Theoretical Insights into the Electron Capture Behavior of H2SO4···N2O Complex: A DFT and Molecular Dynamics Study

Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
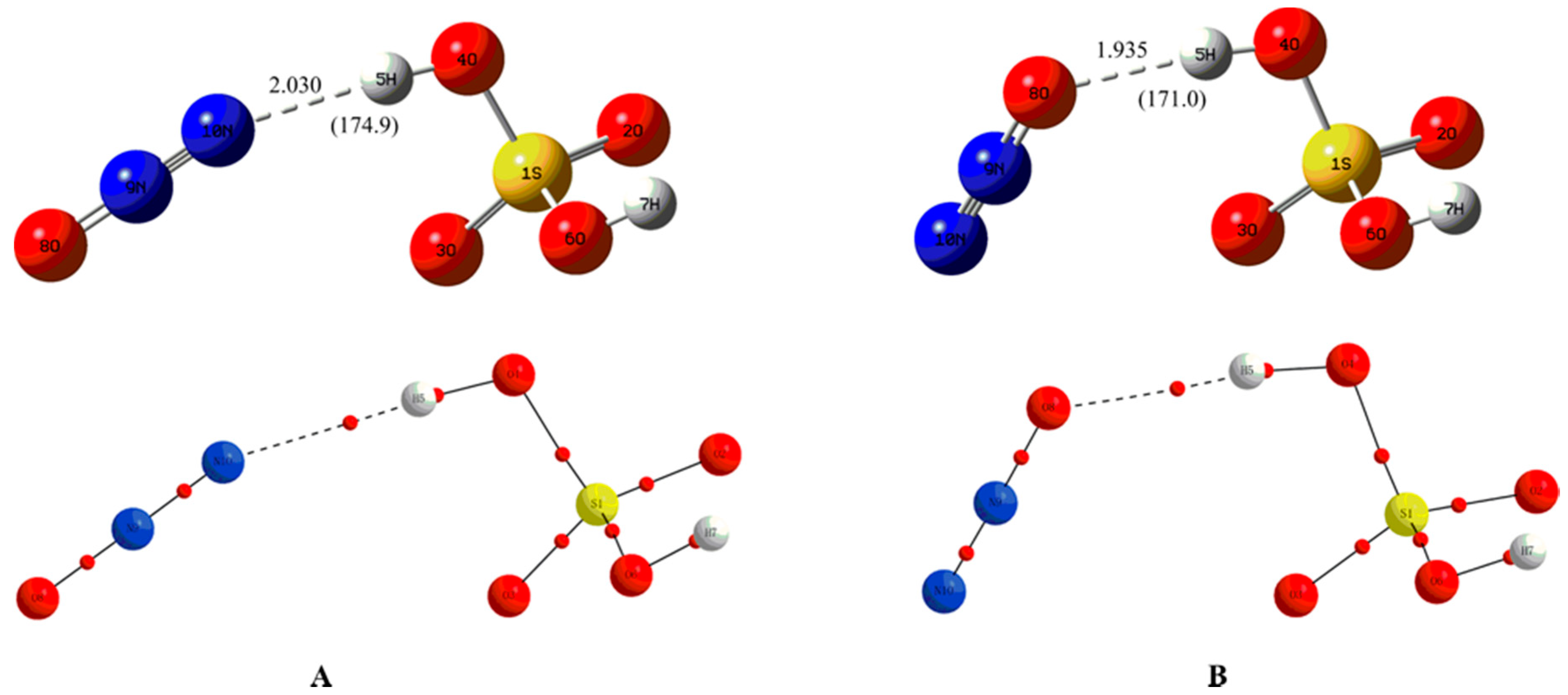
3.1. Formation of Neutral Molecular Complexes
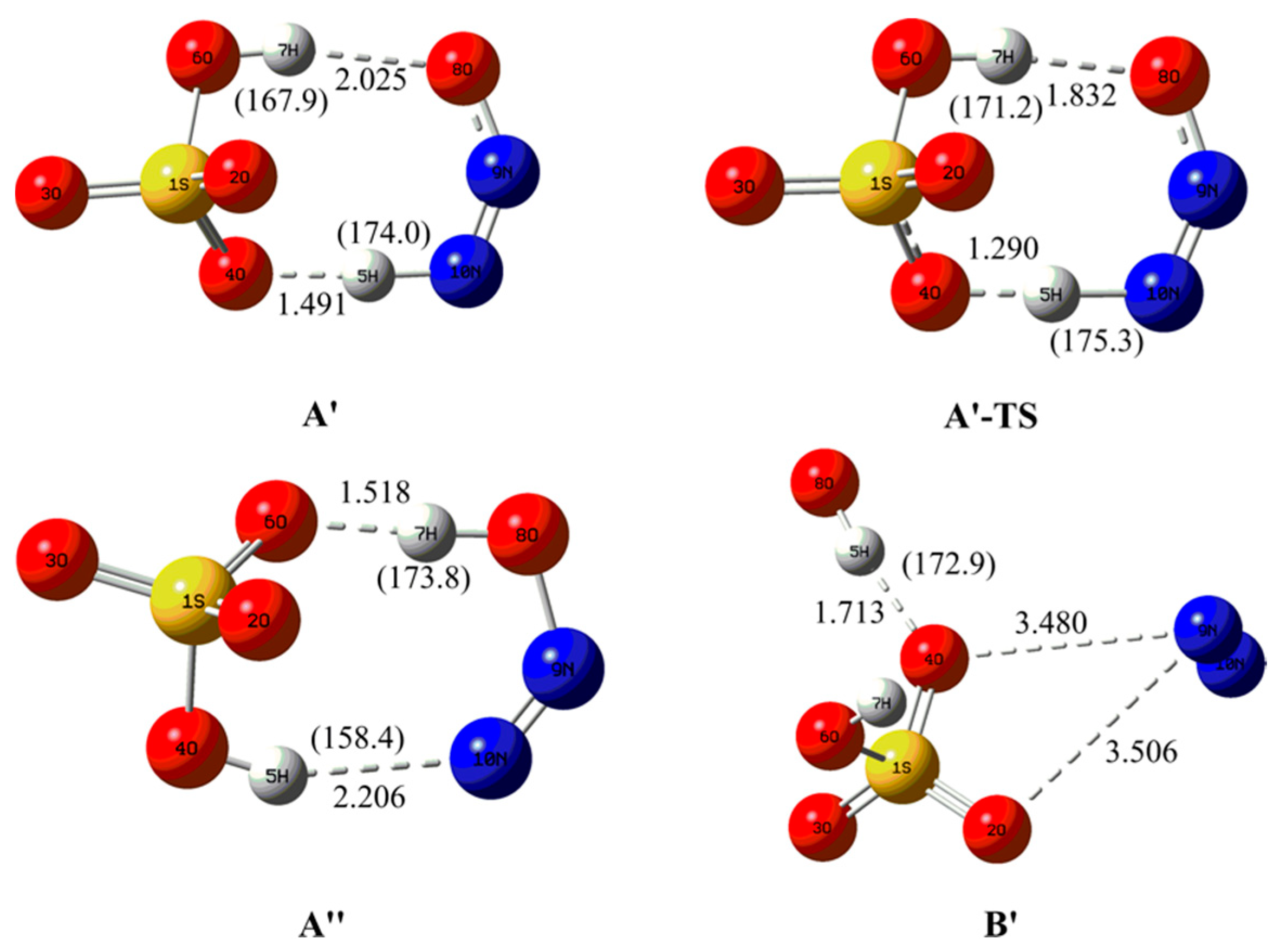
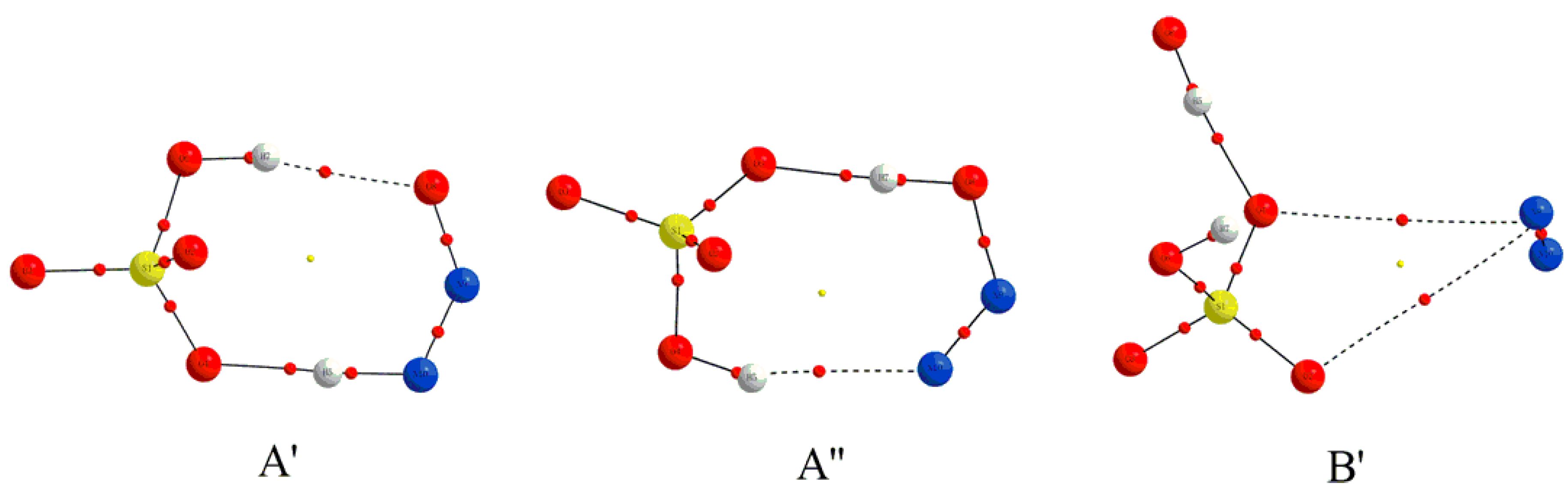
3.1.1. Structural Features
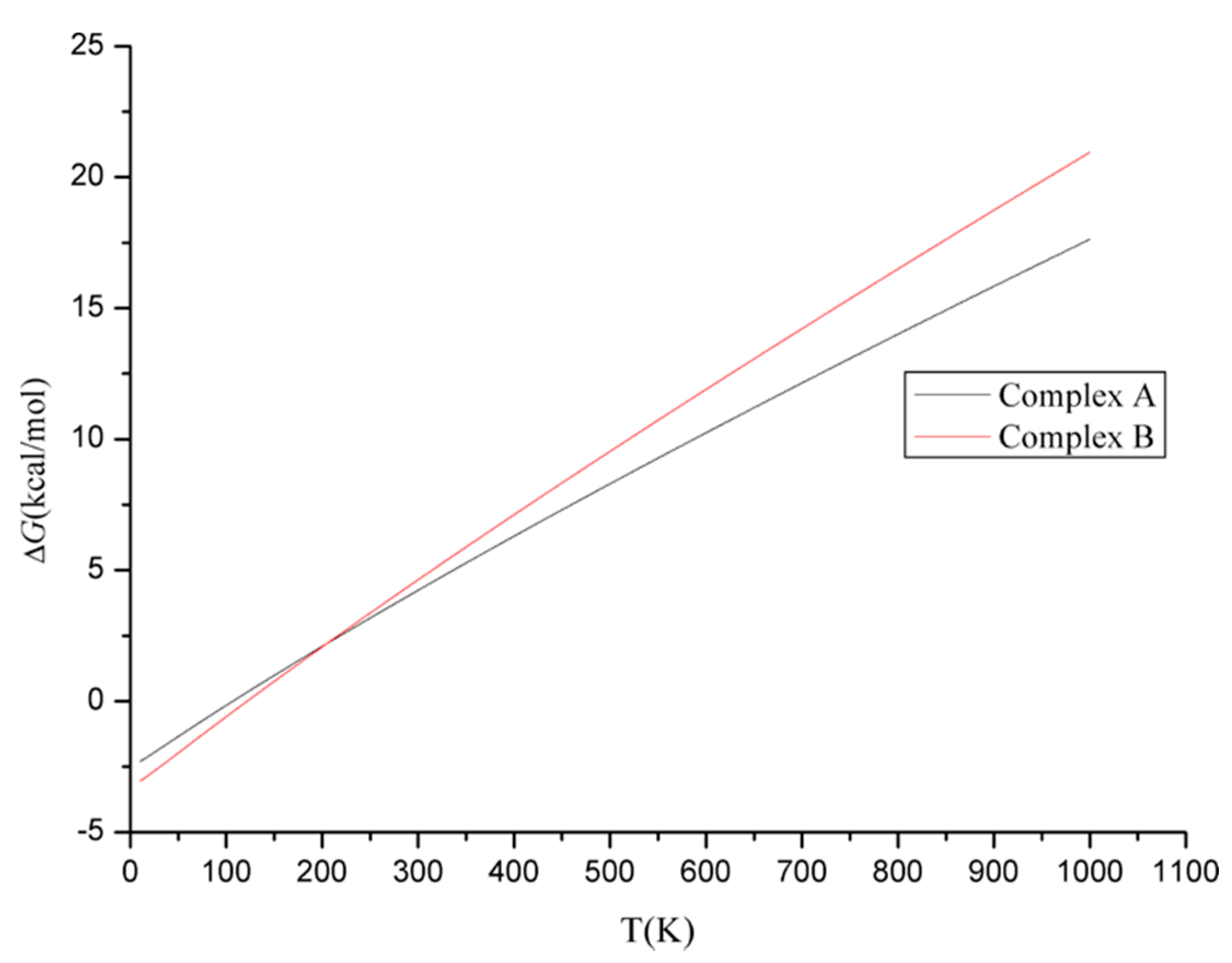
3.1.2. Energy Analyses
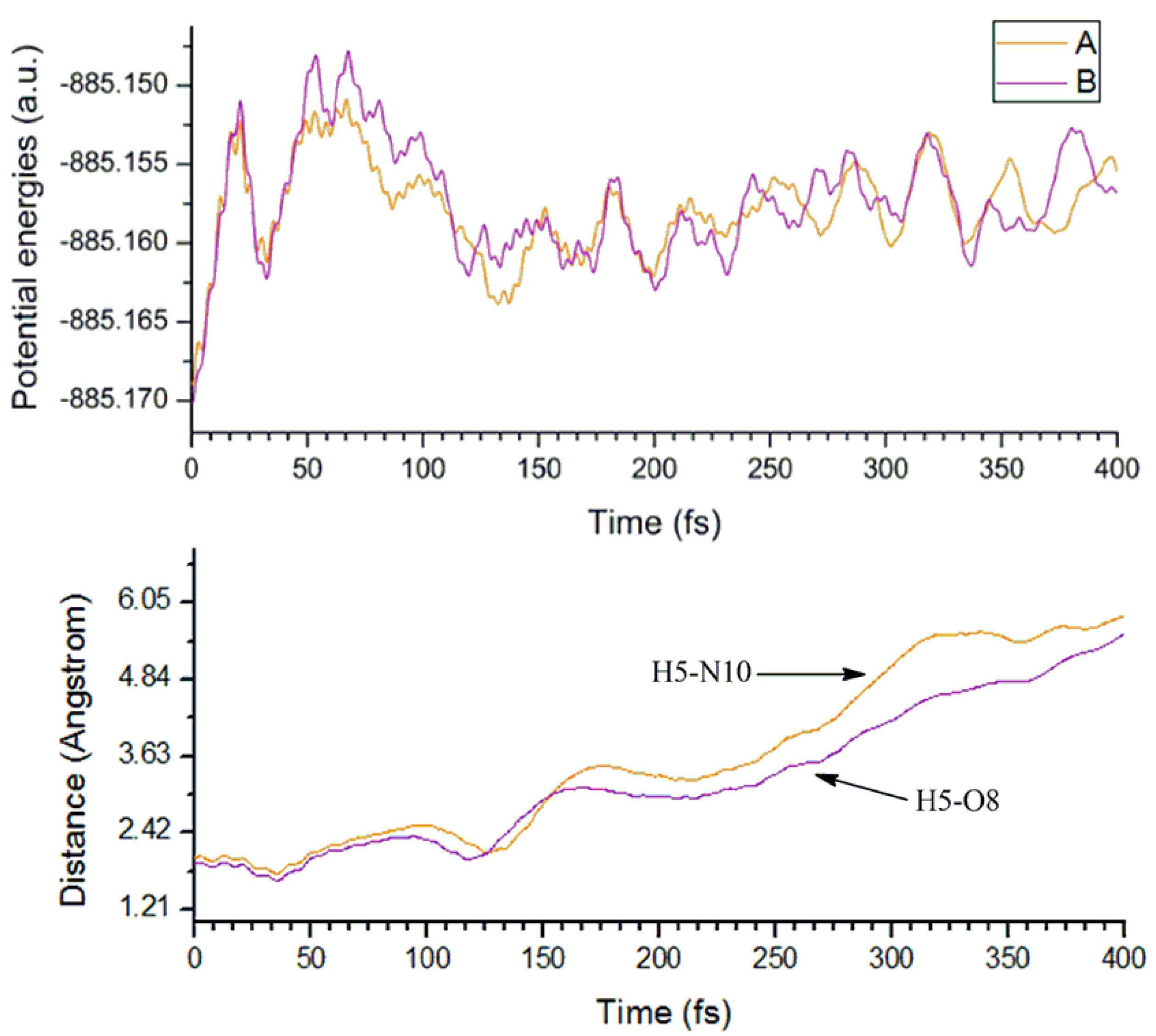
3.1.3. Molecular Dynamics Analyses
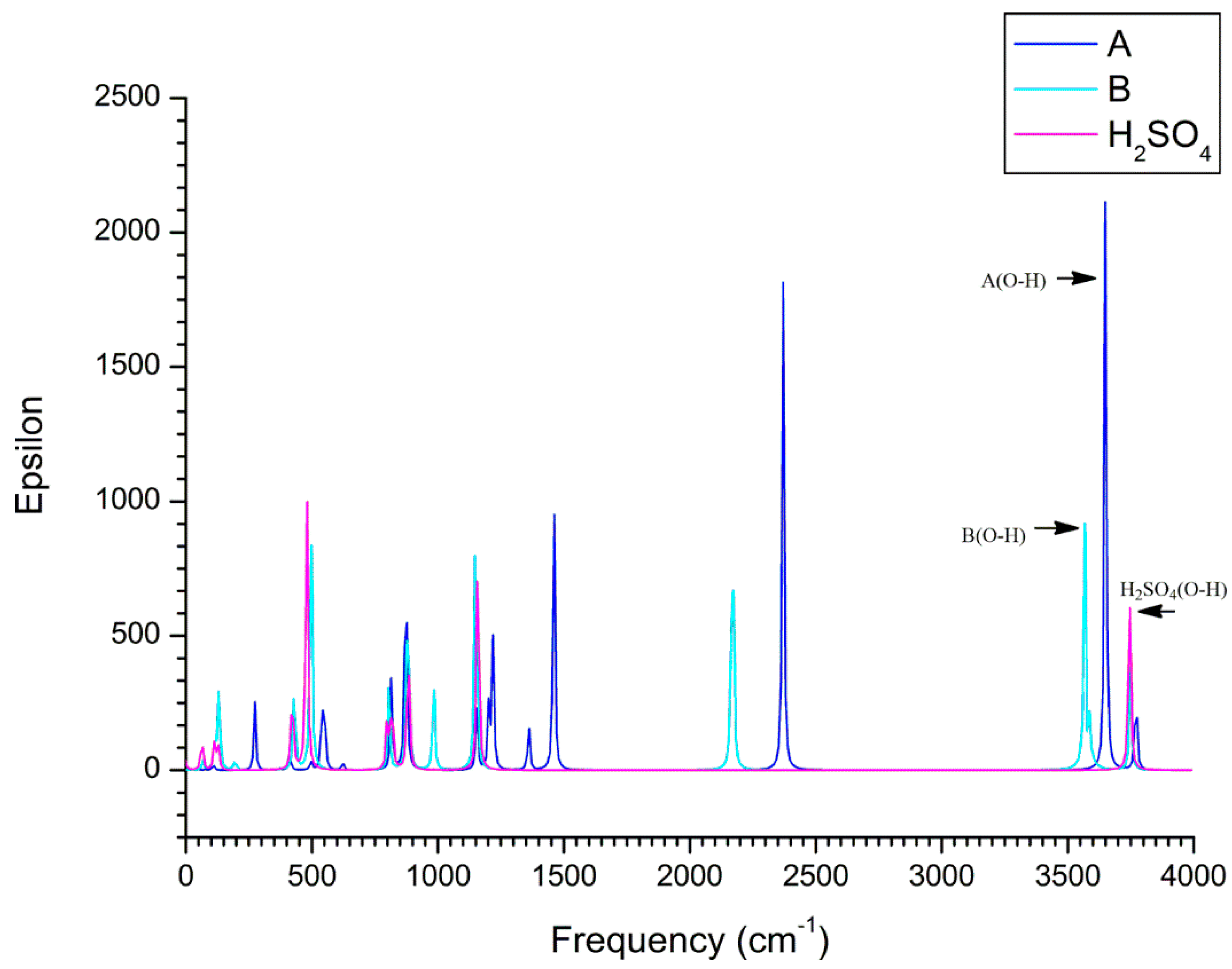
3.2. Electron Capture Process
3.2.1. Structural Features for the Electron Capture Products
3.2.2. Electron Affinity
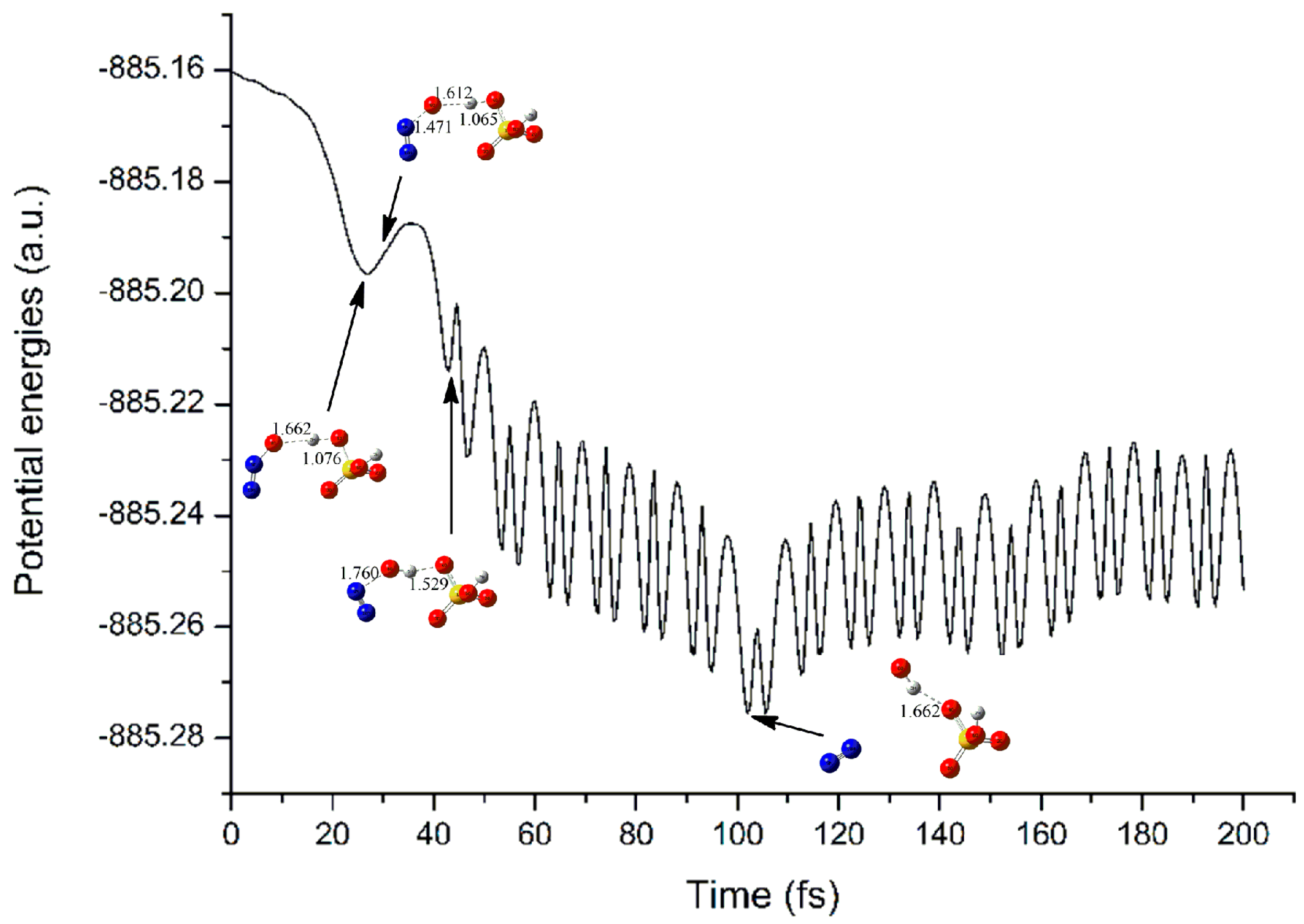
3.2.3. Electron Capture Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Tomicic, M.; Enghoff, M.B.; Svensmark, H. Experimental study of H2SO4 aerosol nucleation at high ionization levels. Atmos. Chem. Phys. 2018, 18, 5921–5930. [Google Scholar] [CrossRef]
- Brus, D.; Škrabalová, L.; Herrmann, E.; Olenius, T.; Trávničková, T.; Makkonen, U.; Merikanto, J. Temperature-dependent diffusion of H2SO4 in air at atmospherically relevant conditions: Laboratory measurements using laminar flow technique. Atmosphere 2017, 8, 132. [Google Scholar] [CrossRef]
- Sipilä, M.; Berndt, T.; Petäjä, T.; Brus, D.; Vanhanen, J.; Stratmann, F.; Patokoski, J.; Mauldin, R.L., III; Hyvärinen, A.; Lihavainen, H.; et al. The role of sulfuric acid in atmospheric nucleation. Science 2010, 327, 1243–1246. [Google Scholar]
