
Molecules that Inhibit Bacterial Resistance Enzymes


Abstract
:1. Introduction
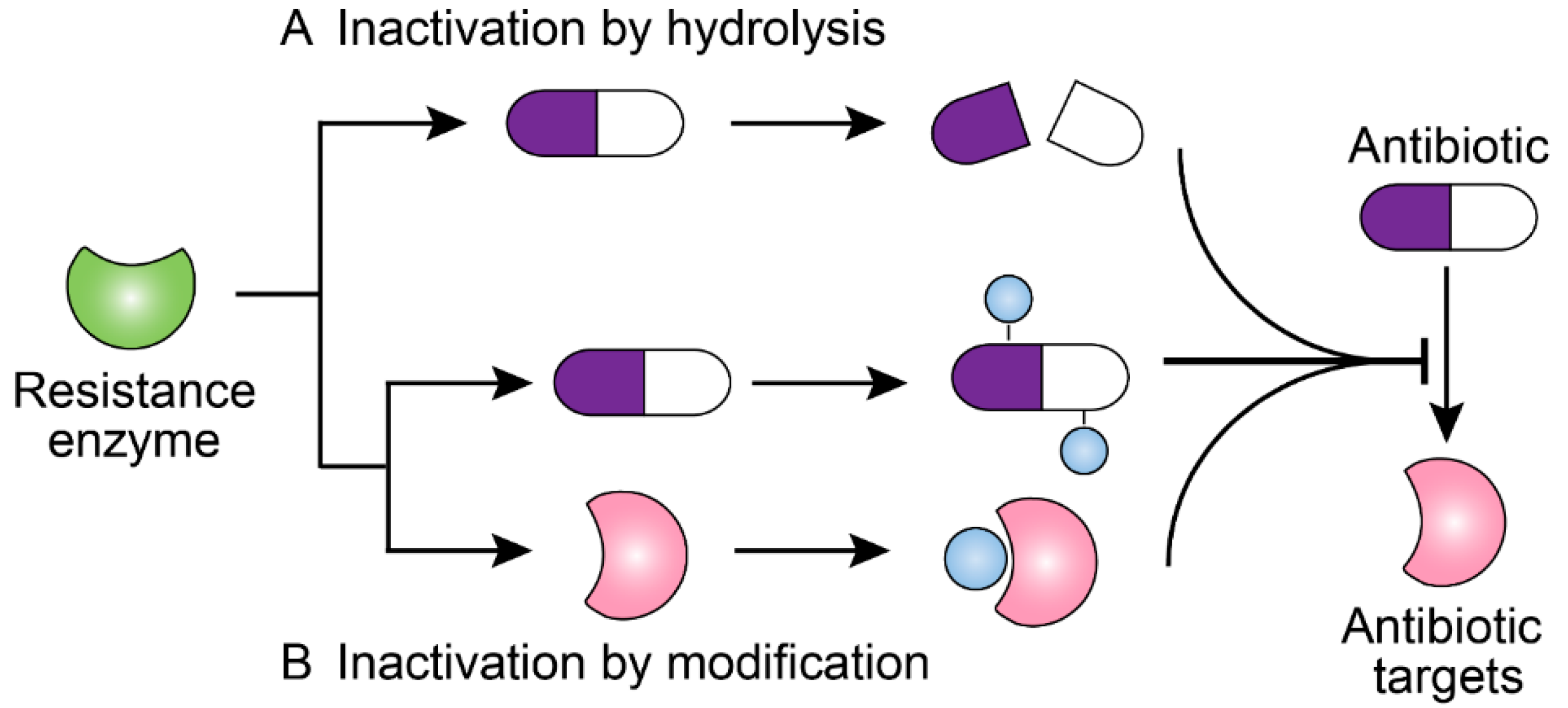
2. Antibiotic Resistance Mediated by Bacterial Enzymes
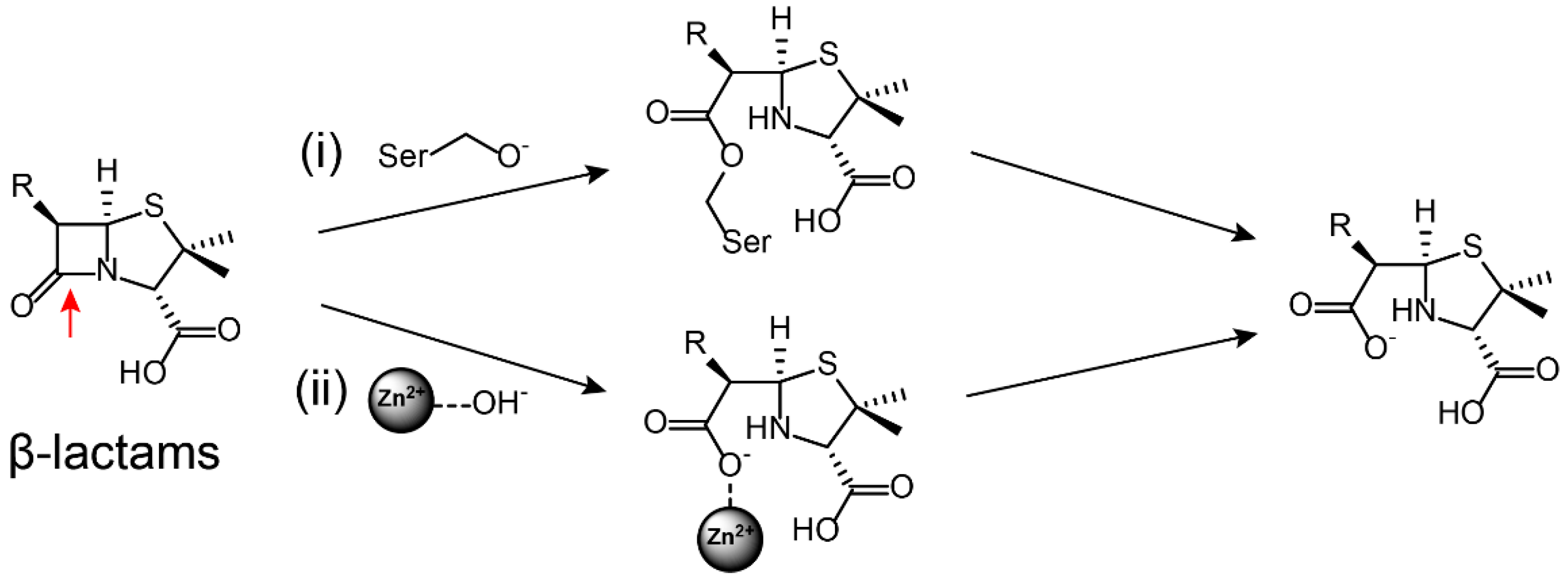
2.1. Inactivation of Antibiotics by Hydrolytic Enzymes
2.2. Inactivation of Antibiotic by Modifying Enzymes
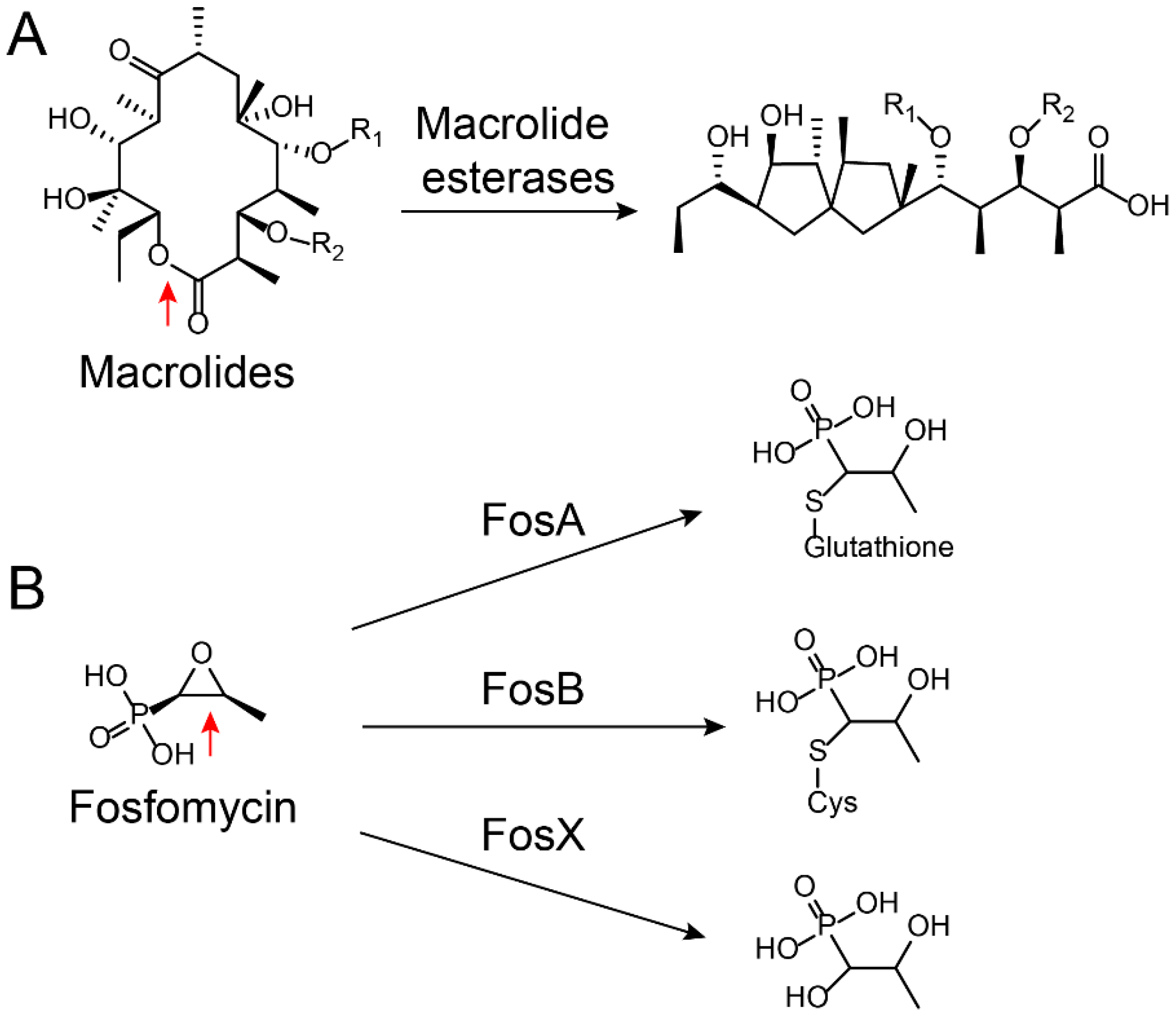
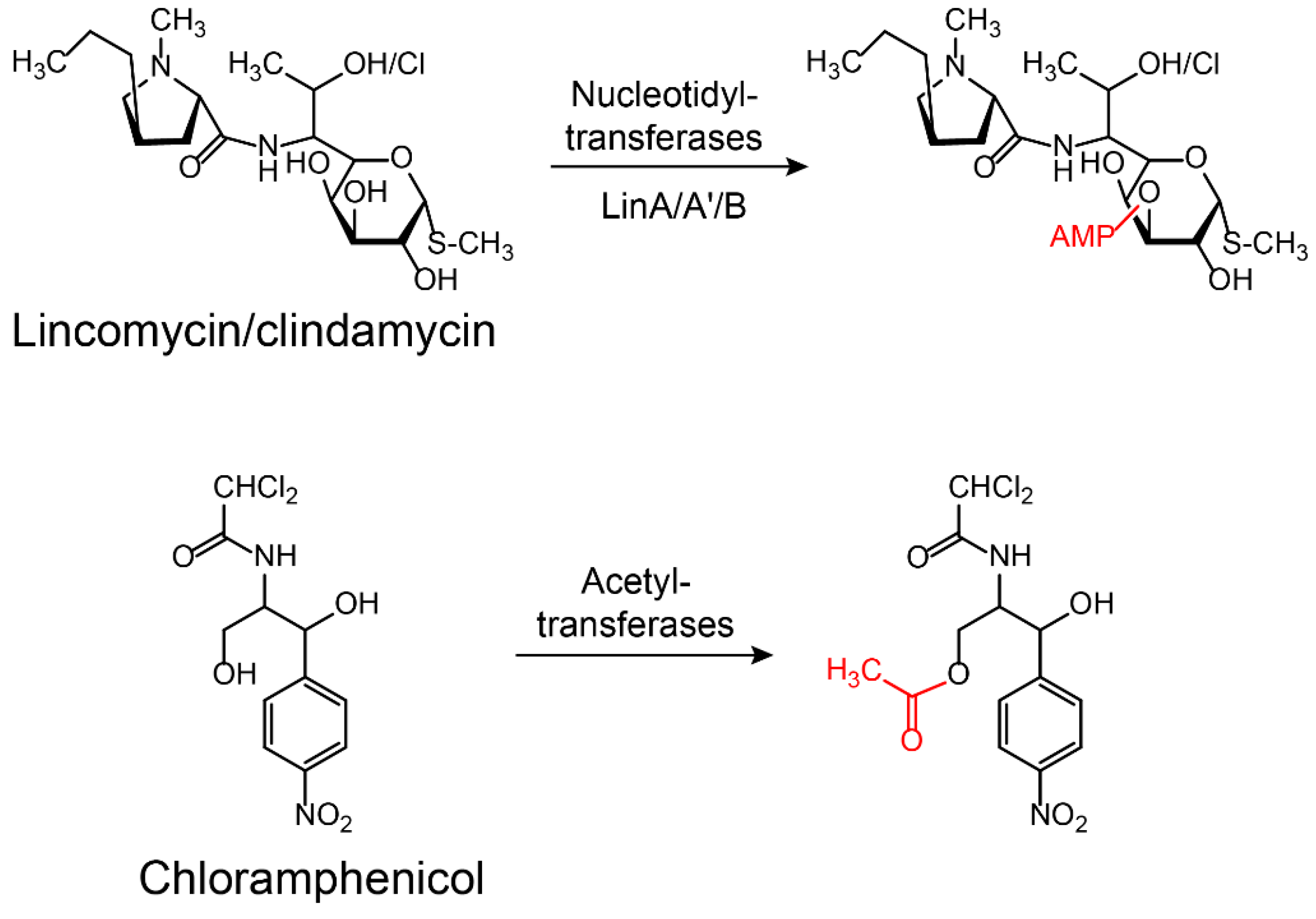
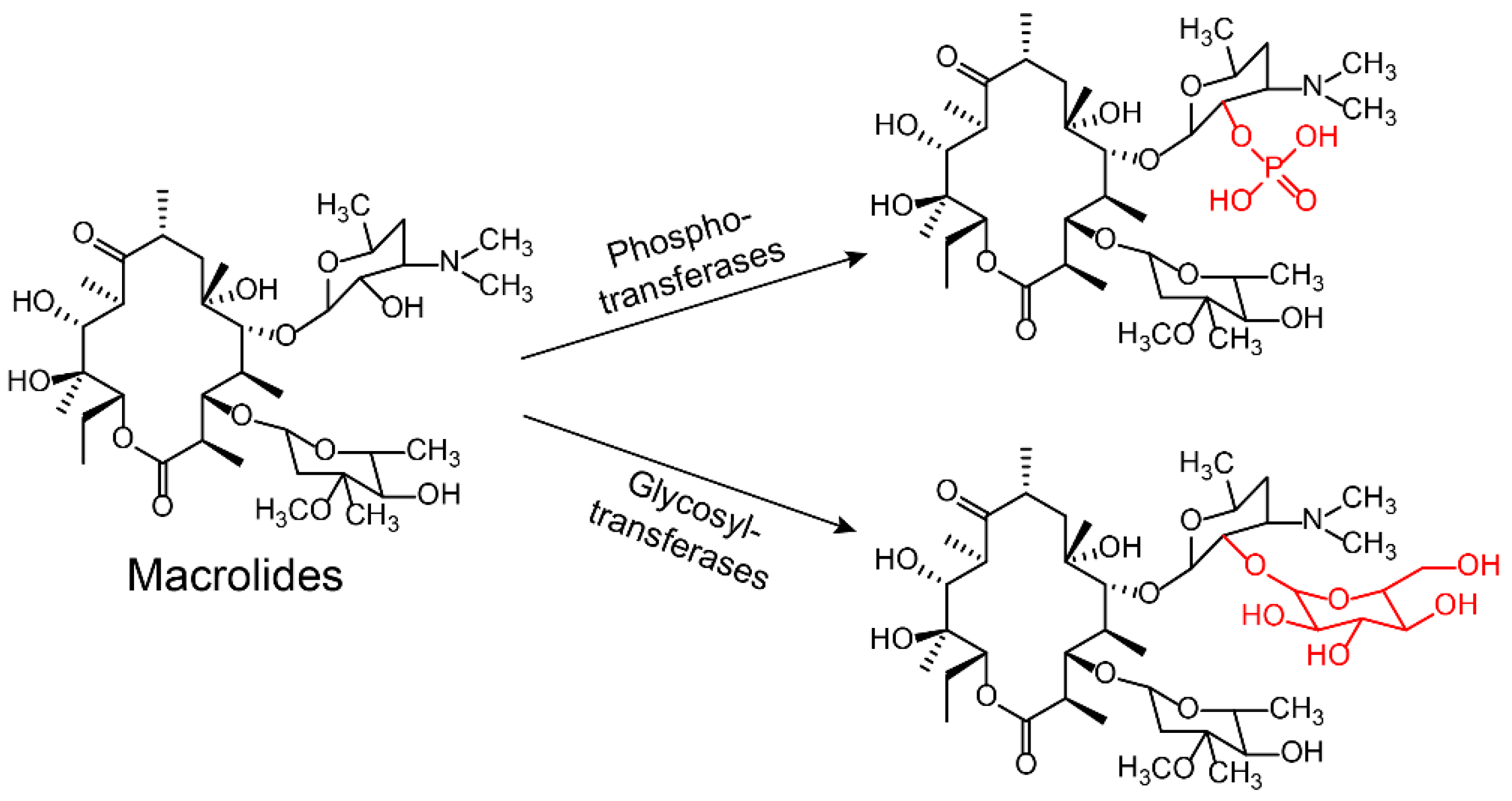
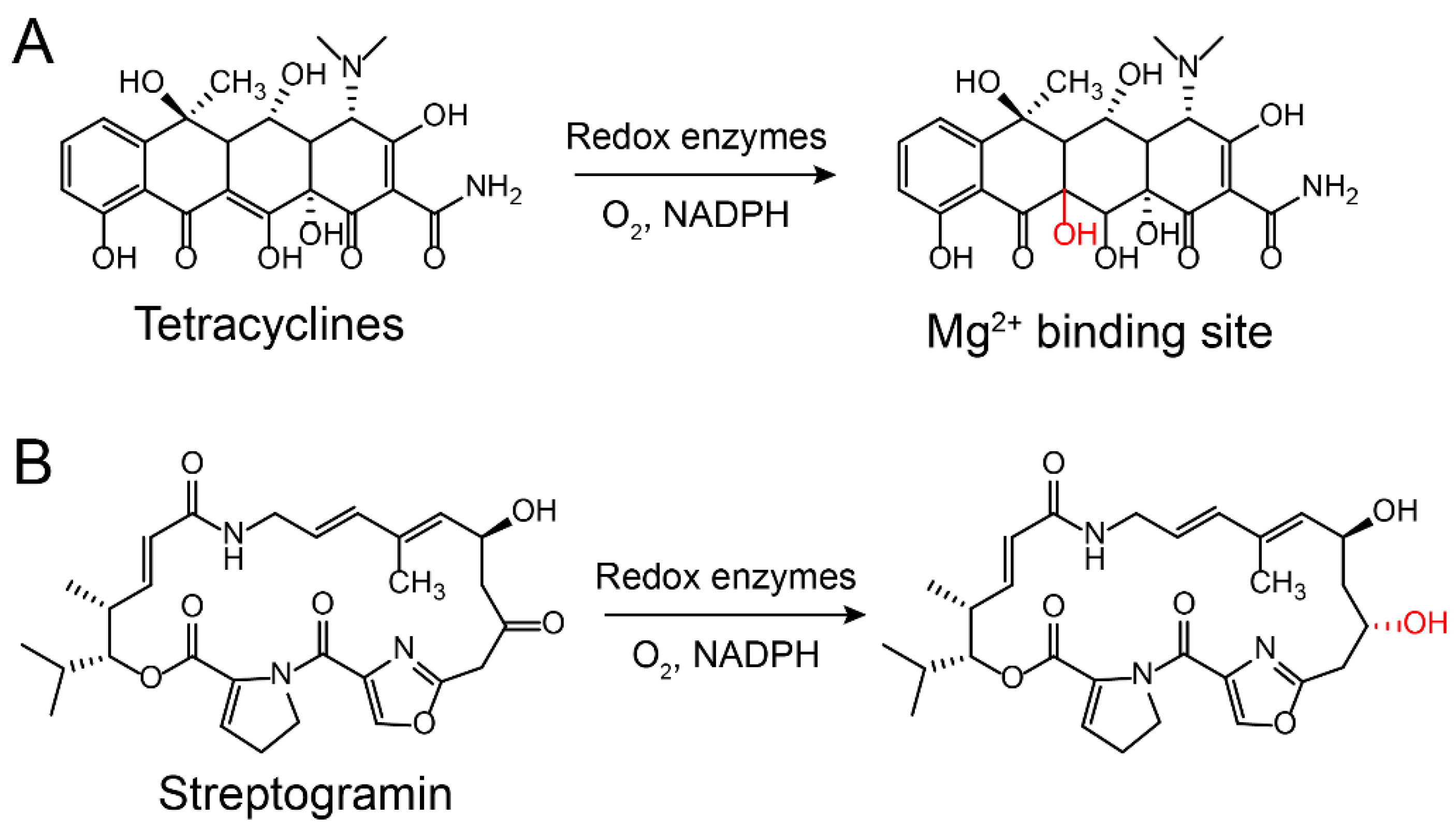
2.2.1. Modification on Antibiotics
2.2.2. Modification of Antibiotic Targets
