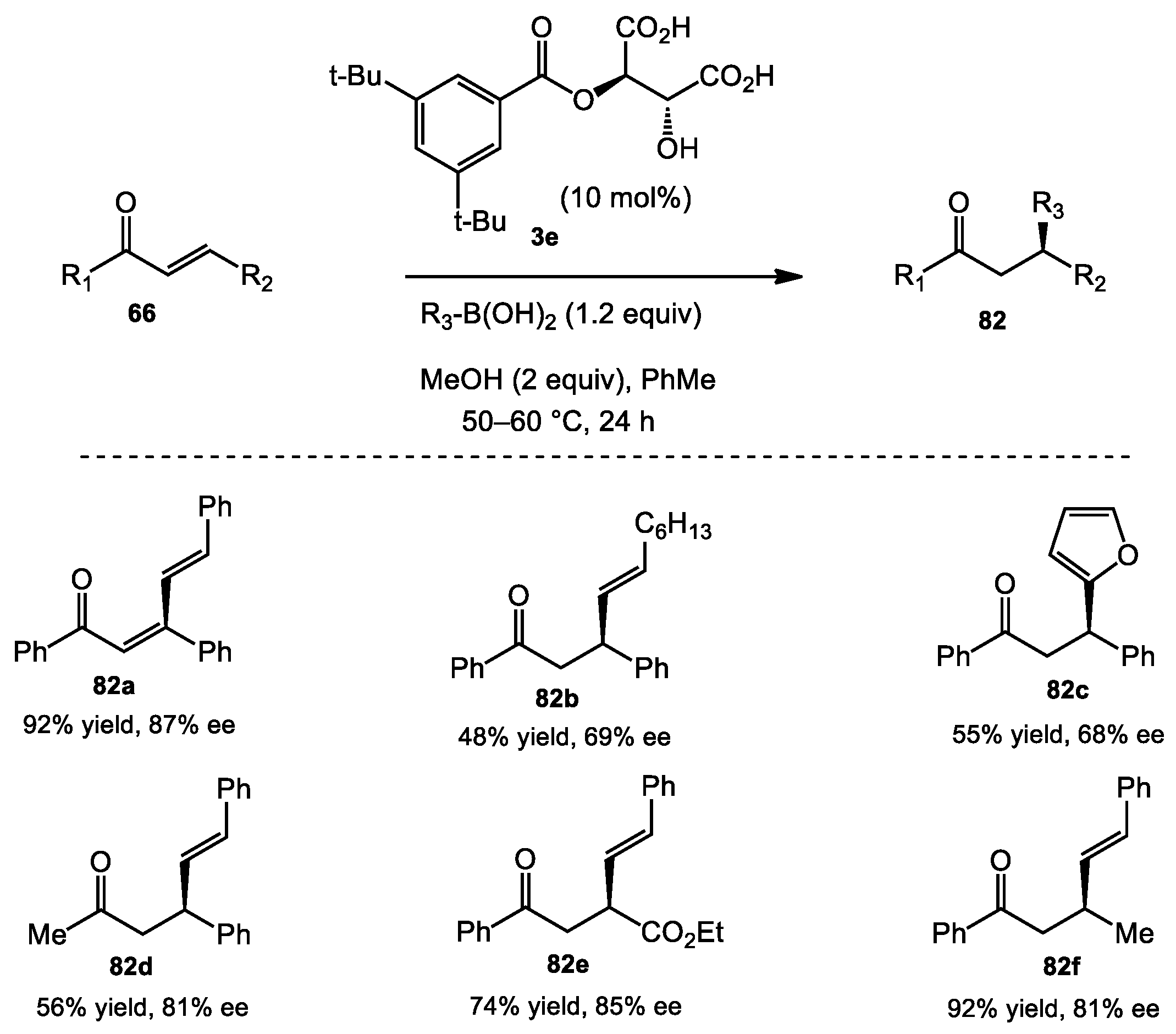
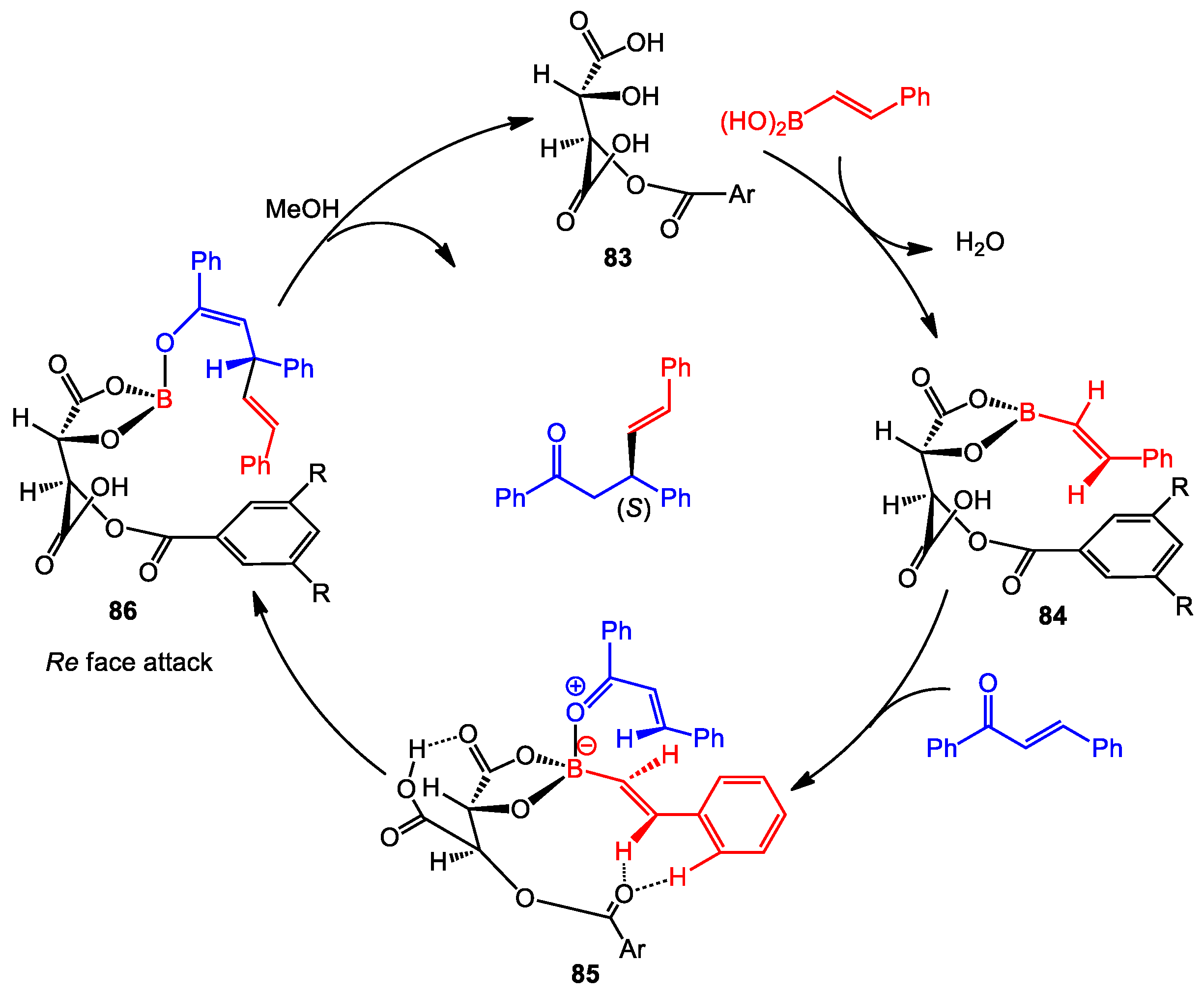
3.3. Asymmetric Conjugate Addition
4. VANOL/VAPOL
4.1. Petasis Reaction
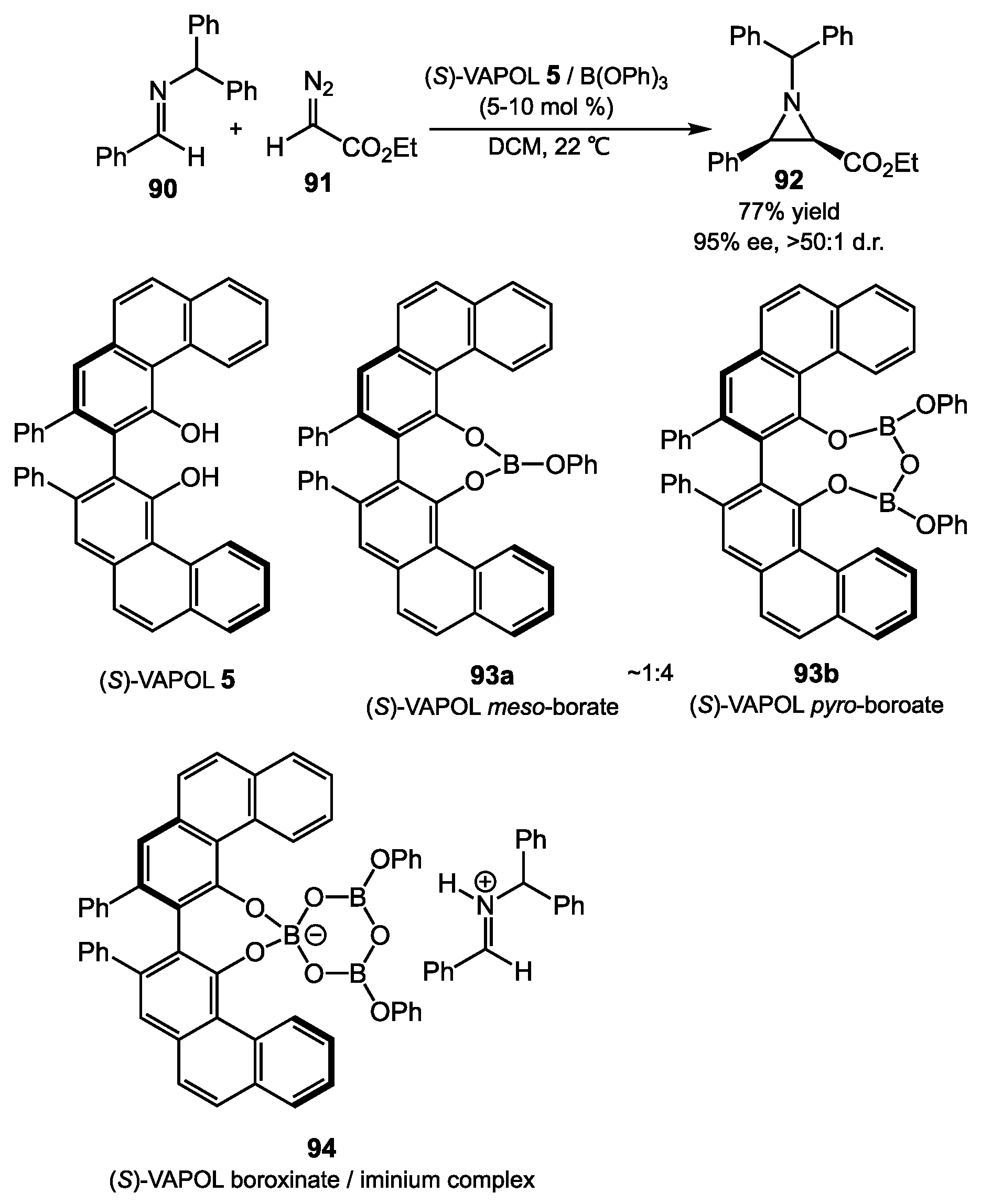
4.2. Aziridination
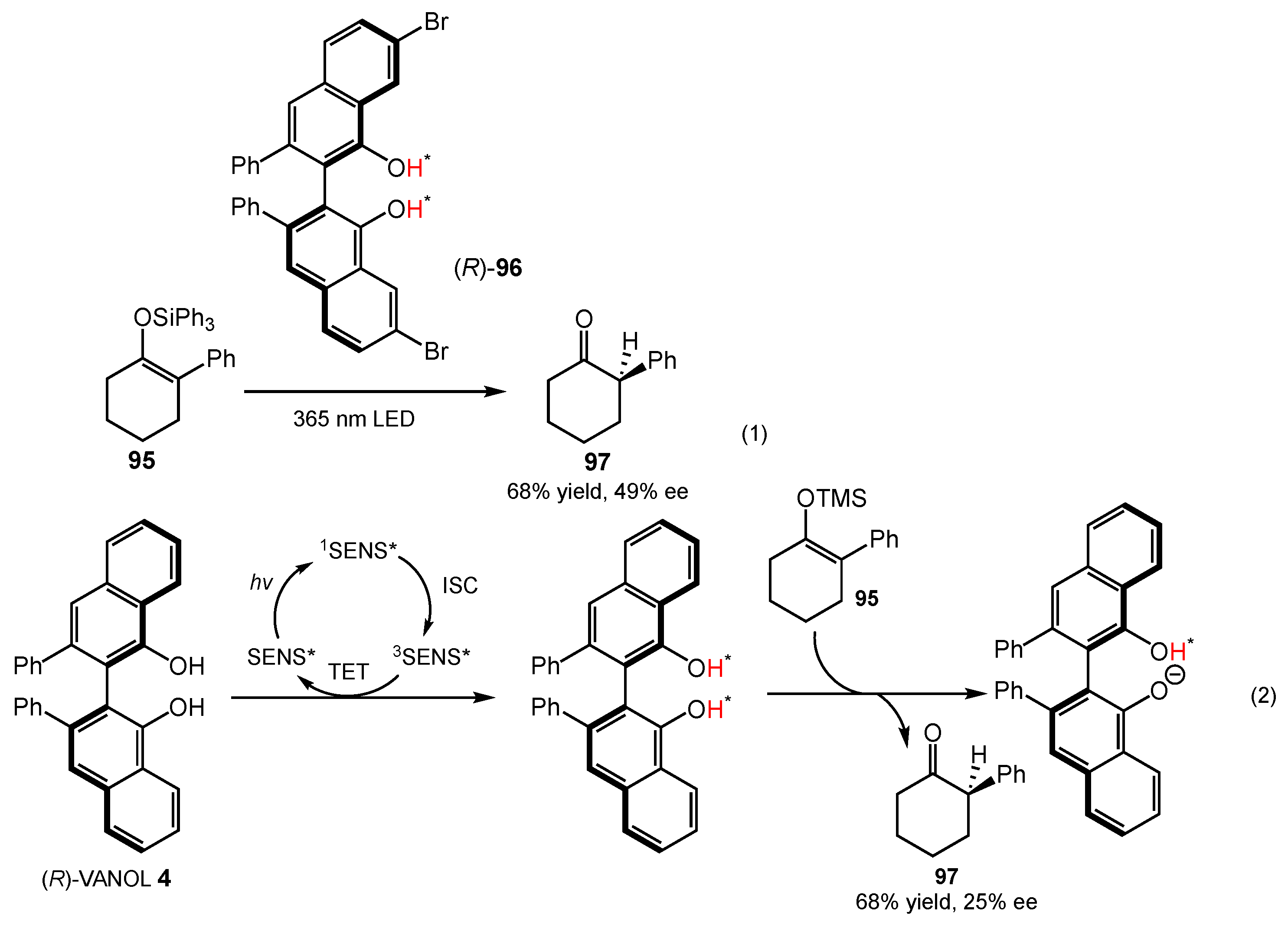
4.3. Protonation of Silyl Enol Ethers
5. Silanediol
5.1. N-acyl Mannich Reaction
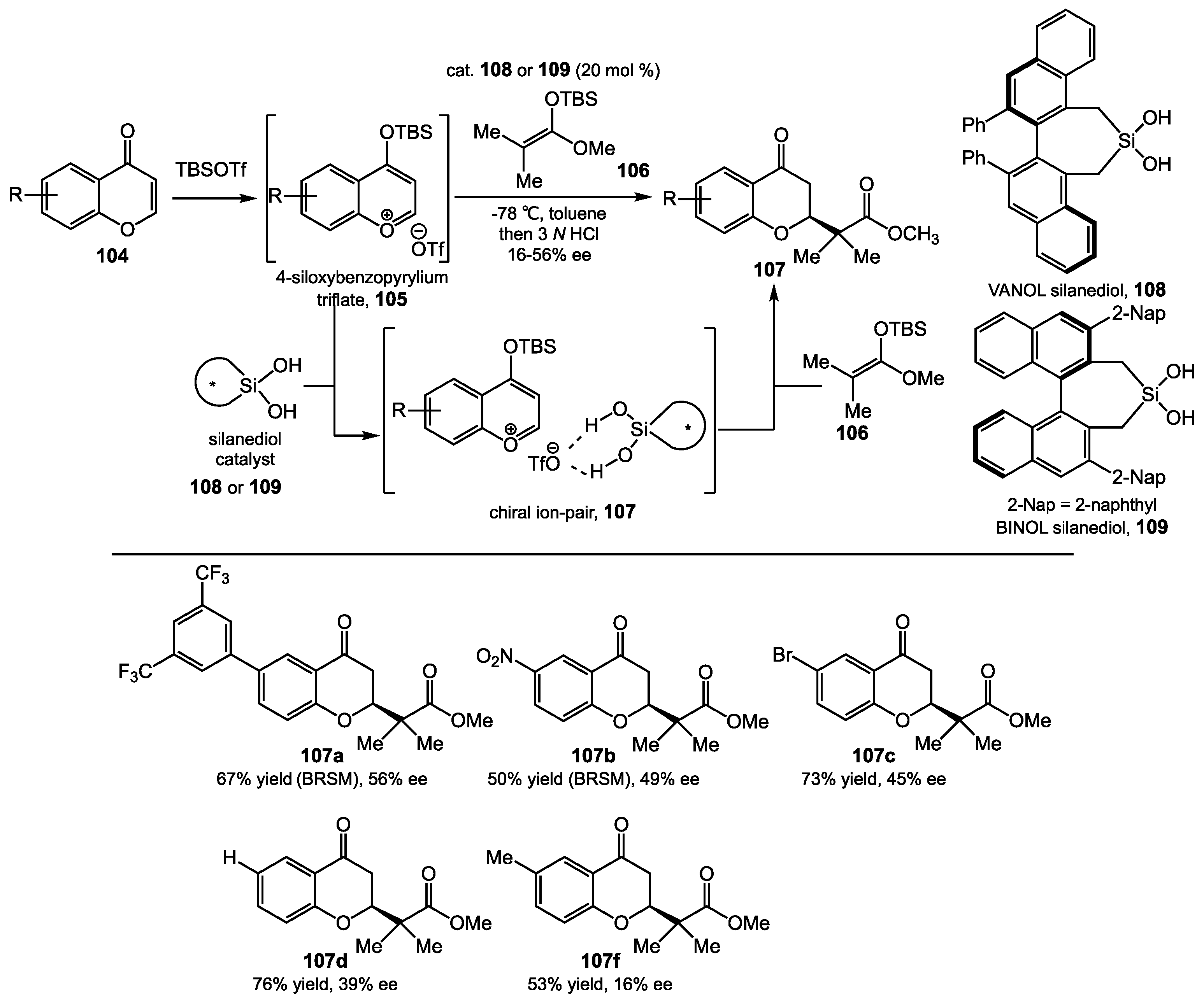
5.2. Chromenone Functionalization
6. TADDOL Derivatives
6.1. Hydrocyanation of Aldimides
6.2. Diels–Alder Reaction
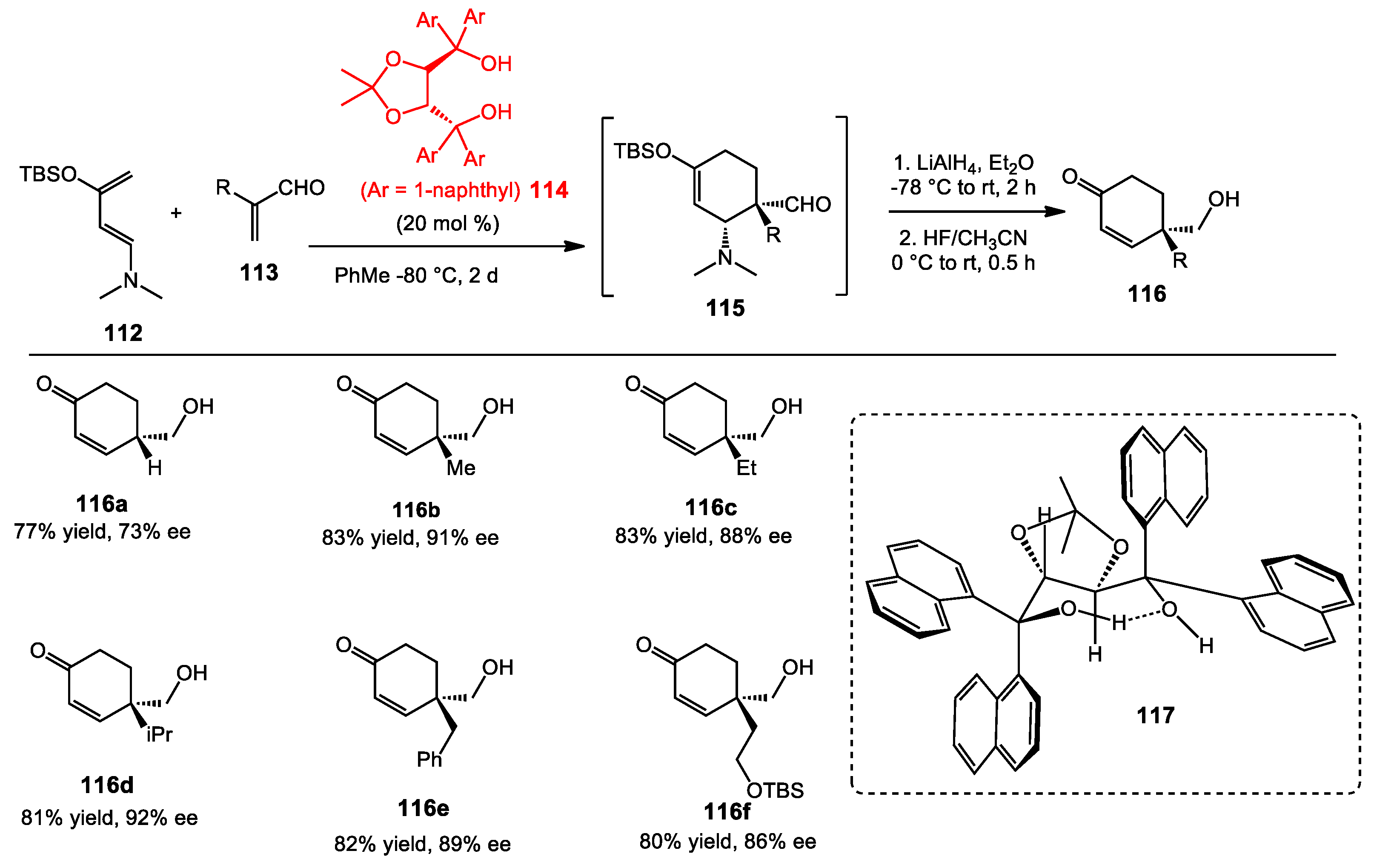
6.3. Vinylogous Mukaiyama Aldol Reaction
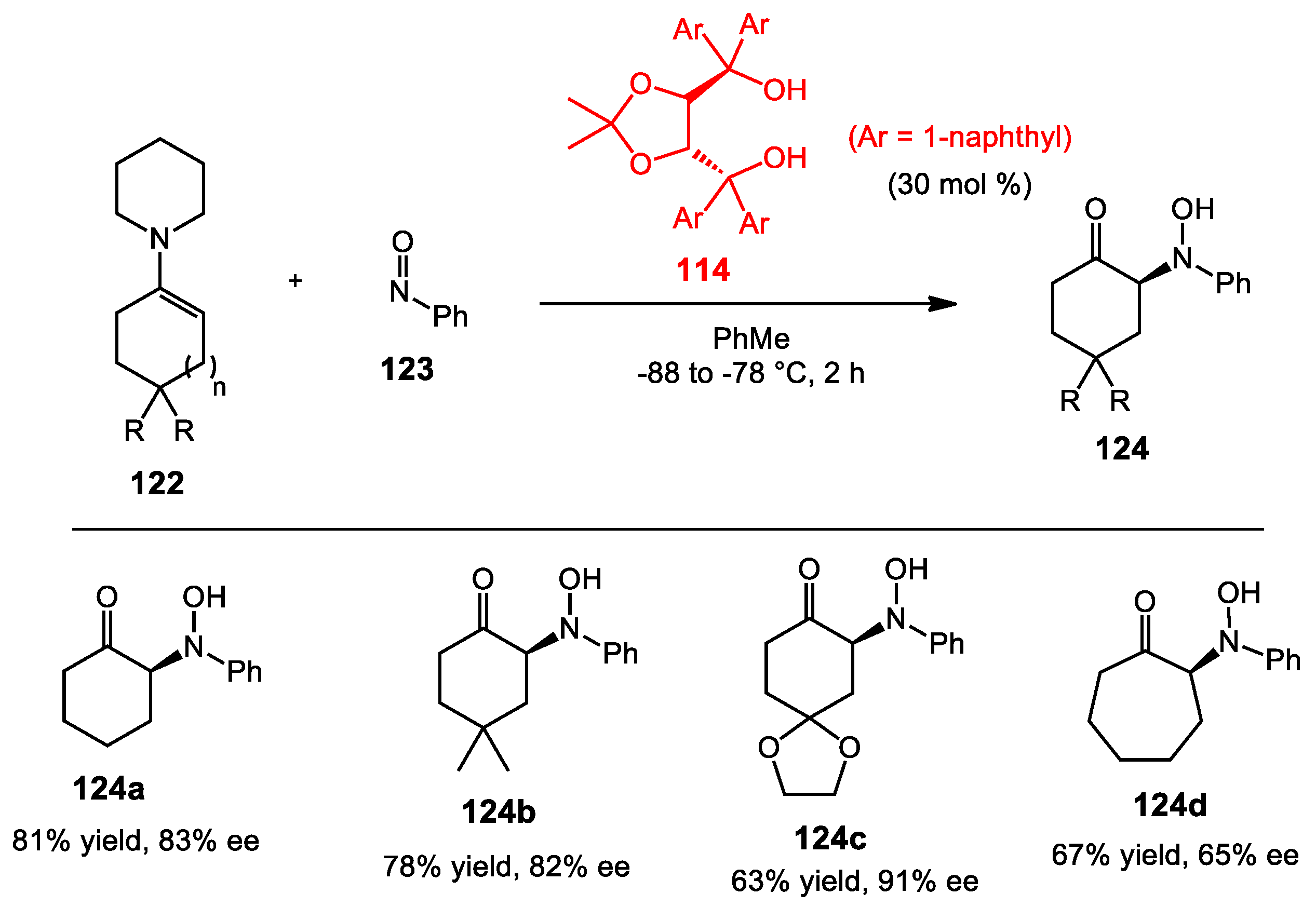
6.4. Nitroso Aldol Reaction
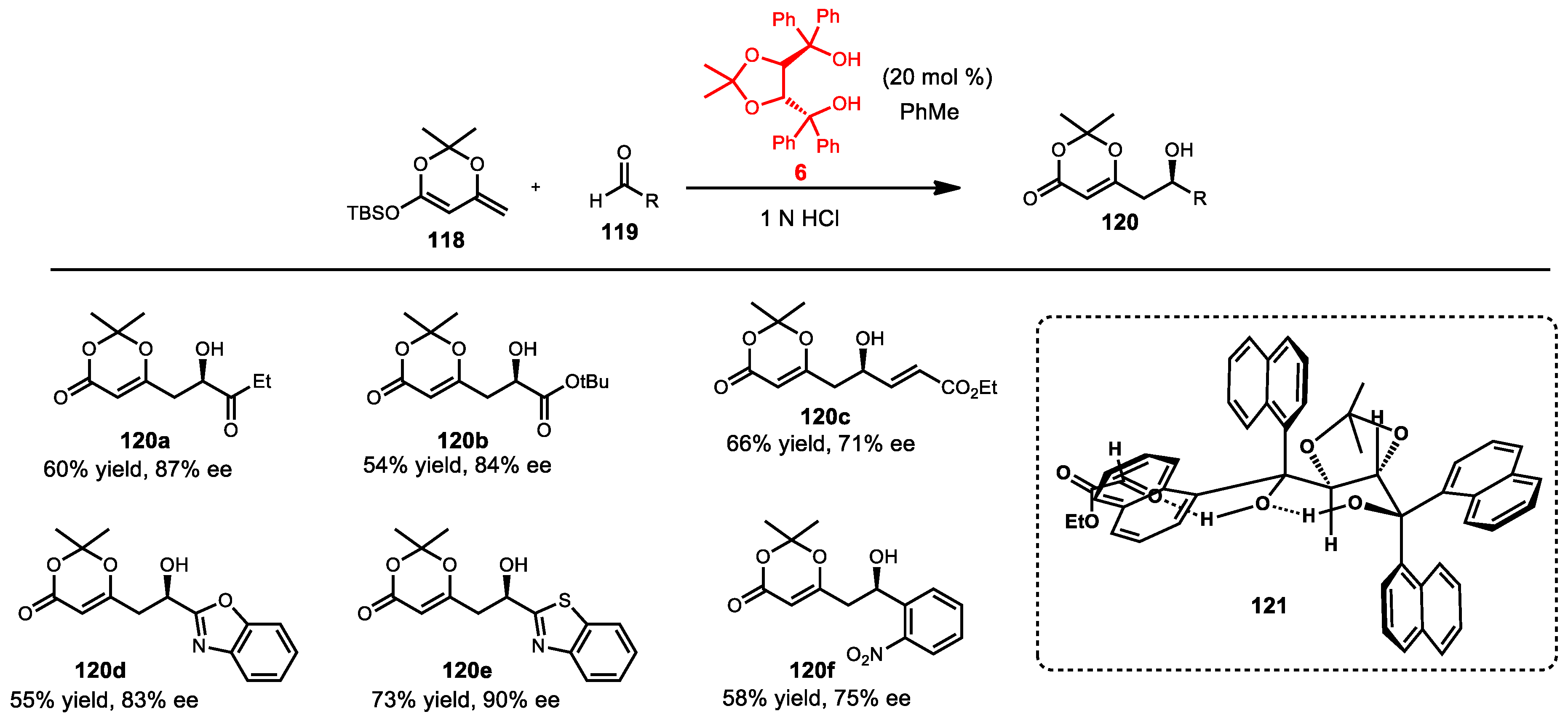
6.5. Mukaiyama Aldol Reaction
6.6. Mukaiyama Aldol Reaction of Ketoesters
7. BAMOL
8. Recent Developed Chiral Diols
8.1. Chiral Ferrocenyl Diols
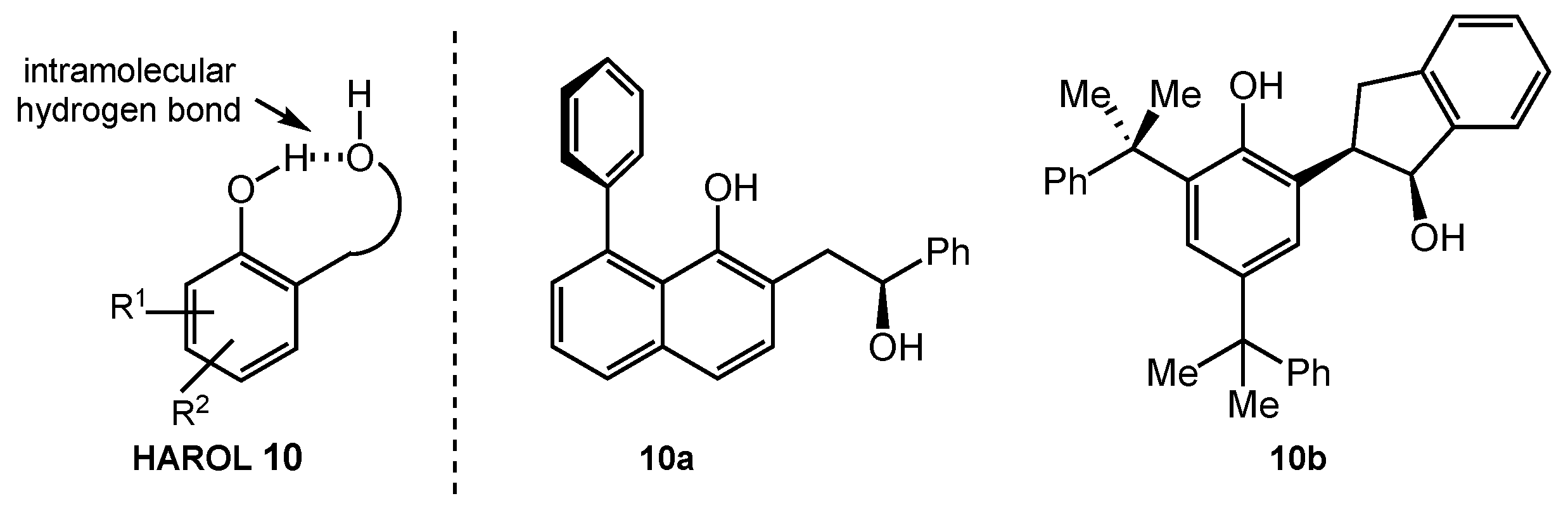
8.2. HAROLs
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hajos, Z.G.; Parrish, D.R. Asymmetric Synthesis of Bicyclic Intermediates of Natural Product Chemistry. J. Org. Chem. 1974, 39, 1615–1621. [Google Scholar] [CrossRef]
- Taylor, M.S.; Jacobsen, E.N. Asymmetric Catalysis by Chiral Hydrogen-Bond Donors. Angew. Chem. Int. Ed. 2006, 45, 1520–1543. [Google Scholar] [CrossRef] [PubMed]
- Doyle, A.G.; Jacobsen, E.N. Small-Molecule H-Bond Donors in Asymmetric Catalysis. Chem. Rev. 2007, 107, 5713–5743. [Google Scholar] [CrossRef] [PubMed]
- Schenker, S.; Zamfir, A.; Freund, M.; Tsogoeva, S.B. Developments in Chiral Binaphthyl-Derived Brønsted/Lewis Acids and Hydrogen-Bond-Donor Organocatalysis. Eur. J. Org. Chem. 2011, 2011, 2209–2222. [Google Scholar] [CrossRef]
- Parmar, D.; Sugiono, E.; Raja, S.; Rueping, M. Complete Field Guide to Asymmetric BINOL-Phosphate Derived Brønsted Acid and Metal Catalysis: History and Classification by Mode of Activation; Brønsted Acidity, Hydrogen Bonding, Ion Pairing, and Metal Phosphates. Chem. Rev. 2014, 114, 9047–9153. [Google Scholar] [CrossRef] [PubMed]
- Min, C.; Seidel, D. Asymmetric Brønsted Acid Catalysis with Chiral Carboxylic Acids. Chem. Soc. Rev. 2017, 46, 5889–5902. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.G.; Waltz, K.M.; Garcia, I.F.; Kwiatkowski, D.; Walsh, P.J. Catalytic Asymmetric Allylation of Ketones and a Tandem Asymmetric Allylation/Diastereoselective Epoxidation of Cyclic Enones. J. Am. Chem. Soc. 2004, 126, 12580–12585. [Google Scholar] [CrossRef] [PubMed]
- Wadamoto, M.; Yamamoto, H. Silver-Catalyzed Asymmetric Sakurai-Hosomi Allylation of Ketones. J. Am. Chem. Soc. 2005, 127, 14556–14557. [Google Scholar] [CrossRef] [PubMed]
- Wada, R.; Oisaki, K.; Kanai, M.; Shibasaki, M. Catalytic Enantioselective Allylboration of Ketones. J. Am. Chem. Soc. 2004, 126, 8910–8911. [Google Scholar] [CrossRef] [PubMed]
- Lou, S.; Moquist, P.N.; Schaus, S.E. Asymmetric Allylboration of Ketones Catalyzed by Chiral Diols. J. Am. Chem. Soc. 2006, 128, 12660–12661. [Google Scholar] [CrossRef] [PubMed]
- Lou, S.; Moquist, P.N.; Schaus, S.E. Asymmetric Allylboration of Acyl Imines Catalyzed by Chiral Diols. J. Am. Chem. Soc. 2007, 129, 15398–15404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corey, E.J.; Decicco, C.P.; Newbold, R.C. Highly enantioselective and diastereoselective synthesis of β-amino acid esters and β-lactams from achiral esters and imines. Tetrahedron Lett. 1991, 32, 5287–5290. [Google Scholar] [CrossRef]