- Anglada, J.M.; Olivella, S.; Sole, A. Hydrogen transfer between sulfuric acid and hydroxyl radical in the gas phase: Competition among hydrogen atom transfer, proton-coupled electron-transfer, and double proton transfer. J. Phys. Chem. A 2006, 110, 1982–1990. [Google Scholar] [CrossRef] [PubMed]
- Almeida, J.; Schobesberger, S.; Kürten, A.; Ortega, I.K.; Kupiainen-Määttä, O.; Praplan, A.P.; Adamov, A.; Amorim, A.; Bianchi, F.; Breitenlechner, M.; et al. Molecular understanding of sulphuric acid–amine particle nucleation in the atmosphere. Nature 2013, 502, 359–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Suh, I.; Zhao, J.; Zhang, D.; Fortner, E.C.; Tie, X.; Molina, L.T.; Molina, M.J. Atmospheric new particle formation enhanced by organic acids. Science 2004, 304, 1487–1490. [Google Scholar] [CrossRef] [PubMed]
- Kirkby, J.; Curtius, J.; Almeida, J.; Dunne, E.; Duplissy, J.; Ehrhart, S.; Franchin, A.; Gagné, S.; Ickes, L.; Kürten, A.; et al. Role of sulphuric acid, ammonia and galactic cosmic rays in atmospheric aerosol nucleation. Nature 2011, 476, 429–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehn, M.; Thornton, J.A.; Kleist, E.; Sipilä, M.; Junninen, H.; Pullinen, I.; Springer, M.; Rubach, F.; Tillmann, R.; Lee, B.; et al. A large source of low-volatility secondary organic aerosol. Nature 2014, 506, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Beichert, P.; Schrems, O. Complexes of sulfuric acid with hydrogen chloride, water, nitric acid, chlorine nitrate, and hydrogen peroxide: An ab initio investigation. J. Phys. Chem. A 1998, 102, 10540–10544. [Google Scholar] [CrossRef]