3. Bacterial Resistance Enzymes Inhibitors
3.1. Hydrolytic Enzymes Inhibitors
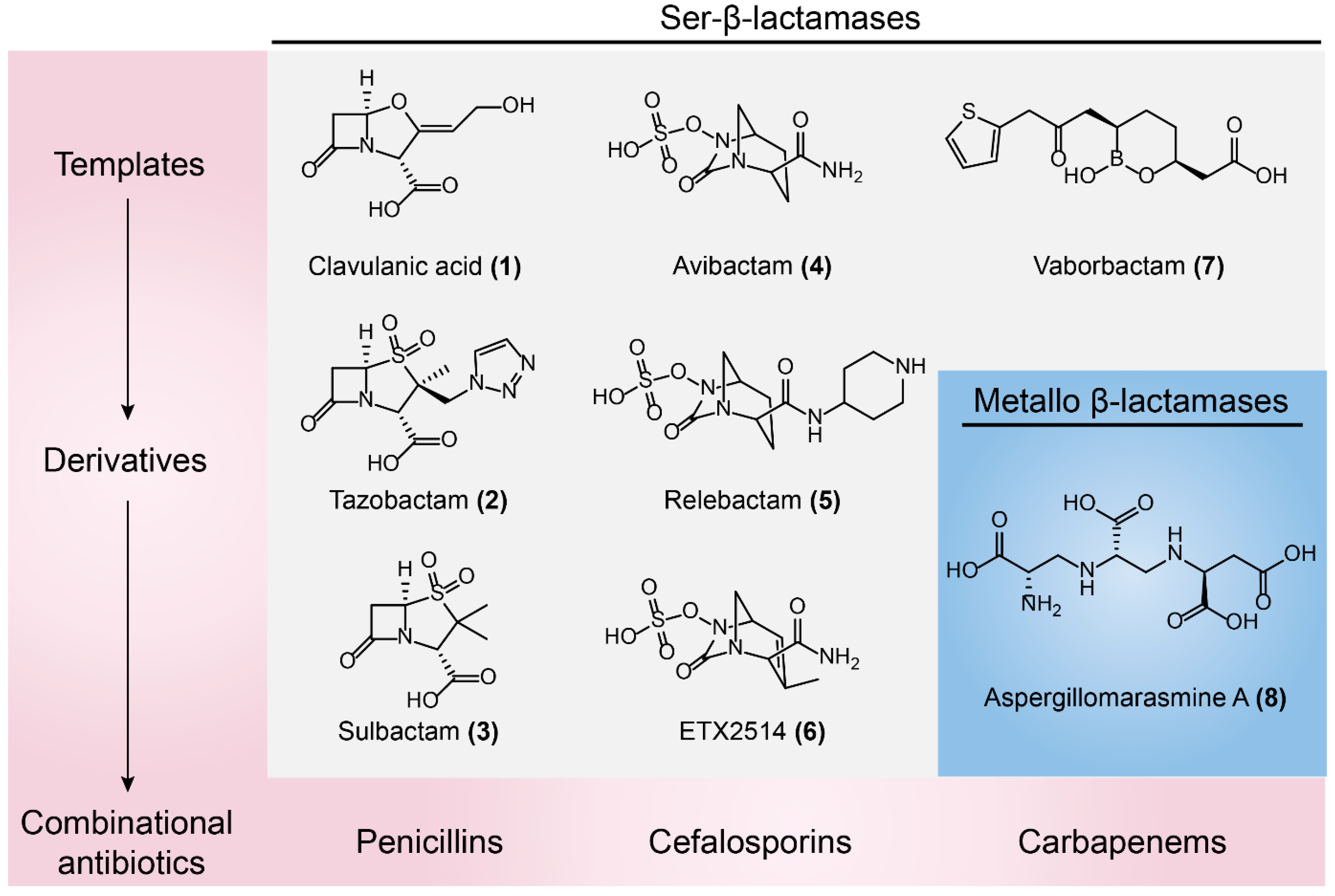
3.1.1. Ser-β-lactamases Inhibitors
3.1.2. Metallo-β-lactamases Inhibitors
3.2. Modifying Enzymes Inhibitors
3.2.1. Aminoglycoside Modifying Enzymes Inhibitors
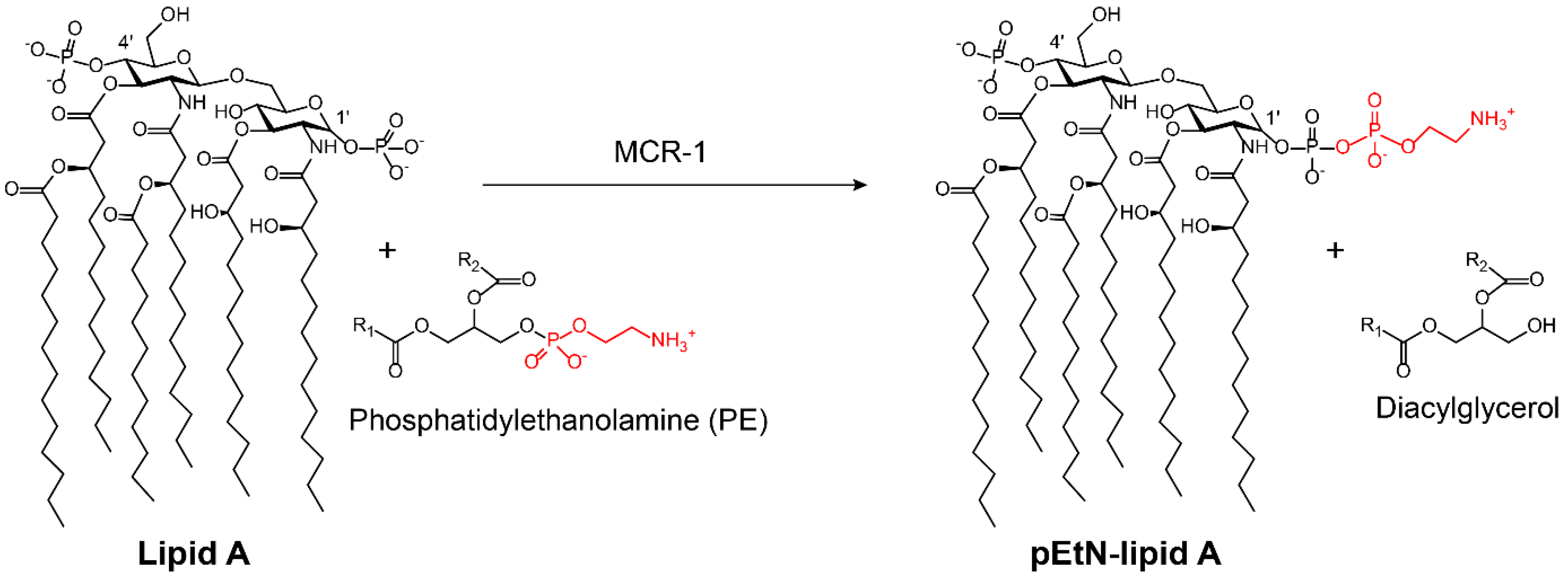
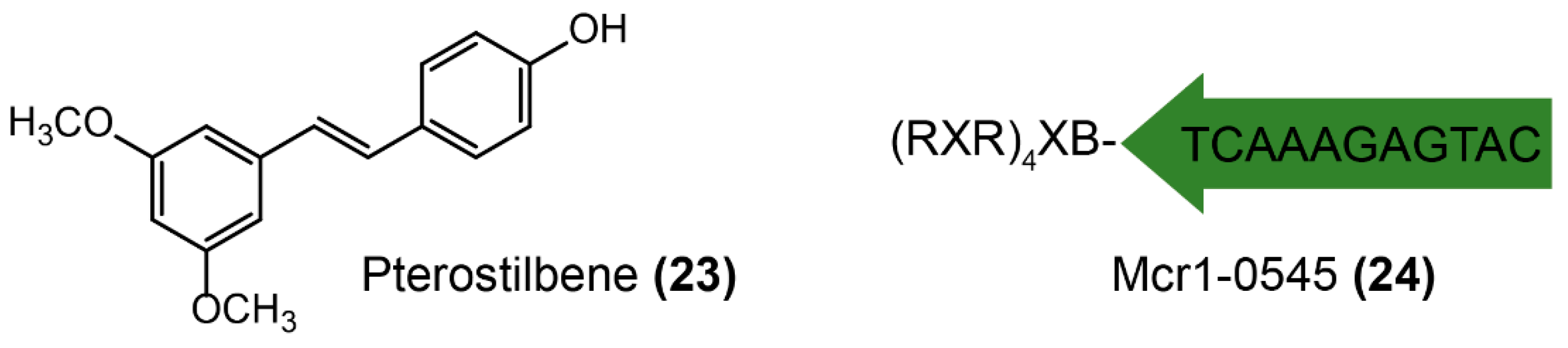
3.2.2. MCR-1 Inhibitors
4. Outlook and Conclusions
Funding
Conflicts of Interest
References
- Kupferschmidt, K. Resistance fighter. Science 2016, 352, 758–761. [Google Scholar] [CrossRef] [PubMed]
- Beatson, S.A.; Walker, M.J. Microbiology. Tracking antibiotic resistance. Science 2014, 345, 1454–1455. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Modarai, M.; Naylor, N.R.; Boyd, S.E.; Atun, R.; Barlow, J.; Holmes, A.H.; Johnson, A.; Robotham, J.V. Quantifying drivers of antibiotic resistance in humans: A. systematic review. Lancet. Infect. Dis. 2018, 18, e368–e378. [Google Scholar] [CrossRef]
- CDC Threat Report: ‘We Will Soon Be in a Post-Antibiotic Era’. Available online: https://www.wired.com/2013/09/cdc-amr-rpt1/ (accessed on 22 December 2018).