- Roush, W.R. Uncatalyzed Additions of Nucleophilic Alkenes to CX pi-Bonds. In Comprehensive Organic Synthesis; Trost, B.M., Fleming, I., Eds.; Pergamon: New York, NY, USA, 1991; Volume 2, p. 1. [Google Scholar]
- Samet, M.; Kass, S.R. Preorganized Hydrogen Bond Donor Catalysts: Acidities and Reactivities. J. Org. Chem. 2015, 80, 7727–7731. [Google Scholar] [CrossRef] [PubMed]
- Shokri, A.; Wang, Y.; O’Doherty, G.A.; Wang, X.B.; Kass, S.R. Hydrogen-Bond Networks: Strengths of Different Types of Hydrogen Bonds and an Alternative to the Low Barrier Hydrogen-Bond Proposal. J. Am. Chem. Soc. 2013, 135, 17919–17924. [Google Scholar] [CrossRef] [PubMed]
- Shokri, A.; Wang, X.B.; Wang, Y.; O’Doherty, G.A.; Kass, S.R. Flexible Acyclic Polyol-Chloride Anion Complexes and Their Characterization by Photoelectron Spectroscopy and Variable Temperature Binding Constant Determinations. J. Phys. Chem. A 2016, 120, 1661–1668. [Google Scholar] [CrossRef] [PubMed]
- Petasis, N.A.; Zavialov, I.A. Highly Stereocontrolled One-Step Synthesis of Anti-β-Amino Alcohols from Organoboronic Acids, Amines, and α-Hydroxy Aldehydes. J. Am. Chem. Soc. 1998, 120, 11798–11799. [Google Scholar] [CrossRef]
- Li, X.; Liu, X.; Fu, Y.; Wang, L.; Zhou, L.; Feng, X. Direct Allylation of Aldimines Catalyzed by C2-Symmetric N,N′-Dioxide-Sc(III) complexes: Highly Enantioselective Synthesis of Homoallylic Amines. Chem. Eur. J. 2008, 14, 4796–4798. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.; List, B. Catalytic Asymmetric Three-Component Synthesis of Homoallylic Amines. Angew. Chem. Int. Ed. 2013, 52, 2573–2576. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Schaus, S.E. Asymmetric Petasis Borono-Mannich Allylation Reactions Catalyzed by Chiral Biphenols. Angew. Chem. Int. Ed. 2017, 56, 1544–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sammis, G.M.; Flamme, E.M.; Xie, H.; Ho, D.M.; Sorensen, E.J. Design, Synthesis, and Reactivity of 1-Hydrazinodienes for Use in Organic Synthesis. J. Am. Chem. Soc. 2005, 127, 8612–8613. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.L.; Porco, J.A.; Taunton, J.; Schreiber, S.L.; Lee, A.Y.; Clardy, J. Application of the Allylic Diazene Rearrangement: Synthesis of the Enediyne-Bridged Tricyclic Core of Dynemicin A. J. Am. Chem. Soc. 1992, 114, 5898–5900. [Google Scholar] [CrossRef]
- Movassaghi, M.; Ahmad, O.K. A Stereospecific Palladium-Catalyzed Route to Monoalkyl Diazenes for Mild Allylic Reduction. Angew. Chem. Int. Ed. 2008, 47, 8909–8912. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; McIntosh, M.C. Acyclic 1,4-Stereocontrol via Reductive 1,3-Transpositions. Org. Lett. 2008, 10, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Thomson, R.J.; Schaus, S.E. Asymmetric Traceless Petasis Borono-Mannich Reactions of Enals: Reductive Transposition of Allylic Diazenes. Angew. Chem. Int. Ed. 2017, 56, 16631–16635. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.G.; Camp, E.H.; Walsh, P.J. Catalytic Asymmetric Methallylation of Ketones with an (H8-BINOLate)Ti-Based Catalyst. Org. Lett. 2006, 8, 4413–4416. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, N.; Qu, B.; Ma, S.; Lee, H.; Gonnella, N.C.; Gao, J.; Li, W.; Tan, Z.; Reeves, J.T.; et al. Asymmetric Methallylation of Ketones Catalyzed by a Highly Active Organocatalyst 3,3′-F2-BINOL. Org. Lett. 2013, 15, 1710–1713. [Google Scholar] [CrossRef] [PubMed]