- Metzger, A.; Verheggen, B.; Dommen, J.; Duplissy, J.; Prevot, A.S.H.; Weingartner, E.; Riipinen, I.; Kulmala, M.; Spracklen, D.V.; Carslaw, K.S.; et al. Evidence for the role of organics in aerosol particle formation under atmospheric conditions. Proc. Natl. Acad. Sci. USA 2010, 107, 6646–6651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, C.E.; Francisco, J.S. The formation of a surprisingly stable HO2-H2SO4 complex. J. Am. Chem. Soc. 2001, 123, 10387–10388. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Ma, Z.; Wang, W.; Zhai, Y.; Sun, H.; Bi, S.; Bu, Y. Theoretical studies on the coupling interactions in H2SO4···HOO●···(H2O)n(n = 0–2) clusters: Toward understanding the role of water molecules in the uptake of HOO● radical by sulfuric acid aerosols. Phys. Chem. Chem. Phys. 2011, 13, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Molina, M.J.; Zhang, R.; Wooldridge, P.J.; McMahon, J.R.; Kim, J.E.; Chang, H.Y.; Beyer, K.D. Physical chemistry of the H2SO4/HNO3/H2O system: Implications for polar stratospheric clouds. Science 1993, 261, 1418–1423. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, A.F.; Van der Hoek, K.W.; Olivier, J.G.J. Uncertainties in the global source distribution of nitrous oxide. J. Geophys. Res. 1995, 100, 2785–2800. [Google Scholar] [CrossRef]