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ding, S.; Shen, J.; Zhu, K. Nonribosomal antibacterial peptides that target multidrug-resistant bacteria. Nat. Prod. Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.D.; Wright, G.D. Antibacterial drug discovery in the resistance era. Nature 2016, 529, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug. Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, M.A.; Walsh, C.T. Antibiotics for emerging pathogens. Science 2009, 325, 1089–1093. [Google Scholar] [CrossRef]
- Zipperer, A.; Konnerth, M.C.; Laux, C.; Berscheid, A.; Janek, D.; Weidenmaier, C.; Burian, M.; Schilling, N.A.; Slavetinsky, C.; Marschal, M.; et al. Human commensals producing a novel antibiotic impair pathogen colonization. Nature 2016, 535, 511–516. [Google Scholar] [CrossRef]
- Johnston, C.W.; Skinnider, M.A.; Dejong, C.A.; Rees, P.N.; Chen, G.M.; Walker, C.G.; French, S.; Brown, E.D.; Berdy, J.; Liu, D.Y.; et al. Assembly and clustering of natural antibiotics guides target identification. Nat. Chem. Biol. 2016, 12, 233–239. [Google Scholar] [CrossRef]
- Chu, J.; Vila-Farres, X.; Inoyama, D.; Ternei, M.; Cohen, L.J.; Gordon, E.A.; Reddy, B.V.; Charlop-Powers, Z.; Zebroski, H.A.; Gallardo-Macias, R.; et al. Discovery of MRSA active antibiotics using primary sequence from the human microbiome. Nat. Chem. Biol. 2016, 12, 1004–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schaberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, H.; Urai, M.; Ishii, K.; Yasukawa, J.; Paudel, A.; Murai, M.; Kaji, T.; Kuranaga, T.; Hamase, K.; Katsu, T.; et al. Lysocin E is a new antibiotic that targets menaquinone in the bacterial membrane. Nat. Chem. Biol. 2015, 11, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Cociancich, S.; Pesic, A.; Petras, D.; Uhlmann, S.; Kretz, J.; Schubert, V.; Vieweg, L.; Duplan, S.; Marguerettaz, M.; Noell, J.; et al. The gyrase inhibitor albicidin consists of p-aminobenzoic acids and cyanoalanine. Nat. Chem. Biol. 2015, 11, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Reynolds, K.A.; Kersten, R.D.; Ryan, K.S.; Gonzalez, D.J.; Nizet, V.; Dorrestein, P.C.; Moore, B.S. Direct cloning and refactoring of a silent lipopeptide biosynthetic gene cluster yields the antibiotic taromycin A. Proc. Natl. Acad. Sci. USA 2014, 111, 1957–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, P.A.; Koehler, M.F.T.; Girgis, H.S.; Yan, D.; Chen, Y.; Chen, Y.; Crawford, J.J.; Durk, M.R.; Higuchi, R.I.; Kang, J.; et al. Optimized arylomycins are a new class of Gram-negative antibiotics. Nature 2018, 561, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-X.; Zhong, Z.; Zhang, W.-P.; Qian, P.-Y. Discovery of cationic nonribosomal peptides as Gram-negative antibiotics through global genome mining. Nat. Commun. 2018, 9, 3273. [Google Scholar] [CrossRef]
- Hover, B.; Kim, S.; Katz, M.; Charlop-Powers, Z.; Owen, J.; Ternei, M.; Maniko, J.; Estrela, A.; Molina, H.; Park, S.; et al. Culture-independent discovery of the malacidins as calcium-dependent antibiotics with activity against multidrug-resistant Gram-positive pathogens. Nat. Microbiol. 2018, 3, 415–422. [Google Scholar] [CrossRef]
- Liu, Y.; Song, M.; Ding, S.; Zhu, K. Discovery of linear low-cationic peptides to target methicillin-resistant Staphylococcus aureus in vivo. ACS Infect. Dis. 2018. [Google Scholar] [CrossRef]
- Wright, G.D. Antibiotic adjuvants: Rescuing antibiotics from resistance. Trends Microbiol. 2016, 24, 862–871. [Google Scholar] [CrossRef]
- Farha, M.A.; Brown, E.D. Discovery of antibiotic adjuvants. Nat. Biotechnol. 2013, 31, 120. [Google Scholar] [CrossRef] [PubMed]
- Czaplewski, L.; Bax, R.; Clokie, M.; Dawson, M.; Fairhead, H.; Fischetti, V.A.; Foster, S.; Gilmore, B.F.; Hancock, R.E.; Harper, D. Alternatives to antibiotics—A pipeline portfolio review. Lancet Infect. Dis. 2016, 16, 239–251. [Google Scholar] [CrossRef]
- Johnson, B.; Abramovitch, R. Small molecules that sabotage bacterial virulence. Trends Pharmacol. Sci. 2017, 38, 339–362. [Google Scholar] [CrossRef] [PubMed]
- Garland, M.; Loscher, S.; Bogyo, M. Chemical strategies to target bacterial virulence. Chem. Rev. 2017, 117, 4422–4461. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.D.; Sutherland, A.D. New strategies for combating multidrug-resistant bacteria. Trends Mol. Med. 2007, 13, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Gill, E.E.; Franco, O.L.; Hancock, R.E. Antibiotic adjuvants: Diverse strategies for controlling drug-resistant pathogens. Chem. Biol. Drug Des. 2015, 85, 56–78. [Google Scholar] [CrossRef] [PubMed]
- Blair, J.M.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol 2015, 13, 42–51. [Google Scholar] [CrossRef]
- Alekshun, M.N.; Levy, S.B. Molecular mechanisms of antibacterial multidrug resistance. Cell 2007, 128, 1037–1050. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Goswami, S.; Gorityala, B.K.; Domalaon, R.; Lyu, Y.; Kumar, A.; Zhanel, G.G.; Schweizer, F.M. A tobramycin vector enhances synergy and efficacy of efflux pump inhibitors against multidrug-resistant Gram-negative bacteria. J. Med. Chem. 2017, 60, 3913–3932. [Google Scholar] [CrossRef]
- Lamers, R.; Cavallari, J.; Burrows, L. The efflux inhibitor phenylalanine-arginine beta-naphthylamide (PAβN) permeabilizes the outer membrane of Gram-negative bacteria. PLoS ONE 2013, 8, e60666. [Google Scholar] [CrossRef]
- Abdali, N.; Parks, J.M.; Haynes, K.M.; Chaney, J.L.; Green, A.T.; Wolloscheck, D.; Walker, J.K.; Rybenkov, V.V.; Baudry, J.; Smith, J.C. Reviving antibiotics: Efflux pump inhibitors that interact with AcrA, a membrane fusion protein of the AcrAB-TolC multidrug efflux pump. ACS Infect. Dis. 2016, 3, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.T.; Kwasny, S.M.; Ding, X.; Cardinale, S.C.; McCarthy, C.T.; Kim, H.-S.; Nikaido, H.; Peet, N.P.; Williams, J.D.; Bowlin, T.L. Structure–activity relationships of a novel pyranopyridine series of Gram-negative bacterial efflux pump inhibitors. Bioorg. Med. Chem. 2015, 23, 2024–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hequet, A.; Burchak, O.N.; Jeanty, M.; Guinchard, X.; Le Pihive, E.; Maigre, L.; Bouhours, P.; Schneider, D.; Maurin, M.; Paris, J.M. 1-(1H-Indol-3-yl) ethanamine derivatives as potent Staphylococcus aureus NorA efflux pump inhibitors. ChemMedChem 2014, 9, 1534–1545. [Google Scholar] [CrossRef] [PubMed]
- Opperman, T.J.; Nguyen, S.T. Recent advances toward a molecular mechanism of efflux pump inhibition. Front. Microbiol. 2015, 6, 421. [Google Scholar] [CrossRef] [PubMed]
- Pagès, J.-M.; Masi, M.; Barbe, J. Inhibitors of efflux pumps in Gram-negative bacteria. Trends Mol. Med. 2005, 11, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Venter, H.; Ma, S. Efflux pump inhibitors: A novel approach to combat efflux-mediated drug resistance in bacteria. Curr. Drug Targets 2016, 17, 702–719. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.R.; Weeks, J.; Livermore, D.M.; Toleman, M.A. Dissemination of NDM-1 positive bacteria in the New Delhi environment and its implications for human health: An environmental point prevalence study. Lancet Infect. Dis. 2011, 11, 355–362. [Google Scholar] [CrossRef]
- Liu, Y.-Y.; Wang, Y.; Walsh, T.R.; Yi, L.-X.; Zhang, R.; Spencer, J.; Doi, Y.; Tian, G.; Dong, B.; Huang, X.; et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: A microbiological and molecular biological study. Lancet Infect. Dis. 2016, 16, 161–168. [Google Scholar] [CrossRef]
- Gao, R.; Hu, Y.; Li, Z.; Sun, J.; Wang, Q.; Lin, J.; Ye, H.; Liu, F.; Srinivas, S.; Li, D. Dissemination and mechanism for the MCR-1 colistin resistance. PLoS Pathog. 2016, 12, e1005957. [Google Scholar] [CrossRef]
- Reading, C.; Cole, M. Clavulanic acid: A beta-lactamase-inhibiting beta-lactam from Streptomyces clavuligerus. Antimicrob. Agents Chemother. 1977, 11, 852–857. [Google Scholar] [CrossRef]
- Wright, G.D. Bacterial resistance to antibiotics: Enzymatic degradation and modification. Adv. Drug Deliv. Rev. 2005, 57, 1451–1470. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, G.A.; Munoz-Price, L.S. The new β-lactamases. N. Engl. J. Med. 2005, 352, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.P.; Chain, E. An enzyme from bacteria able to destroy penicillin. 1940. Rev. Infect. Dis. 1940, 10, 677. [Google Scholar]
- Paterson, D.L.; Bonomo, R.A. Extended-spectrum β-lactamases: A. clinical update. Clin. Microbiol. Rev. 2005, 18, 657–686. [Google Scholar] [CrossRef] [PubMed]
- Rd, L.J.; Clark, N.M.; Zhanel, G.G. Evolution of antimicrobial resistance among Enterobacteriaceae (focus on extended spectrum β-lactamases and carbapenemases). Expert Opin. Pharmacother. 2013, 14, 199–210. [Google Scholar]
- Fishbain, J.T.; Sinyavskiy, O.; Riederer, K.; Hujer, A.M.; Bonomo, R.A. Detection of extended spectrum β-lactamase (ESBL) and Klebsiella pneumoniae Carbapenemase (KPC) genes directly from blood cultures using a nucleic acid microarray. J. Clin. Microbiol. 2012. [Google Scholar] [CrossRef]
- Liu, X.; Thungrat, K.; Boothe, D.M. Occurrence of OXA-48 carbapenemase and other β-lactamase genes in ESBL-producing multidrug resistant Escherichia coli from dogs and cats in the United States, 2009–2013. Front. Microbiol. 2016, 7, 1057. [Google Scholar] [CrossRef] [PubMed]
- Chereau, F.; Herindrainy, P.; Garin, B.; Huynh, B.-T.; Randrianirina, F.; Padget, M.; Piola, P.; Guillemot, D.; Delarocque-Astagneau, E. ESBL–and NDM-1-producing Enterobacteriaceae colonization among pregnant women in the community in a low income country: A potential reservoir for transmission of multi-resistant Enterobacteriaceae to neonates. Antimicrob. Agents Chemother. 2015, 59, 3652–3655. [Google Scholar]