- Alam, R.; Vollgraff, T.; Eriksson, L.; Szabó, K.J. Synthesis of Adjacent Quaternary Stereocenters by Catalytic Asymmetric Allylboration. J. Am. Chem. Soc. 2015, 137, 11262–11265. [Google Scholar] [CrossRef] [PubMed]
- Barnett, D.S.; Schaus, S.E. Asymmetric Propargylation of Ketones Using Allenylboronates Catalyzed by Chiral Biphenols. Org. Lett. 2011, 13, 4020–4023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishop, J.A.; Lou, S.; Schaus, S.E. Enantioselective Addition of Boronates to Acyl Imines Catalyzed by Chiral Biphenols. Angew. Chem. Int. Ed. 2009, 48, 4337–4340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrinet, S.C.; Goodman, J.M. Asymmetric Conjugate Addition of Alkynylboronates to Enones: Rationale for the Intriguing Catalysis Exerted by Binaphthols. J. Am. Chem. Soc. 2006, 128, 3116–3117. [Google Scholar] [CrossRef] [PubMed]
- Paton, R.S.; Goodman, J.M.; Pellegrinet, S.C. Theoretical Study of the Asymmetric Conjugate Alkenylation of Enones Catalyzed by Binaphthols. J. Org. Chem. 2008, 73, 5078–5089. [Google Scholar] [CrossRef] [PubMed]
- Boger, D.L.; Weinreb, S.M. Hetero Diels−Alder Methodology in Organic Synthesis; Academic Press: San Diego, CA, USA, 1987. [Google Scholar]
- Córdova, A. (Ed.) Catalytic Asymmetric Conjugate Reactions; Wiley: New York, NY, USA, 2010. [Google Scholar]
- Luan, Y.; Schaus, S.E. Enantioselective Addition of Boronates to o-Quinone Methides Catalyzed by Chiral Biphenols. J. Am. Chem. Soc. 2012, 134, 19965–19968. [Google Scholar] [CrossRef] [PubMed]
- Barbato, K.S.; Luan, Y.; Ramella, D.; Panek, J.S.; Schaus, S.E. Enantioselective Multicomponent Condensation Reactions of Phenols, Aldehydes, and Boronates Catalyzed by Chiral Biphenols. Org. Lett. 2015, 17, 5812–5815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mundal, D.A.; Lutz, K.E.; Thomson, R.J. A direct synthesis of allenes by a traceless Petasis reaction. J. Am. Chem. Soc. 2012, 134, 5782–5785. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.A.; May, J.A. Hydrazone-Initiated Reaction Cascades. Asian J. Org. Chem. 2016, 5, 1296–1303. [Google Scholar] [CrossRef]
- Jiang, Y.; Diagne, A.B.; Thomson, R.J.; Schaus, S.E. Enantioselective Synthesis of Allenes by Catalytic Traceless Petasis Reactions. J. Am. Chem. Soc. 2017, 139, 1998–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, A.G.; Movassaghi, M. Highly Efficient Methodology for the Reductive Coupling of Aldehyde Tosylhydrazones with Alkyllithium Reagents. J. Am. Chem. Soc. 1998, 120, 8891–8892. [Google Scholar] [CrossRef]
- Nguyen, T.N.; May, J.A. Enantioselective Organocatalytic Conjugate Addition of Organoboron Nucleophiles. Tetrahedron Lett. 2017, 58, 1535–1544. [Google Scholar] [CrossRef]
- Wu, T.R.; Chong, J.M. Ligand-catalyzed asymmetric alkynylboration of enones: A new paradigm for asymmetric synthesis using organoboranes. J. Am. Chem. Soc. 2005, 127, 3244–3245. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.R.; Chong, J.M. Asymmetric conjugate alkenylation of enones catalyzed by chiral diols. J. Am. Chem. Soc. 2007, 129, 4908–4909. [Google Scholar] [CrossRef] [PubMed]
- Lundy, B.J.; Jansone-Popova, S.; May, J.A. Enantioselective conjugate addition of alkenylboronic acids to indole-appended enones. Org. Lett. 2011, 13, 4958–4961. [Google Scholar] [CrossRef] [PubMed]