- Crutzen, P.J.; Mosier, A.R.; Smith, K.A.; Winiwarter, W. N2O release from agro-biofuel production negates global warming reduction by replacing fossil fuels. Atmos. Chem. Phys. 2008, 8, 389–395. [Google Scholar] [CrossRef] [Green Version]
- World Meteorological Organization. The state of greenhouse gases in the atmosphere based on global observations through 2016. WMO Greenh. Gas Bull. 2017, 13, 1–8. [Google Scholar]
- Ravishankara, A.R.; Daniel, J.S.; Portmann, R.W. Nitrous oxide (N2O): The dominant ozone-depleting substance emitted in the 21st century. Science 2009, 326, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Tachikawa, H.; Abe, S. Reaction dynamics following electron capture of chlorofluorocarbon adsorbed on water cluster: A direct density functional theory molecular dynamics study. J. Chem. Phys. 2007, 126, 194310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tachikawa, H. Dissociative electron capture of halocarbon caused by the internal electron transfer from water trimer anion. Phys. Chem. Chem. Phys. 2008, 10, 2200–2206. [Google Scholar] [CrossRef] [PubMed]
- Tachikawa, H. Direct ab initio MD study on the electron capture dynamics of hydroperoxy radical (HOO)-water complexes. J. Phys. Chem. A 2010, 114, 4951–4956. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.B.; Sanche, L. Effects of cosmic rays on atmospheric chlorofluorocarbon dissociation and ozone depletion. Phys. Rev. Lett. 2001, 87, 078501. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.B. Correlation between cosmic rays and ozone depletion. Phys. Rev. Lett. 2009, 102, 118501. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.B. Cosmic-ray-driven electron-induced reactions of halogenated molecules adsorbed on ice surfaces: Implications for atmospheric ozone depletion and global climate change. Phys. Rep. 2010, 487, 141–167. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Wang, W.; Wei, W.; Feng, W.; Sun, Q.; Li, P. Insights into the reaction mechanism of Criegee intermediate CH2OO with methane and implications for the formation of methanol. J. Phys. Chem. A 2017, 121, 7236–7245. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Wang, W.; Feng, W.; Li, P. Insights into the one-electron reduction behavior of tetrachloro-o-benzoquinone: A DFT and molecular dynamics study. RSC Adv. 2017, 7, 12775–12782. [Google Scholar] [CrossRef]
- Li, P.; Guo, C.; Feng, W.; Sun, Q.; Wang, W. A DFT study on the reaction mechanism between tetrachloro-o-benzoquinone and H2O2 and an alternative reaction approach to produce the hydroxyl radical. RSC Adv. 2017, 7, 22919–22926. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, X.; Li, P.; Sun, Q.; Li, Z.; Ren, C.; Guo, C. CO2 capture and separation from N2/CH4 mixtures by Co@B8/Co@B8– and M@B9/M@B9– (M = Ir, Rh, Ru) clusters: A theoretical study. J. Phys. Chem. A 2015, 119, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, W.; Sun, Q.; Li, Z.; Du, A.; Bi, S.; Zhao, Y. Insights into the mechanism of the reaction between tetrachloro-p-benzoquinone and hydrogen peroxide and their implications in the catalytic role of water molecules in producing the hydroxyl radical. Chem. Phys. Chem. 2013, 14, 2737–2743. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Ma, Z.; Wang, W.; Song, R.; Zhai, Y.; Bi, S.; Sun, H.; Bu, Y. Theoretical studies on the electron capture properties of the H2SO4…HOO● complex and its implications as an alternative source of HOOH. Phys. Chem. Phys. Chem. 2011, 13, 5931–5939. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Schlegel, H.B.; Millam, J.M.; Iyengar, S.S.; Voth, G.A.; Daniels, A.D.; Scuseria, G.E.; Frisch, M.J. Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals. J. Chem. Phys. 2001, 114, 9758–9763. [Google Scholar] [CrossRef]
- Iyengar, S.S.; Schlegel, H.B.; Millam, J.M.; Voth, G.A.; Scuseria, G.E.; Frisch, M.J. Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals. II. Generalizations based on mass-weighting, idempotency, energy conservation and choice of initial conditions. J. Chem. Phys. 2001, 115, 10291–10302. [Google Scholar] [CrossRef]
- Schlegel, H.B.; Iyengar, S.S.; Li, X.; Millam, J.M.; Voth, G.A.; Scuseria, G.E.; Frisch, M.J. Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals. III. Comparison with Born-Oppenheimer dynamics. J. Chem. Phys. 2002, 117, 8694–8704. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. Gaussian, 9th ed.; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Li, P.; Ma, Z.; Wang, W.; Shen, Z.; Bi, S.; Sun, H.; Bu, Y. Coupling interactions between sulfurous acid and the hydroperoxyl radical and implications for the formation of highly stable intermediates. ChemPhysChem 2010, 11, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Torrent-Sucarrat, M.; Anglada, J.M. On the gas phase hydrogen bond complexes between formic acid and hydroperoxyl radical. A theoretical study. J. Phys. Chem. A 2006, 110, 9718–9726. [Google Scholar] [CrossRef] [PubMed]
- Fonseca Guerra, C.; Bickelhaupt, F.M.; Snijders, J.G.; Baerends, E.J. The nature of the hydrogen bond in DNA base pairs: The role of charge transfer and resonance assistance. Chem. Eur. J. 1999, 5, 3581–3594. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |














{kind=link}
{kind=link}
{kind=link}
{kind=link}
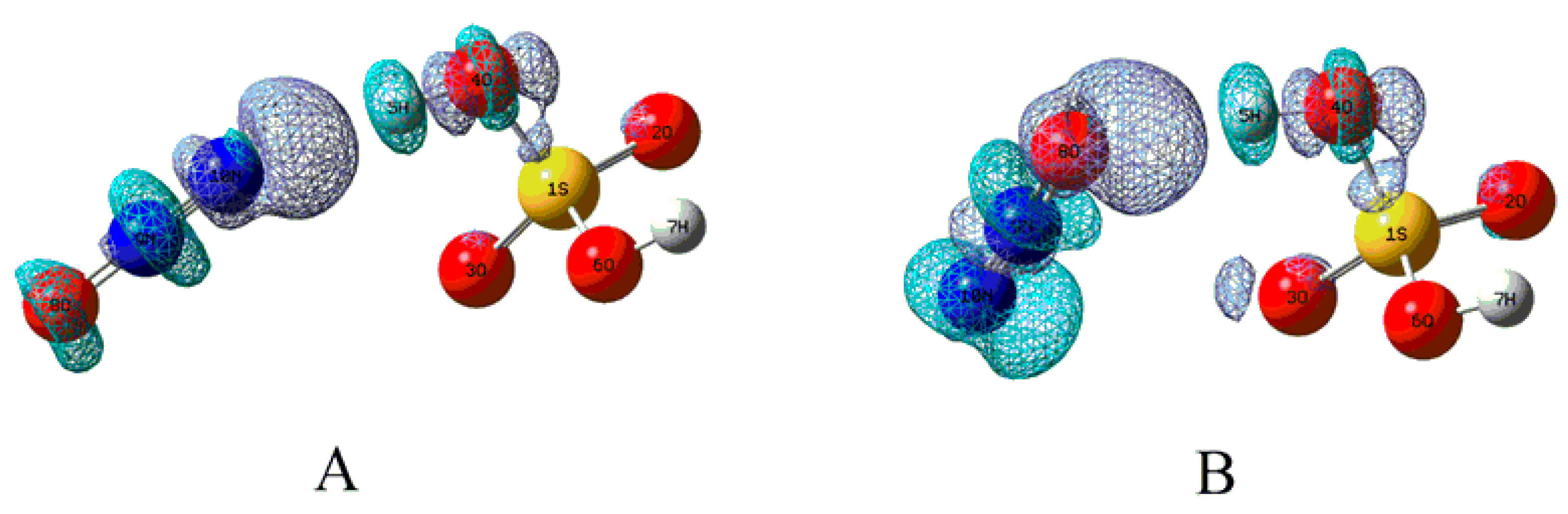{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complexes | BCP | ρbcp | ∇2ρbcp | Gbcp | Vbcp | Hbcp |
|---|---|---|---|---|---|---|
| A | O4-H5···N10 | 0.0198 | 0.0694 | 0.0155 | −0.0137 | 0.0018 |
| B | O4-H5···O8 | 0.0230 | 0.0813 | 0.0190 | −0.0177 | 0.0013 |
| A′ | O6-H7···O8 | 0.0201 | 0.0681 | 0.0156 | −0.0142 | 0.0014 |
| N10-H5···O4 | 0.0816 | 0.0897 | 0.0550 | −0.0876 | −0.0326 | |
| A″ | O8-H7···O6 | 0.0727 | 0.1029 | 0.0523 | −0.0788 | −0.0266 |
| O4-H5···N10 | 0.0147 | 0.0494 | 0.0103 | −0.0083 | 0.0020 | |
| B′ | O8-H5···O4 | 0.0424 | 0.1090 | 0.0340 | −0.0408 | −0.0068 |
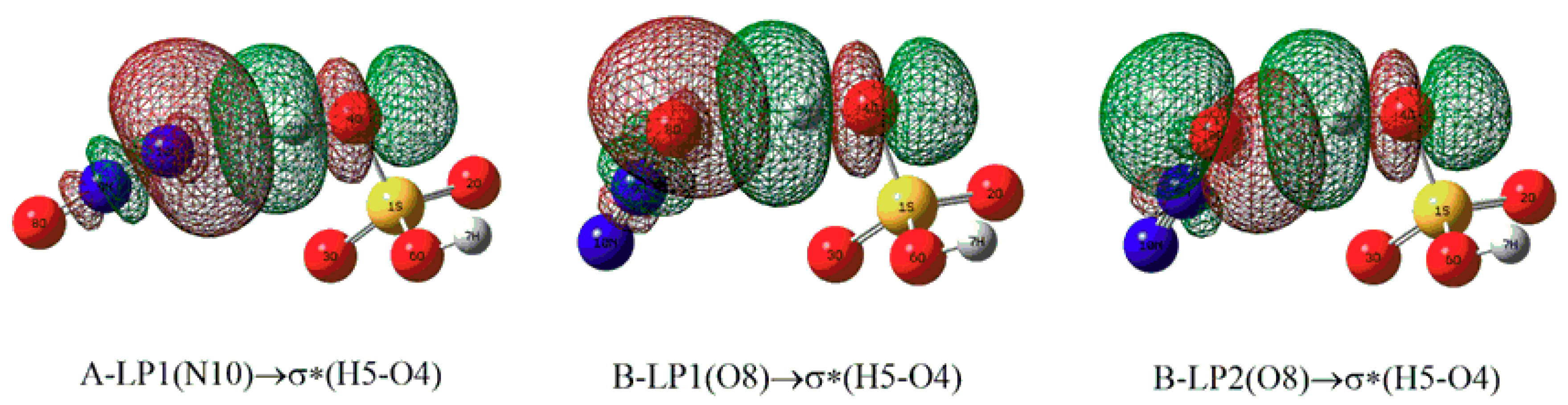