- Woodford, N.; Turton, J.F.; Livermore, D.M. Multiresistant Gram-negative bacteria: The role of high-risk clones in the dissemination of antibiotic resistance. FEMS Microbiol. Rev. 2011, 35, 736–755. [Google Scholar] [CrossRef]
- Livermore, D.M. beta-lactamases in laboratory and clinical resistance. Clin. Microbiol. Rev. 1995, 8, 557–584. [Google Scholar] [CrossRef]
- Ambler, R.P. The structure of β-lactamases. Phil. Trans. R. Soc. Lond. B 1980, 289, 321–331. [Google Scholar] [CrossRef]
- Bush, K.; Jacoby, G.A.; Medeiros, A.A. A functional classification scheme for beta-lactamases and its correlation with molecular structure. Antimicrob. Agents Chemother. 1995, 39, 1211. [Google Scholar] [CrossRef] [PubMed]
- Ambler, R.; Coulson, A.; Frere, J.-M.; Ghuysen, J.-M.; Joris, B.; Forsman, M.; Levesque, R.; Tiraby, G.; Waley, S. A standard numbering scheme for the class A beta-lactamases. Biochem. J. 1991, 276, 269. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.R.; Toleman, M.A.; Poirel, L.; Nordmann, P. Metallo-β-lactamases: The quiet before the storm? Clin. Microbiol. Rev. 2005, 18, 306–325. [Google Scholar] [CrossRef] [PubMed]
- Lisa, M.-N.; Palacios, A.R.; Aitha, M.; González, M.M.; Moreno, D.M.; Crowder, M.W.; Bonomo, R.A.; Spencer, J.; Tierney, D.L.; Llarrull, L.I. A general reaction mechanism for carbapenem hydrolysis by mononuclear and binuclear metallo-β-lactamases. Nat. Commun. 2017, 8, 538. [Google Scholar] [CrossRef] [PubMed]
- Kumarasamy, K.K.; Toleman, M.A.; Walsh, T.R.; Bagaria, J.; Butt, F.; Balakrishnan, R.; Chaudhary, U.; Doumith, M.; Giske, C.G.; Irfan, S. Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: A molecular, biological, and epidemiological study. Lancet Infect. Dis. 2010, 10, 597–602. [Google Scholar] [CrossRef]
- Patrice, N.; Laurent, P.; Walsh, T.R.; Livermore, D.M. The emerging NDM carbapenemases. Trends Microbiol. 2011, 19, 588–595. [Google Scholar]
- Johnson, A.P.; Woodford, N. Global spread of antibiotic resistance: The example of New Delhi metallo-β-lactamase (NDM)-mediated carbapenem resistance. J. Med. Microbiol. 2013, 62, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Dortet, L.; Poirel, L.; Nordmann, P. Worldwide dissemination of the NDM-type carbapenemases in Gram-negative bacteria. Biomed Res. Int. 2014, 2014, 249856. [Google Scholar] [CrossRef]
- Nordmann, P.; Naas, T.; Poirel, L. Global spread of carbapenemase-producing Enterobacteriaceae. Emerg. Infect. Dis. 2011, 17, 1791. [Google Scholar] [CrossRef]
- Mohibur, R.; Sanket Kumar, S.; Kashi Nath, P.; Ovejero, C.M.; Binod Kumar, P.; Aparna, T.; Avinash, S.; Srivastava, A.K.; Bruno, G.Z. Prevalence and molecular characterisation of New Delhi metallo-β-lactamases NDM-1, NDM-5, NDM-6 and NDM-7 in multidrug-resistant Enterobacteriaceae from India. Int. J. Antimicrob. Agents 2014, 44, 30–37. [Google Scholar]
- Laurent, D.; Laurent, P.; Nadia, A.; Patrice, N. New Delhi Metallo-β-Lactamase 4–producingEscherichia coliin Cameroon. Emerg. Infect. Dis. 2012, 18, 1540–1542. [Google Scholar]
- Tada, T.; Miyoshi-Akiyama, T.; Dahal, R.K.; Sah, M.K.; Ohara, H.; Kirikae, T.; Pokhrel, B.M. NDM-8 metallo-β-lactamase in a multidrug-resistant Escherichia coli strain isolated in Nepal. Antimicrob. Agents Chemother. 2013, 57, 2394–2396. [Google Scholar] [CrossRef]
- Tada, T.; Shrestha, B.; Miyoshiakiyama, T.; Shimada, K.; Ohara, H.; Kirikae, T.; Pokhrel, B.M. NDM-12, a novel New Delhi metallo-β-Lactamase variant from a carbapenem-resistant Escherichia coli clinical isolate in Nepal. Antimicrob. Agents Chemother. 2014, 58, 6302–6305. [Google Scholar] [CrossRef]
- Shrestha, B.; Tada, T.; Miyoshiakiyama, T.; Shimada, K.; Ohara, H.; Kirikae, T.; Pokhrel, B.M. Identification of a Novel NDM Variant, NDM-13, from a multidrug-resistant Escherichia coli clinical isolate in Nepal. Antimicrob. Agents Chemother. 2015, 59, 5847. [Google Scholar] [CrossRef] [PubMed]
- Zou, D.; Huang, Y.; Zhao, X.; Liu, W.; Dong, D.; Li, H.; Wang, X.; Huang, S.; Wei, X.; Yan, X. A novel New Delhi metallo-β-lactamase variant, NDM-14, isolated in a Chinese hospital possesses increased enzymatic activity against carbapenems. Antimicrob. Agents Chemother. 2015, 59, 2450–2453. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, J.; Wang, X.; Liu, D.; Ke, Y.; Wang, Y.; Shen, J. Novel variant of New Delhi metallo-β-lactamase, NDM-20, in Escherichia coli. Front. Microbiol. 2018, 9, 248. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, Y.; Walsh, T.R.; Liu, D.; Shen, Z.; Zhang, R.; Yin, W.; Yao, H.; Li, J.; Shen, J. Plasmid-mediated novel blaNDM-17 gene encoding a carbapenemase with enhanced activity in a ST48 Escherichia coli strain. Antimicrob. Agents Chemother. 2018, 61, e02233-16. [Google Scholar]
- Nordmann, P.; Poirel, L.; Carrër, A.; Toleman, M.A.; Walsh, T.R. How to detect NDM-1 producers. J. Clin. Microbiol. 2011, 49, 718–721. [Google Scholar] [CrossRef]
- Seiple, I.B.; Zhang, Z.; Jakubec, P.; Langlois-Mercier, A.; Wright, P.M.; Hog, D.T.; Yabu, K.; Allu, S.R.; Fukuzaki, T.; Carlsen, P.N. A platform for the discovery of new macrolide antibiotics. Nature 2016, 533, 338. [Google Scholar] [CrossRef]
- Golkar, T.; Zieliński, M.; Berghuis, A.M. Look and outlook on enzyme-mediated macrolide resistance. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef]
- Morar, M.; Pengelly, K.; Koteva, K.; Wright, G.D. Mechanism and diversity of the erythromycin esterase family of enzymes. Biochemistry 2012, 51, 1740–1751. [Google Scholar] [CrossRef] [PubMed]
- Dinos, G.P. The macrolide antibiotic renaissance. Br. J. Pharmacol. 2017, 174, 2967–2983. [Google Scholar] [CrossRef] [PubMed]
- Falagas, M.E.; Vouloumanou, E.K.; Samonis, G.; Vardakas, K.Z. Fosfomycin. Clin. Microbiol. Rev. 2016, 29, 321–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahan, F.M.; Kahan, J.S.; Cassidy, P.J.; Kropp, H. The mechanism of action of fosfomycin (phosphonomycin). Ann. N. Y. Acad. Sci. 1974, 235, 364–386. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.K.; Keithly, M.E.; Sulikowski, G.A.; Armstrong, R.N. Diversity in fosfomycin resistance proteins. Perspect. Sci. 2015, 4, 17–23. [Google Scholar] [CrossRef]
- Rigsby, R.E.; Fillgrove, K.L.; Beihoffer, L.A.; Armstrong, R.N. Fosfomycin resistance proteins: A nexus of glutathione transferases and epoxide hydrolases in a metalloenzyme superfamily. Methods Enzymol. 2005, 401, 367–379. [Google Scholar] [PubMed]
- Green, K.D.; Chen, W.; Houghton, J.L.; Fridman, M.; Garneau-Tsodikova, S. Exploring the substrate promiscuity of drug-modifying enzymes for the chemoenzymatic generation of N-acylated aminoglycosides. Chembiochem 2010, 11, 119–126. [Google Scholar] [CrossRef]
- Galimand, M.; Fishovitz, J.; Lambert, T.; Barbe, V.; Zajicek, J.; Mobashery, S.; Courvalin, P. AAC(3)-XI, a new aminoglycoside 3-N-acetyltransferase from Corynebacterium striatum. Antimicrob. Agents Chemother. 2015, 59, 5647–5653. [Google Scholar] [CrossRef]
- Wright, G.D.; Thompson, P.R. Aminoglycoside phosphotransferases: Proteins, structure, and mechanism. Front. Biosci. 1999, 4, D9–D21. [Google Scholar]
- Zhang, G.; Leclercq, S.O.; Tian, J.; Wang, C.; Yahara, K.; Ai, G.; Liu, S.; Feng, J. A new subclass of intrinsic aminoglycoside nucleotidyltransferases, ANT(3′′)-II, is horizontally transferred among Acinetobacter spp. by homologous recombination. PLoS Genet. 2017, 13, e1006602. [Google Scholar] [CrossRef]
- Kocaman, S.; Selvaraj, B.; Wright, E.; Cuneo, M.; Serpersu, E. Investigation of the molecular mechanisms which result in aminoglycoside nucleotidyltransferase 4′(ANT4) variants with different levels of thermostability. Biophys. J. 2018, 114, 52a. [Google Scholar] [CrossRef]
- Wang, C.-M.; Zhao, F.-L.; Zhang, L.; Chai, X.-Y.; Meng, Q.-G. Synthesis and antibacterial evaluation of a series of 11, 12-cyclic carbonate azithromycin-3-o-descladinosyl-3-o-carbamoyl glycosyl derivatives. Molecules 2017, 22, 2146. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Tomich, A.D.; McElheny, C.L.; Cooper, V.S.; Tait-Kamradt, A.; Wang, M.; Hu, F.; Rice, L.B.; Sluis-Cremer, N.; Doi, Y. High-level fosfomycin resistance in vancomycin-resistant Enterococcus faecium. Emerg. Infect. Dis. 2017, 23, 1902. [Google Scholar] [CrossRef] [PubMed]
- Falagas, M.E.; Athanasaki, F.; Voulgaris, G.L.; Triarides, N.A.; Vardakas, K.Z. Resistance to fosfomycin: Mechanisms, frequency and clinical consequences. Int. J. Antimicrob. Agents 2018. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.S.; Tolmasky, M.E. Aminoglycoside Modifying Enzymes. Drug Resist. Update. 2010, 13, 151–171. [Google Scholar] [CrossRef]
- Jana, S.; Deb, J. Molecular understanding of aminoglycoside action and resistance. Appl. Microbiol. Biotechnol. 2006, 70, 140–150. [Google Scholar] [CrossRef]
- Azucena, E.; Mobashery, S. Aminoglycoside-modifying enzymes: Mechanisms of catalytic processes and inhibition. Drug Resist. Update. 2001, 4, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Shaw, K.; Rather, P.; Hare, R.; Miller, G. Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol. Rev. 1993, 57, 138–163. [Google Scholar]
- Wright, G.D. Aminoglycoside-modifying enzymes. Curr. Opin. Microbiol. 1999, 2, 499–503. [Google Scholar] [CrossRef]
- Petinaki, E.; Guérin-Faublée, V.; Pichereau, V.; Villers, C.; Achard, A.; Malbruny, B.; Leclercq, R. Lincomycin resistance gene lnu(D) in Streptococcus uberis. Antimicrob. Agents Chemother. 2008, 52, 626–630. [Google Scholar] [CrossRef]
- Robicsek, A.; Strahilevitz, J.; Jacoby, G.A.; Macielag, M.; Abbanat, D.; Park, C.H.; Bush, K.; Hooper, D.C. Fluoroquinolone-modifying enzyme: A new adaptation of a common aminoglycoside acetyltransferase. Nat. Med. 2006, 12, 83. [Google Scholar] [CrossRef] [PubMed]