- Turner, H.M.; Patel, J.; Niljianskul, N.; Chong, J.M. Binaphthol-catalyzed asymmetric conjugate arylboration of enones. Org. Lett. 2011, 13, 5796–5799. [Google Scholar] [CrossRef] [PubMed]
- Shih, J.L.; Nguyen, T.S.; May, J.A. Organocatalyzed asymmetric conjugate addition of heteroaryl and aryl trifluoroborates: A synthetic strategy for Discoipyrrole D. Angew. Chem. Int. Ed. 2015, 54, 9931–9935. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.H.; McDonald, R.; Hall, D.G. Enantioselective preparation and chemoselective cross-coupling of 1,1-diboron compounds. Nat. Chem. 2011, 3, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Lennox, A.J.J.; Lloyd-Jone, G.C. Organotrifluoroborate hydrolysis: Boronic acid release mechanism and an acid–base paradox in cross-coupling. J. Am. Chem. Soc. 2012, 134, 7431–7441. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.Y.; Sarlah, D.; Carreira, E.M. Iridium-catalyzed enantioselective allylic vinylation. J. Am. Chem. Soc. 2013, 135, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.Y.; Sarlah, D.; Carreira, E.M. Iridium-catalyzed enantioselective allylic alkynylation. Angew. Chem. Int. Ed. 2013, 52, 7532–7535. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Quach, T.D.; Batey, R.A. Copper-catalyzed nondecarboxylative cross coupling of alkenyltrifluoroborate salts with carboxylic acids or carboxylates: Synthesis of enol esters. Org. Lett. 2013, 15, 3150–3153. [Google Scholar] [CrossRef] [PubMed]
- Mallouk, G.L.; Rosenthal, G.L.; Mueller, G.; Brusasco, R.; Bartlett, N. Fluoride ion affinities of germanium tetrafluoride and boron trifluoride from thermodynamic and structural data for (SF3)2GeF6, ClO2GeF5, and ClO2BF4. Inorg. Chem. 1984, 23, 3167–3173. [Google Scholar] [CrossRef]
- Haartz, J.C.; McDaniel, D.H. Fluoride ion affinity of some Lewis acids. J. Am. Chem. Soc. 1973, 95, 8562–8565. [Google Scholar] [CrossRef]
- Kodama, T.; Moquist, P.N.; Schaus, S.E. Enantioselective Boronate Additions to N-Acyl Quinoliniums Catalyzed by Tartaric Acid. Org. Lett. 2011, 13, 6316–6319. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Meng, Z.; Li, C.; Lou, H.; Liu, L. Organocatalytic Enantioselective Oxidative C-H Alkenylation and Arylation of N-Carbamoyl Tetrahydropyridines and Tetrahydro-β-Carbolines. Angew. Chem. Int. Ed. 2015, 54, 6012–6015. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, M.; Tokudomi, M.; Nakajima, M. Enantioselective conjugate addition of boronic acids to enones catalyzed by O-monoacyltartaric acids. Chem. Commun. 2010, 46, 7799–7800. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, M.; Kinoshita, R.; Nakajima, M. O-Monoacyltartaric Acid Catalyzed Enantioselective Conjugate Addition of a Boronic Acid to Dienones: Application to the Synthesis of Optically Active Cyclopentenones. Org. Lett. 2014, 16, 5172–5175. [Google Scholar] [CrossRef] [PubMed]
- Grimblat, N.; Sugiura, M.; Pellegrinet, S.C. A hydrogen bond rationale for the enantioselective β-alkenylboration of enones catalyzed by O-monoacyltartaric acids. J. Org. Chem. 2014, 79, 6754–6758. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Wulff, W.D. Vaulted Biaryls as Chiral Ligands for Asymmetric Catalytic Diels-Alder Reactions. J. Am. Chem. Soc. 1993, 115, 3814–3815. [Google Scholar] [CrossRef]