| Complexes | Donor NBO | Acceptor NBO | E(2) |
|---|---|---|---|
| A | LP(N10) | σ*(H5-O4) | 6.38 |
| B | LP1(O8) | σ*(H5-O4) | 2.91 |
| LP2(O8) | σ*(H5-O4) | 4.79 |
| Complexes | ΔEInt | ΔERel | ΔH | ΔG |
|---|---|---|---|---|
| A | −2.14(−2.72) | 0.76(1.72) | −2.07(−2.34) | 4.19(3.92) |
| B | −2.77(−4.22) | 0.00(0.00) | −2.92(−3.93) | 4.58(3.57) |
| Complexes | AEA | VEA | VEDE |
|---|---|---|---|
| A | 1.80 | −0.27 | 5.22 |
| B | 3.18 | −0.27 | 7.44 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.-H.; Feng, W.-L.; Wang, W.-L.; Li, P. Theoretical Insights into the Electron Capture Behavior of H2SO4···N2O Complex: A DFT and Molecular Dynamics Study. Molecules 2018, 23, 2349. https://doi.org/10.3390/molecules23092349
Wang W-H, Feng W-L, Wang W-L, Li P. Theoretical Insights into the Electron Capture Behavior of H2SO4···N2O Complex: A DFT and Molecular Dynamics Study. Molecules. 2018; 23(9):2349. https://doi.org/10.3390/molecules23092349
Chicago/Turabian StyleWang, Wei-Hua, Wen-Ling Feng, Wen-Liang Wang, and Ping Li. 2018. "Theoretical Insights into the Electron Capture Behavior of H2SO4···N2O Complex: A DFT and Molecular Dynamics Study" Molecules 23, no. 9: 2349. https://doi.org/10.3390/molecules23092349
APA StyleWang, W. -H., Feng, W. -L., Wang, W. -L., & Li, P. (2018). Theoretical Insights into the Electron Capture Behavior of H2SO4···N2O Complex: A DFT and Molecular Dynamics Study. Molecules, 23(9), 2349. https://doi.org/10.3390/molecules23092349