- Speer, B.S.; Salyers, A.A. Characterization of a novel tetracycline resistance that functions only in aerobically grown Escherichia coli. J. Bacteriol. 1988, 170, 1423–1429. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; LaPara, T.M.; Sadowsky, M.J. Transformation of tetracycline by TetX and its subsequent degradation in a heterologous host. FEMS Microbiol. Ecol. 2015, 91. [Google Scholar] [CrossRef] [Green Version]
- Volkers, G.; Palm, G.J.; Weiss, M.S.; Wright, G.D.; Hinrichs, W. Structural basis for a new tetracycline resistance mechanism relying on the TetX monooxygenase. FEBS Lett. 2011, 585, 1061–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naamala, J.; Jaiswal, S.K.; Dakora, F.D. Antibiotics resistance in Rhizobium: Type, process, mechanism and benefit for agriculture. Curr Microbiol 2016, 72, 804–816. [Google Scholar] [CrossRef] [PubMed]
- Ainsworth, G.; Brown, A.M.; Brownlee, G. ‘Aerosporin’, an antibiotic produced by Bacillus aerosporus. Nature 1947, 160, 263. [Google Scholar] [CrossRef]
- Benedict, R.; Langlykke, A. Antibiotic activity of Bacillus polymyxa. J. Bacteriol. 1947, 54, 24. [Google Scholar]
- Wolinsky, E.; Hines, J.D. Neurotoxic and nephrotoxic effects of colistin in patients with renal disease. N. Engl. J. Med. 1962, 266, 759–762. [Google Scholar] [CrossRef]
- Dai, C.; Li, J.; Li, J. New insight in colistin induced neurotoxicity with the mitochondrial dysfunction in mice central nervous tissues. Exp. Toxicol. Pathol. 2013, 65, 941–948. [Google Scholar] [CrossRef]
- Dai, C.S.; Li, J.C.; Tang, S.S.; Li, J.; Xiao, X.L. Colistin-induced nephrotoxicity in mice involves the mitochondrial, death receptor, and endoplasmic reticulum pathways. Antimicrob. Agents Chemother. 2014, 58, 4075–4085. [Google Scholar] [CrossRef]
- Falagas, M.E.; Kasiakou, S.K.; Saravolatz, L.D. Colistin: The revival of polymyxins for the management of multidrug-resistant Gram-negative bacterial infections. Clin. Infect. Dis. 2005, 40, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Nation, R.L.; Turnidge, J.D.; Milne, R.W.; Coulthard, K.; Rayner, C.R.; Paterson, D.L. Colistin: The re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet. Infect. Dis. 2006, 6, 589–601. [Google Scholar] [CrossRef]
- Velkov, T.; Dai, C.; Ciccotosto, G.D.; Cappai, R.; Hoyer, D.; Li, J. Polymyxins for Cns infections: Pharmacology and neurotoxicity. Pharmacol. Ther. 2018, 181, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Al-Tawfiq, J.A.; Laxminarayan, R.; Mendelson, M. How should we respond to the emergence of plasmid-mediated colistin resistance in humans and animals? Int. J. Infect. Dis. 2017, 54, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Liu, F.; Lin, I.Y.; Gao, G.F.; Zhu, B. Dissemination of the mcr-1 colistin resistance gene. Lancet. Infect. Dis. 2016, 16, 146–147. [Google Scholar] [CrossRef]
- Yang, Y.Q.; Li, Y.X.; Lei, C.W.; Zhang, A.Y.; Wang, H.N. Novel plasmid-mediated colistin resistance gene mcr-7.1 in Klebsiella pneumoniae. J. Antimicrob. Chemother. 2018, 73, 1791–1795. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Zhou, Y.; Li, J.; Yin, W.; Wang, S.; Zhang, S.; Shen, J.; Shen, Z.; Wang, Y. Emergence of a novel mobile colistin resistance gene, mcr-8, in NDM-producing Klebsiella pneumoniae. Emerg. Microbes Infec. 2018, 7, 122. [Google Scholar] [CrossRef]
- Yin, W.; Li, H.; Shen, Y.; Liu, Z.; Wang, S.; Shen, Z.; Zhang, R.; Walsh, T.R.; Shen, J.; Wang, Y. Novel plasmid-mediated colistin resistance gene mcr-3 in Escherichia coli. mBio 2017, 8, e00543-17. [Google Scholar] [CrossRef]
- Carattoli, A.; Villa, L.; Feudi, C.; Curcio, L.; Orsini, S.; Luppi, A.; Pezzotti, G.; Magistrali, C.F. Novel plasmid-mediated colistin resistance mcr-4 gene in Salmonella and Escherichia coli, Italy 2013, Spain and Belgium, 2015 to 2016. Eurosurveillance 2017, 22. [Google Scholar] [CrossRef]
- Borowiak, M.; Fischer, J.; Hammerl, J.A.; Hendriksen, R.S.; Szabo, I.; Malorny, B. Identification of a novel transposon-associated phosphoethanolamine transferase gene, mcr-5, conferring colistin resistance in d-tartrate fermenting Salmonella enterica subsp. enterica serovar Paratyphi B. J. Antimicrob. Chemother. 2017, 72, 3317–3324. [Google Scholar] [CrossRef]
- Abuoun, M.; Stubberfield, E.J.; Duggett, N.A.; Kirchner, M.; Dormer, L.; Nunezgarcia, J.; Randall, L.P.; Lemma, F.; Crook, D.W.; Teale, C. mcr-1 and mcr-2 variant genes identified in Moraxella species isolated from pigs in Great Britain from 2014 to 2015. J. Antimicrob. Chemother. 2017, 72, 2745–2749. [Google Scholar] [CrossRef] [PubMed]
- Xavier, B.B.; Lammens, C.; Ruhal, R.; Kumar-Singh, S.; Butaye, P.; Goossens, H.; Malhotra-Kumar, S. Identification of a novel plasmid-mediated colistin-resistance gene, mcr-2, in Escherichia coli, Belgium, June 2016. Eurosurveillance 2016, 21, 30280. [Google Scholar] [CrossRef] [PubMed]
- Kaminska, K.H.; Purta, E.; Hansen, L.H.; Bujnicki, J.M.; Vester, B.; Long, K.S. Insights into the structure, function and evolution of the radical-SAM 23S rRNA methyltransferase Cfr that confers antibiotic resistance in bacteria. Nucleic Acids Res. 2009, 38, 1652–1663. [Google Scholar] [CrossRef] [Green Version]
- Arias, C.A.; Vallejo, M.; Reyes, J.; Panesso, D.; Moreno, J.; Castañeda, E.; Villegas, M.V.; Murray, B.E.; Quinn, J.P. Clinical and microbiological aspects of linezolid resistance mediated by the cfr gene encoding a 23S rRNA methyltransferase. J. Clin. Microbiol. 2008, 46, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Long, K.S.; Poehlsgaard, J.; Kehrenberg, C.; Schwarz, S.; Vester, B. The Cfr rRNA methyltransferase confers resistance to phenicols, lincosamides, oxazolidinones, pleuromutilins, and streptogramin A antibiotics. Antimicrob. Agents Chemother. 2006, 50, 2500–2505. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.; Kiratisin, P.; Mendes, R.; Panesso, D.; Singh, K.V.; Arias, C.A. Transferable plasmid-mediated resistance to linezolid due to cfr in a human clinical isolate of Enterococcus faecalis. Antimicrob. Agents Chemother. 2012, 56, 3917–3922. [Google Scholar] [CrossRef]
- Shen, J.; Wang, Y.; Schwarz, S. Presence and dissemination of the multiresistance gene cfr in Gram-positive and Gram-negative bacteria. J. Antimicrob. Chemother. 2013, 68, 1697–1706. [Google Scholar] [CrossRef]
- Locke, J.B.; Finn, J.; Hilgers, M.; Morales, G.; Rahawi, S.; Kedar, G.; Picazo, J.J.; Im, W.; Shaw, K.J.; Stein, J.L. Structure-activity relationships of diverse oxazolidinones for linezolid-resistant Staphylococcus aureus strains possessing the cfr methyltransferase gene or ribosomal mutations. Antimicrob. Agents Chemother. 2010, 54, 5337–5343. [Google Scholar] [CrossRef]
- Yang, Q.; Li, M.; Spiller, O.B.; Andrey, D.O.; Hinchliffe, P.; Li, H.; MacLean, C.; Niumsup, P.; Powell, L.; Pritchard, M. Balancing mcr-1 expression and bacterial survival is a delicate equilibrium between essential cellular defence mechanisms. Nat. Commun. 2017, 8, 2054. [Google Scholar] [CrossRef]
- Howarth, T.T.; Brown, A.G.; King, T.J. Clavulanic acid, a novel β-lactam isolated from Streptomyces clavuligerus; X-ray crystal structure analysis. Chem. Commun. 1976, 266–267. [Google Scholar] [CrossRef]
- Neu, H.C.; Fu, K.P. Clavulanic acid, a novel inhibitor of β-lactamases. Antimicrob. Agents Chemother. 1978, 14, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Vons, C.; Barry, C.; Maitre, S.; Pautrat, K.; Leconte, M.; Costaglioli, B.; Karoui, M.; Alves, A.; Dousset, B.; Valleur, P. Amoxicillin plus clavulanic acid versus appendicectomy for treatment of acute uncomplicated appendicitis: An open-label, non-inferiority, randomised controlled trial. Lancet 2011, 377, 1573–1579. [Google Scholar] [CrossRef]
- Lodise Jr, T.P.; Lomaestro, B.; Drusano, G.L. Piperacillin-tazobactam for Pseudomonas aeruginosa infection: Clinical implications of an extended-infusion dosing strategy. Clin. Infect. Dis. 2007, 44, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.S.; Levy, C.E.; Manrique, A.E.I.; Medeiros, E.A.; Costa, S.F. Severe nosocomial infections with imipenem-resistant Acinetobacter baumannii treated with ampicillin/sulbactam. Int. J. Antimicrob. Agents 2003, 21, 58–62. [Google Scholar] [CrossRef]
- Jacoby, G.A. AmpC β-lactamases. Clin. Microbiol. Rev. 2009, 22, 161–182. [Google Scholar] [CrossRef] [PubMed]
- BA, E.; SG, A. OXA β-Lactamases. Clin. Microbiol. Rev. 2014, 27, 241–263. [Google Scholar]
- Coleman, K. Diazabicyclooctanes (DBOs): A potent new class of non-β-lactam β-lactamase inhibitors. Curr. Opin. Microbiol. 2011, 14, 550–555. [Google Scholar] [CrossRef]
- Ehmann, D.E.; Jahić, H.; Ross, P.L.; Gu, R.-F.; Hu, J.; Kern, G.; Walkup, G.K.; Fisher, S.L. Avibactam is a covalent, reversible, non–β-lactam β-lactamase inhibitor. Proc. Natl. Acad. Sci. USA 2012, 109, 11663–11668. [Google Scholar] [CrossRef]
- Bonnet, R. Growing group of extended-spectrum β-lactamases: The CTX-M enzymes. Antimicrob. Agents Chemother. 2004, 48, 1–14. [Google Scholar] [CrossRef]
- Cantón, R.; Coque, T.M. The CTX-M β-lactamase pandemic. Curr. Opin. Microbiol. 2006, 9, 466–475. [Google Scholar] [CrossRef]
- Cantón, R.; González-Alba, J.M.; Galán, J.C. CTX-M enzymes: Origin and diffusion. Front. Microbiol. 2012, 3, 110. [Google Scholar] [CrossRef] [PubMed]
- Yigit, H.; Queenan, A.M.; Anderson, G.J.; Domenech-Sanchez, A.; Biddle, J.W.; Steward, C.D.; Alberti, S.; Bush, K.; Tenover, F.C. Novel carbapenem-hydrolyzing β-lactamase, KPC-1, from a carbapenem-resistant strain of Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2001, 45, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, P.; Cuzon, G.; Naas, T. The real threat of Klebsiella pneumoniae carbapenemase-producing bacteria. Lancet. Infect. Dis. 2009, 9, 228–236. [Google Scholar] [CrossRef]
- US Food and Drug Administration, FDA Approves New Antibacterial Drug Avycaz. US FDA 2015. Available online: https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugInnovation/ucm474696.htm (accessed on 22 December 2018).