- Zhang, Y.; Desai, A.; Lu, Z.; Hu, G.; Ding, Z.; Wulff, W.D. Catalytic Asymmetric Aziridination with Borate Catalysts Derived from VANOL and VAPOL Ligands: Scope and Mechanistic Studies. Chem. Eur. J. 2008, 14, 3785–3803. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, Z.; Desai, A.; Wulff, W.D. Mapping the Active Site in a Chemzyme: Diversity in the N-Substituent in the Catalytic Asymmetric Aziridination of Imines. Org. Lett. 2008, 10, 5429–5432. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, M.; Gupta, A.K.; Lu, Z.; Zhang, Y.; Wulff, W.D. Seeking Passe-Partout in the Catalytic Asymmetric Aziridination of Imines: Evolving Toward Substrate Generality for a Single Chemzyme. J. Org. Chem. 2010, 75, 5643–5660. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Gupta, A.K.; Huang, R.H.; Mukherjee, M.; Wulff, W.D. Substrate-Induced Covalent Assembly of a Chemzyme and Crystallographic Characterization of a Chemzyme-Substrate Complex. J. Am. Chem. Soc. 2010, 132, 14669–14675. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, F.; Zhao, L.; He, Q.; Chen, F.; Zheng, C. An Efficient Enantioselective Synthesis of Florfenicol via Asymmetric Aziridination. Tetrahedron 2011, 67, 9199–9203. [Google Scholar] [CrossRef]
- Zhao, W.; Yin, X.; Gupta, A.K.; Zhang, X.; Wulff, W.D. The Nature of meso- and pyro-Borate Precatalysts to the VANOL and VAPOL BOROX Catalysts. Synletter 2015, 26, 1606–1614. [Google Scholar] [CrossRef]
- Vetticatt, M.J.; Desai, A.A.; Wulff, W.D. Isotope Effects and Mechanism of the Asymmetric BOROX Brønsted Acid Catalyzed Aziridination Reaction. J. Org. Chem. 2013, 78, 5142–5152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Y.; Ding, Z.; Wulff, W.D. Vaulted Biaryls in Catalysis: A Structure–Activity Relationship Guided Tour of the Immanent Domain of the VANOL Ligand. Chem. Eur. J. 2013, 19, 15565–15571. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.A.; Wulff, W.D. Controlled Diastereo- and Enantioselection in a Catalytic Asymmetric Aziridination. J. Am. Chem. Soc. 2010, 123, 13100–13103. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Wulff, W.D. Direct Catalytic Asymmetric Aminoallylation of Aldehydes: Synergism of Chiral and Nonchiral Brønsted Acid. J. Am. Chem. Soc. 2011, 133, 5656–5659. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Huang, L.; Guan, Y.; Wulff, W.D. Three-Component Asymmetric Catalytic Ugi Raction-Concinnity from Diversity by Substrate-Mediated Catalyst Assembly. Angew. Chem. Int. Ed. 2014, 53, 3436–3441. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Gupta, A.K.; Huang, L.; Zhao, W.; Yin, X.; Osminski, W.E.G.; Huang, R.H.; Wulff, W.D.; Izzo, J.A.; Vetticatt, M.J. Pyro-Borates, spiro-Borates, and Boroxinates of BINOL-Assembly, Structures, and Reactivity. J. Am. Chem. Soc. 2017, 139, 10267–10285. [Google Scholar] [CrossRef] [PubMed]
- Petasis, N.A.; Goodman, A.; Zavialov, I. A new synthesis of α-arylglycines from aryl boronic acids. Tetrahedron 1997, 53, 16463–16470. [Google Scholar] [CrossRef]
- Petasis, N.A.; Boral, S. One-step three-component reaction among organoboronic acids, amines, and salicylaldehydes. Tetrahedron Lett. 2001, 42, 539–542. [Google Scholar] [CrossRef]
- Petasis, N.A.; Butkevich, A.N. Synthesis of 2H-chromenes and 1,2-dihydroquinolines from aryl aldehydes, amines, and alkenylboron compounds. J. Organomet. Chem. 2009, 694, 1747–1753. [Google Scholar] [CrossRef] [PubMed]
- Schaus, S.E.; Lou, S. Asymmetric Petasis reactions catalyzed by chiral biphenols. J. Am. Chem. Soc. 2008, 130, 6922–6923. [Google Scholar] [CrossRef]
- Hu, G.; Huang, L.; Huang, R.H.; Wulff, W.D. Evidence for a Boroxinate Based Brønsted Acid Derivative of VAPOL as the Active Catalyst in the Catalytic Asymmetric Aziridination Reaction. J. Am. Chem. Soc. 2009, 131, 15615–15617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, M.; Zhou, Y.; Dai, Y.; Gupta, A.K.; Pulgam, V.R.; Staples, R.J.; Wulff, W.D. Catalyst-Controlled Multicomponent Aziridination of Chiral Aldehydes. Chem. Eur. J. 2017, 23, 2552–2556. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Ayad, S.; Hanson, K. Enantioselective Protonateon of Silyl Enol Ether Using Excited State Proton Transfer Dyes. Org. Lett. 2016, 18, 5416–5419. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Banerjee, T.; Hanson, K. Protonation of Silylenol Ether via Excited State Proton Transfer Catalysis. Chem. Commun. 2016, 52, 1350–1353. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekhar, V.; Boomishankar, R.; Nagendran, S. Recent developments in the synthesis and structure of organosilanols. Chem. Rev. 2004, 104, 5847–5910. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Harada, T.; Tanaka, R.; Unno, M. Anion recognition by a silanediol-based receptor. Org. Lett. 2006, 8, 4621–4624. [Google Scholar] [CrossRef] [PubMed]
- Mattson, A.E.; Schafer, A.G.; Wieting, J.M. Silanediols: A new class of hydrogen bond donor catalysts. Org. Lett. 2011, 13, 5228–5231. [Google Scholar] [CrossRef]
- Franz, A.K.; Tran, N.T.; Min, T. Silanediol hydrogen bonding activation of carbonyl compounds. Chem. Eur. J. 2011, 17, 9897–9900. [Google Scholar] [CrossRef]
- Mattson, A.E.; Andrew, A.G.; Wieting, J.M.; Fisher, T.J. Chiral silanediols in anion-binding catalysts. Angew. Chem. Int. Ed. 2013, 52, 11321–11324. [Google Scholar] [CrossRef]
- Mattson, A.E.; Wieting, J.M.; Fisher, T.J.; Schafer, A.G.; Visco, M.D.; Gallucci, J.C. Preparation and catalytic activety of BINOL-derived silanediols. Eur. J. Org. Chem. 2015, 525–533. [Google Scholar] [CrossRef]
- Mattson, A.E.; Visco, M.D.; Attard, J.; Guan, Y. Anion-binding catalyst designs for enantioselective synthesis. Tetrahedron Lett. 2017, 58, 2623–2628. [Google Scholar] [CrossRef]
- Mattson, A.E.; Kondo, S.I.; Hardman-Baldwin, A.M.; Visco, M.D.; Wieting, J.M.; Stern, C. Silanediol-catalyzed chromenone functionalization. Org. Lett. 2016, 18, 3766–3769. [Google Scholar] [CrossRef]
- Rueping, M.; Sugiono, E.; Azap, C. A highly enantioselective Brønsted acid catalyst for the Strecker reaction. Angew. Chem. Int. Ed. 2006, 45, 2617–2619. [Google Scholar] [CrossRef] [PubMed]
- Rueping, M.; Sugiono, E.; Moreth, S.A. Metal-free, enantioselective strecker reactions catalyzed by chiral BINOL and TADDOL catalysts. Adv. Synth. Catal. 2007, 349, 759–764. [Google Scholar] [CrossRef]
- Etter, M.C.; Urbanczyk-Lipkowska, Z.; Zia-Ebrahimi, M.; Panunto, T.W. Hydrogen bond-directed cocrystallization and molecular recognition properties of diarylureas. J. Am. Chem. Soc. 1990, 112, 8415–8426. [Google Scholar] [CrossRef]
- Etter, M.C.; Reutzel, S.M. Hydrogen bond directed cocrystallization and molecular recognition properties of acyclic imides. J. Am. Chem. Soc. 1991, 113, 2586–2598. [Google Scholar] [CrossRef]
- Curran, D.P.; Kuo, L.H. Altering the stereochemistry of allylation reactions of cyclic alpha-sulfinyl radicals with diarylureas. J. Org. Chem. 1994, 59, 3259–3261. [Google Scholar] [CrossRef]