- Lucasti, C.; Vasile, L.; Sandesc, D.; Venskutonis, D.; McLeroth, P.; Lala, M.; Rizk, M.L.; Brown, M.L.; Losada, M.C.; Pedley, A. Phase 2, dose-ranging study of relebactam with imipenem/cilastatin in subjects with complicated intra-abdominal infection. Antimicrob. Agents Chemother. 2016, 60, 6234–6243. [Google Scholar] [CrossRef] [PubMed]
- Lapuebla, A.; Abdallah, M.; Olafisoye, O.; Cortes, C.; Urban, C.; Landman, D.; Quale, J. Activity of imipenem with relebactam against Gram-negative pathogens from New York City. Antimicrob. Agents Chemother. 2015, 59, 5029–5031. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, S.D.; Mangani, S.; Jahić, H.; Benvenuti, M.; Durand-Reville, T.F.; De Luca, F.; Ehmann, D.E.; Rossolini, G.M.; Alm, R.A.; Docquier, J.-D. Molecular basis of selective inhibition and slow reversibility of avibactam against class D carbapenemases: A structure-guided study of OXA-24 and OXA-48. ACS Chem. Biol. 2014, 10, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Durandréville, T.F.; Guler, S.; Comitaprevoir, J.; Chen, B.; Bifulco, N.; Huynh, H.; Lahiri, S.; Shapiro, A.B.; Mcleod, S.M.; Carter, N.M. ETX2514 is a broad-spectrum β-lactamase inhibitor for the treatment of drug-resistant Gram-negative bacteria including Acinetobacter baumannii. Nat. Microbiol. 2017, 2, 17104. [Google Scholar] [CrossRef]
- Castanheira, M.; Huband, M.D.; Mendes, R.E.; Flamm, R.K. Meropenem-vaborbactam tested against contemporary Gram-negative isolates collected worldwide during 2014, including carbapenem-resistant, KPC-producing, multidrug-resistant, and extensively drug-resistant Enterobacteriaceae. Antimicrob. Agents Chemother. 2017, 62, e01904-17. [Google Scholar] [CrossRef]
- Brem, J.; van Berkel, S.S.; Zollman, D.; Lee, S.Y.; Gileadi, O.; McHugh, P.J.; Walsh, T.R.; McDonough, M.A.; Schofield, C.J. Structural basis of metallo-β-lactamase inhibition by captopril stereoisomers. Antimicrob. Agents Chemother. 2016, 60, 142–150. [Google Scholar] [CrossRef]
- García-Sáez, I.; Hopkins, J.; Papamicael, C.; Franceschini, N.; Amicosante, G.; Rossolini, G.M.; Galleni, M.; Frère, J.-M.; Dideberg, O. The 1.5-Å structure of Chryseobacterium meningosepticum zinc β-lactamase in complex with the inhibitor, D-captopril. J. Biol. Chem. 2003, 278, 23868–23873. [Google Scholar] [CrossRef]
- Klingler, F.-M.; Wichelhaus, T.A.; Frank, D.; Cuesta-Bernal, J.; El-Delik, J.; Möller, H.F.; Sjuts, H.; Göttig, S.; Koenigs, A.; Pos, K.M. Approved drugs containing thiols as inhibitors of metallo-β-lactamases: Strategy to combat multidrug-resistant bacteria. J. Med. Chem. 2015, 58, 3626–3630. [Google Scholar] [CrossRef] [PubMed]
- Li, G.-B.; Abboud, M.I.; Brem, J.; Someya, H.; Lohans, C.T.; Yang, S.-Y.; Spencer, J.; Wareham, D.W.; McDonough, M.A.; Schofield, C.J. NMR-filtered virtual screening leads to non-metal chelating metallo-β-lactamase inhibitors. Chem. Sci. 2017, 8, 928–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CM, R.; GD, W. Inhibitors of metallo-β-lactamases. Curr. Opin. Microbiol. 2017, 39, 96. [Google Scholar]
- Bach, E.; Christensen, S.; Dalgaard, L.; Larsen, P.; Olsen, C.; Smedegård-Petersen, V. Structures, properties and relationship to the aspergillomarasmines of toxins produced by Pyrenophora teres. Physiol. Plant Pathol. 1979, 14, 41–46. [Google Scholar] [CrossRef]
- Kalinka, K.; King, A.M.; Capretta, A.; Wright, G.D. Total synthesis and activity of the metallo-β-lactamase inhibitor Aspergillomarasmine A. Angew. Chem. Int. Ed. 2016, 55, 2210–2212. [Google Scholar]
- King, A.M.; Reid-Yu, S.A.; Wang, W.; King, D.T.; De Pascale, G.; Strynadka, N.C.; Walsh, T.R.; Coombes, B.K.; Wright, G.D. Aspergillomarasmine A overcomes metallo-β-lactamase antibiotic resistance. Nature 2014, 510, 503–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
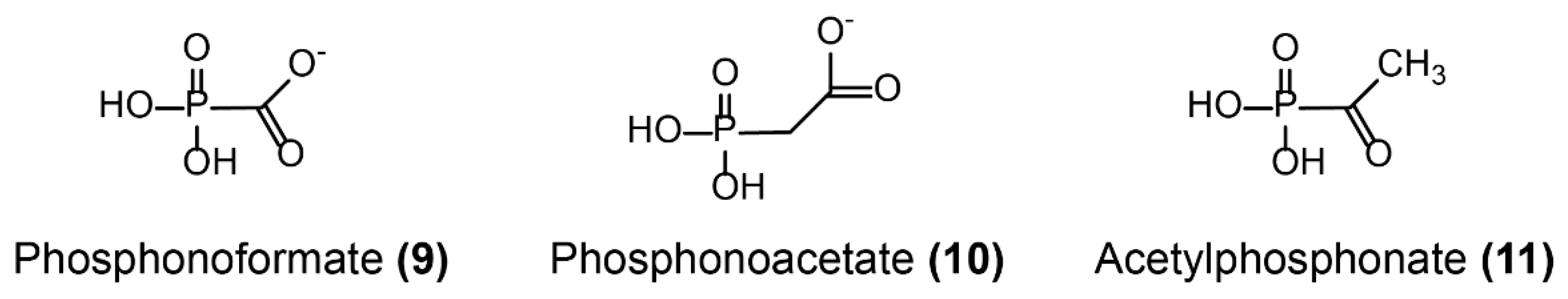
- Rigsby, R.E.; Rife, C.L.; Fillgrove, K.L.; Newcomer, M.E.; Armstrong, R.N. Phosphonoformate: A minimal transition state analogue inhibitor of the fosfomycin resistance protein, FosA. Biochemistry 2004, 43, 13666–13673. [Google Scholar] [CrossRef]
- Fourmy, D.; Recht, M.I.; Blanchard, S.C.; Puglisi, J.D. Structure of the A site of Escherichia coli 16S ribosomal RNA complexed with an aminoglycoside antibiotic. Science 1996, 274, 1367–1371. [Google Scholar] [CrossRef]
- Benveniste, R.; Davies, J. Structure-activity relationships among the aminoglycoside antibiotics: Role of hydroxyl and amino groups. Antimicrob. Agents Chemother. 1973, 4, 402–409. [Google Scholar] [CrossRef]
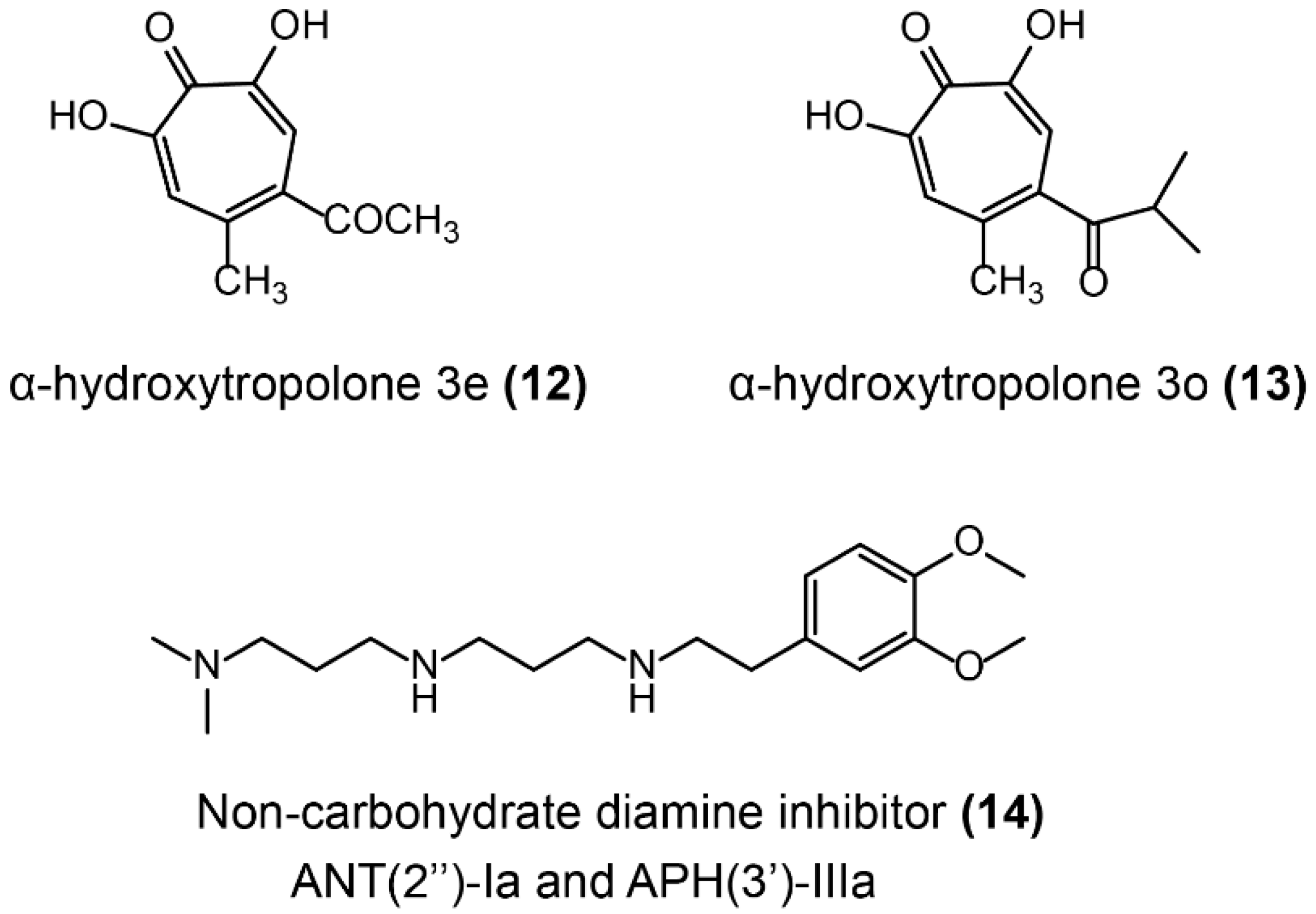