- Schreiner, P.R.; Wittkopp, A. H-Bonding additives act like Lewis acid catalysts. Org. Lett. 2002, 4, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Schuster, T.; Kurz, M.; Göbel, M.W. Catalysis of a Diels−Alder Reaction by Amidinium Ions. J. Org. Chem. 2000, 65, 1697–1701. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.R.; Meghani, P.; Ekkundi, V.S. Diels-alder reactions: Rate acceleration promoted by a biphenylenediol. Tetrahedron Lett. 1990, 31, 3381–3384. [Google Scholar] [CrossRef]
- Schuster, T.; Bauch, M.; Dürner, G.; Göbel, M.W. Axially chiral amidinium ions as inducers of enantioselectivity in Diels−Alder reactions. Org. Lett. 2000, 2, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Thadani, A.N.; Stnkovic, A.R.; Rawal, V.H. Enantioselective Diels-Alder reactions catalyzed by hydrogen bonding. Proc. Natl. Acad. Sci. USA 2004, 101, 5846–5850. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Sunami, S.; Sugita, Y.; Kaneko, C. Use of 1, 3-dioxin-4-ones and related compounds in synthesis. XLIV. Asymmetric aldol reaction of 4-trimethylsiloxy-6-methylene-1, 3-dioxines: Use of tartaric acid-derived (acyloxy)borane complex as the catalyst. Chem. Pharm. Bull. 1994, 42, 839–845. [Google Scholar] [CrossRef]
- Sato, M.; Sunami, S.; Sugita, Y.; Kaneko, C. An efficient asymmetric aldol reaction of 4-trimethylsiloxy-6-methylene-1,3-dioxines by chiral binaphthol-titanium complex catalysis. Heterocycles 1995, 41, 1435–1444. [Google Scholar] [CrossRef]
- Singer, R.A.; Carreira, E.M. Catalytic, enantioselective dienolate additions to aldehydes: Preparation of optically active acetoacetate aldol adducts. J. Am. Chem. Soc. 1995, 117, 12360–12361. [Google Scholar] [CrossRef]
- De Rosa, M.; Acocella, M.R.; Villano, R.; Soriente, A.; Scettri, A. A convenient catalytic procedure for the highly enantioselective aldol condensation of O-silyldienolates. Tetrahedron Asymmetry 2003, 14, 2499–2502. [Google Scholar] [CrossRef]
- Evans, D.A.; Murry, J.A.; Kozlowski, M.C. C2-Symmetric copper(II) complexes as chiral Lewis acids. Catalytic enantioselective aldol additions of silylketene acetals to (benzyloxy)acetaldehyde. J. Am. Chem. Soc. 1996, 118, 5814–5815. [Google Scholar] [CrossRef]
- Evans, D.A.; Kozlowski, M.C.; Murry, J.A.; Burgey, C.S.; Campos, K.R.; Connell, B.T.; Staples, R.J. C2-Symmetric copper(II) complexes as chiral Lewis acids. Scope and mechanism of catalytic enantioselective aldol additions of enolsilanes to (benzyloxy)acetaldehyde. J. Am. Chem. Soc. 1999, 121, 669–685. [Google Scholar] [CrossRef]
- Krüger, J.; Carreira, E.M. Apparent catalytic generation of chiral metal enolates: enantioselective dienolate additions to aldehydes mediated by tol-BINAP·Cu(II) fluoride complexes. J. Am. Chem. Soc. 1998, 120, 837–838. [Google Scholar] [CrossRef]
- Shimada, Y.; Matsuoka, Y.; Irie, R.; Katsuki, T. Highly enantioselective Cr(salen)-catalyzed Mukaiyama aldol reaction: Construction of δ-hydroxy-β-keto ester derivatives. Synletter 2004, 1, 57–60. [Google Scholar] [CrossRef]
- Gondi, V.B.; Gravel, M.; Rawal, V.H. Hydrogen bond catalyzed enantioselective vinylogous Mukaiyama aldol reaction. Org. Lett. 2005, 7, 5657–5660. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.W.; Myers, P.L.; Ormerod, J.A. The reaction of aromatic nitroso-compounds with enamines. Part I. The reaction of nitrosobenzene with 1-morpholin-1-ylcyclohexene. J. Chem. Soc. Perkin Trans. 1972, 20, 2521–2524. [Google Scholar] [CrossRef]
- Momiyama, N.; Yamamoto, H. Brønsted acid catalysis of achiral enamine for regio- and enantioselective nitroso adol synthesis. J. Am. Chem. Soc. 2005, 127, 1080–1081. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.G. Catalyzed enantioselective aldol additions of latent enolate equivalents. Tetrahedron Asymmetry 1998, 9, 357–389. [Google Scholar] [CrossRef]
- Carreira, E.M. Modern Carbonyl Chemistry; Wiley-VCH: Weinheim, Germany, 2000; pp. 227–248. [Google Scholar]
- Palomo, C.; Oiarbide, M.; Garcia, J.M. Current progress in the asymmetric aldol addition reaction. Chem. Soc. Rev. 2004, 33, 65–75. [Google Scholar] [CrossRef] [PubMed]
- McGilvra, J.D.; Unni, A.K.; Modi, K.; Rawal, V.H. Highly diastereo- and enantioselective Mukaiyama aldol reactions catalyzed by hydrogen bonding. Angew. Chem. Int. Ed. 2006, 45, 6130–6133. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.A.; Kozlowski, M.C.; Burgey, C.S.; MacMillan, D.W.C. C2-Symmetric copper(II) complexes as chiral Lewis acids. Catalytic enantioselective aldol additions of enolsilanes to pyruvate esters. J. Am. Chem. Soc. 1997, 119, 7893–7894. [Google Scholar] [CrossRef]
- Gondi, V.B.; Hagihara, K.; Rawal, V.H. Diastereoselective and enantioselective Mukaiyama aldol reaction of α-ketoesters using hydrogen bond catalysis. Chem. Commun. 2010, 46, 904–906. [Google Scholar] [CrossRef] [PubMed]
- Unni, A.K.; Takenaka, N.; Yamamoto, H.; Rawal, V.H. Axially chiral biaryl diols catalyze highly enantioselective Hetero-Diels-Alder reactions through hydrogen bonding. J. Am. Chem. Soc. 2005, 127, 1336–1337. [Google Scholar] [CrossRef] [PubMed]
- Nottingham, C.; Müller-Bunz, H.; Guiry, P.J. A family of chiral ferrocenyl diols: Modular synthesis, solid-state characterization, and application in asymmetric organocatalysis. Angew. Chem. Int. Ed. 2016, 55, 11115–11119. [Google Scholar] [CrossRef] [PubMed]
- Dilek, Ö.; Tezeren, M.A.; Tilki, T.; Ertürk, E. Chiral 2-(2-hydroxyaryl)alcohols (HAROLs) with a 1,4-diol scaffold as a new family of ligands and organocatalyst. Tetrahedron 2018, 74, 268–286. [Google Scholar] [CrossRef]
- McDougal, N.T.; Schaus, S.E. Asymmetric Morita-Baylis-Hillman Reactions Catalyzed by Chiral Brønsted Acids. J. Am. Chem. Soc. 2003, 125, 12094–12095. [Google Scholar] [CrossRef] [PubMed]

















































© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.N.; Chen, P.-A.; Setthakarn, K.; May, J.A. Chiral Diol-Based Organocatalysts in Enantioselective Reactions. Molecules 2018, 23, 2317. https://doi.org/10.3390/molecules23092317
Nguyen TN, Chen P-A, Setthakarn K, May JA. Chiral Diol-Based Organocatalysts in Enantioselective Reactions. Molecules. 2018; 23(9):2317. https://doi.org/10.3390/molecules23092317
Chicago/Turabian StyleNguyen, Truong N., Po-An Chen, Krit Setthakarn, and Jeremy A. May. 2018. "Chiral Diol-Based Organocatalysts in Enantioselective Reactions" Molecules 23, no. 9: 2317. https://doi.org/10.3390/molecules23092317
APA StyleNguyen, T. N., Chen, P. -A., Setthakarn, K., & May, J. A. (2018). Chiral Diol-Based Organocatalysts in Enantioselective Reactions. Molecules, 23(9), 2317. https://doi.org/10.3390/molecules23092317