- Hirsch, D.R.; Cox, G.; D’Erasmo, M.P.; Shakya, T.; Meck, C.; Mohd, N.; Wright, G.D.; Murelli, R.P. Inhibition of the ANT(2″)-Ia resistance enzyme and rescue of aminoglycoside antibiotic activity by synthetic α-hydroxytropolones. Bioorg. Med. Chem. Lett. 2014, 24, 4943–4947. [Google Scholar] [CrossRef] [Green Version]
- Welch, K.T.; Virga, K.G.; Whittemore, N.A.; Özen, C.; Wright, E.; Brown, C.L.; Lee, R.E.; Serpersu, E.H. Discovery of non-carbohydrate inhibitors of aminoglycoside-modifying enzymes. Bioorg. Med. Chem. 2005, 13, 6252–6263. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Yan, X.; Shakya, T.; Baettig, O.M.; Ait-Mohand-Brunet, S.; Berghuis, A.M.; Wright, G.D.; Auclair, K. Synthesis and structure-activity relationships of truncated bisubstrate inhibitors of aminoglycoside 6′-N-acetyltransferases. J. Med. Chem. 2006, 49, 5273–5281. [Google Scholar] [CrossRef] [PubMed]
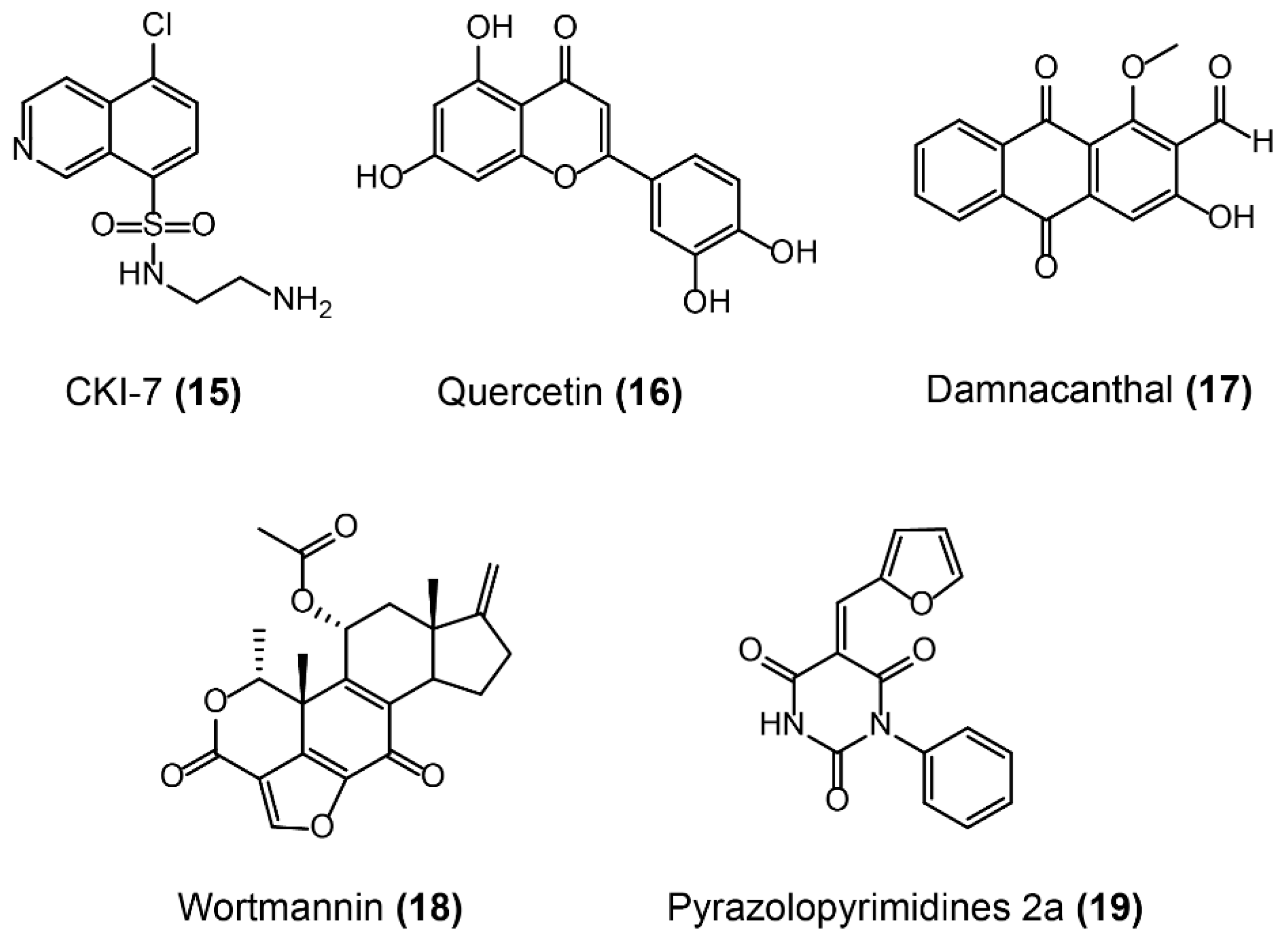
- Daigle, D.M.; Mckay, G.A.; Wright, G.D. Inhibition of aminoglycoside antibiotic resistance enzymes by protein kinase inhibitors. J. Biol. Chem. 1997, 272, 24755. [Google Scholar] [CrossRef] [PubMed]
- Shakya, T.; Stogios, P.J.; Waglechner, N.; Evdokimova, E.; Ejim, L.; Blanchard, J.E.; McArthur, A.G.; Savchenko, A.; Wright, G.D. A small molecule discrimination map of the antibiotic resistance kinome. Chem. Biol. 2011, 18, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Sampei, K.; Nakagawa, M.; Uchiumi, N.; Amanuma, T.; Aiba, S.; Oikawa, M.; Mizuno, K. Damnacanthal, an effective inhibitor of LIM-kinase, inhibits cell migration and invasion. Mol. Biol. Cell 2014, 25, 828–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Jiang, N.; Wu, J.; Dai, W.; Rosenblum, J.S. Polo-like kinases inhibited by wortmannin Labeling site and downstream effects. J. Biol. Chem. 2007, 282, 2505–2511. [Google Scholar] [CrossRef] [PubMed]
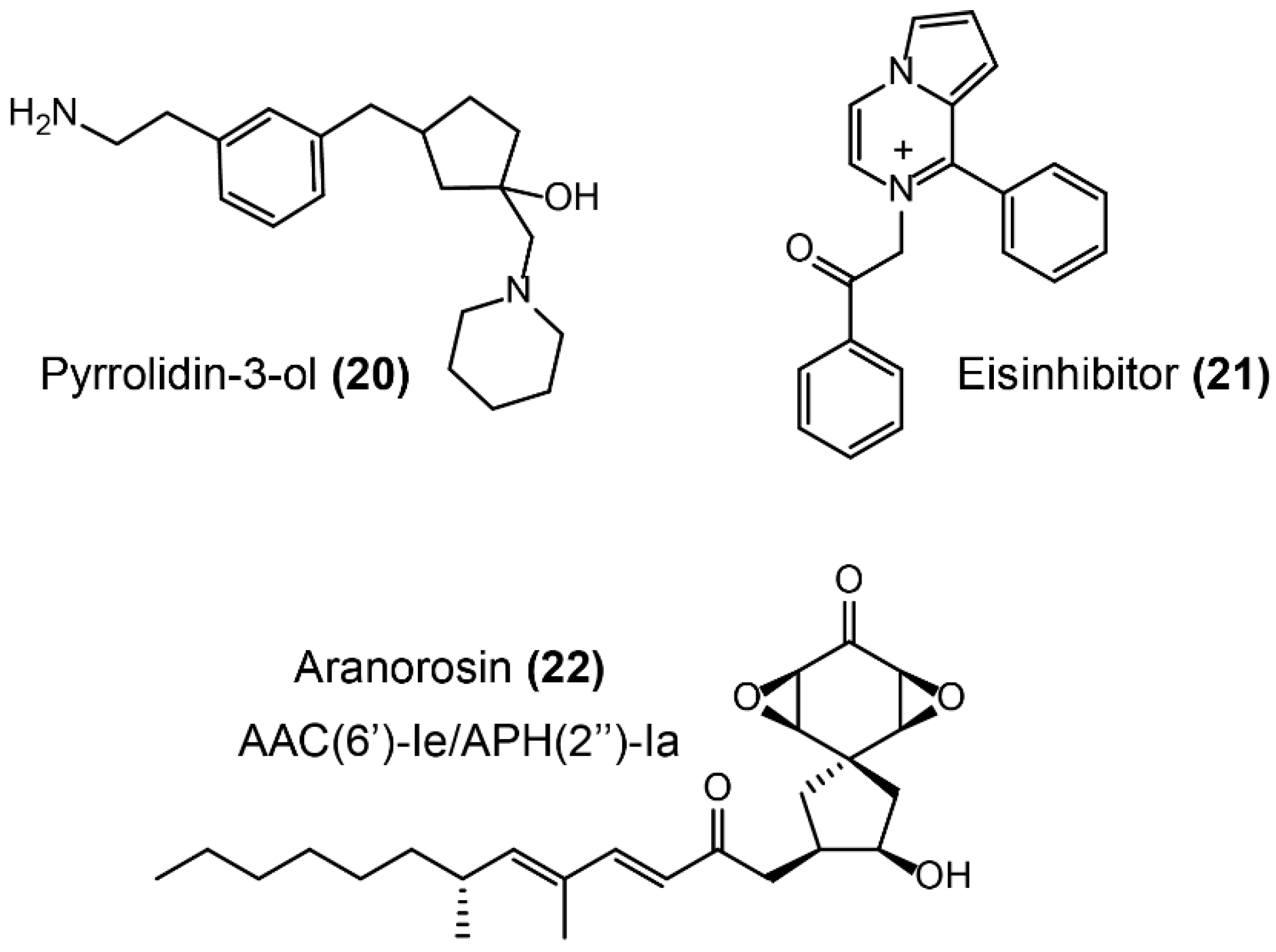
- Stogios, P.J.; Spanogiannopoulos, P.; Evdokimova, E.; Egorova, O.; Shakya, T.; Todorovic, N.; Capretta, A.; Wright, G.D.; Savchenko, A. Structure-guided optimization of protein kinase inhibitors reverses aminoglycoside antibiotic resistance. Biochem. J. 2013, 454, 191–200. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug. Discov. 2004, 3, 935. [Google Scholar] [CrossRef]
- Alonso, H.; Bliznyuk, A.A.; Gready, J.E. Combining docking and molecular dynamic simulations in drug design. Med. Res. Rev. 2006, 26, 531–568. [Google Scholar] [CrossRef]
- Moroy, G.; Martiny, V.Y.; Vayer, P.; Villoutreix, B.O.; Miteva, M.A. Toward in silico structure-based ADMET prediction in drug discovery. Drug Discov. Today 2012, 17, 44–55. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177. [Google Scholar] [CrossRef] [PubMed]
- Rohit, T.; Kiran, M.; Ryan, P.; Chenglong, L.; Werner, T. Carborane clusters in computational drug design: A comparative docking evaluation using AutoDock, FlexX, Glide, and Surflex. J. Chem. Inf. Model. 2009, 49, 1581–1589. [Google Scholar]
- Sato, H.; Shewchuk, L.M.; Tang, J. Prediction of multiple binding modes of the CDK2 inhibitors, anilinopyrazoles, using the automated docking programs GOLD, FlexX, and LigandFit: An evaluation of performance. J. Chem. Inf. Model. 2006, 46, 2552. [Google Scholar] [CrossRef] [PubMed]
- Schellhammer, I.; Rarey, M. FlexX-Scan: Fast, structure-based virtual screening. Proteins 2004, 57, 504–517. [Google Scholar] [CrossRef]
- Stahl, M. Modifications of the scoring function in FlexX for virtual screening applications. Perspect. Drug Discov. 2000, 20, 83–98. [Google Scholar] [CrossRef]
- Jain, A.N. Scoring functions for protein-ligand docking. Curr. Protein Pept. Sci. 2006, 7, 407–420. [Google Scholar]
- Jain, A.N. Surflex: Fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem. 2003, 46, 499–511. [Google Scholar] [CrossRef]
- Chiem, K.; Jani, S.; Fuentes, B.; Lin, D.L.; Rasche, M.E.; Tolmasky, M.E. Identification of an inhibitor of the aminoglycoside 6′-N-acetyltransferase type Ib [AAC(6′)-Ib] by glide molecular docking. Medchemcommun 2016, 7, 184–189. [Google Scholar] [CrossRef]
- Green, K.D.; Chen, W.; Garneautsodikova, S. Identification and characterization of inhibitors of the aminoglycoside resistance acetyltransferase Eis from Mycobacterium tuberculosis. Chemmedchem 2012, 7, 73–77. [Google Scholar] [CrossRef]
- Suga, T.; Ishii, T.; Iwatsuki, M.; Yamamoto, T.; Nonaka, K.; Masuma, R.; Matsui, H.; Hanaki, H.; Ōmura, S.; Shiomi, K. Aranorosin circumvents arbekacin-resistance in MRSA by inhibiting the bifunctional enzyme AAC (6′)/APH (2 ′′). J. Antibiot. 2012, 65, 527. [Google Scholar] [CrossRef]
- Mccormack, D.; Mcfadden, D. Pterostilbene and cancer: Current review. J. Surg. Res. 2012, 173, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.G.; Alosi, J.A.; Mcdonald, D.E.; Mcfadden, D.W. Pterostilbene inhibits lung cancer through induction of apoptosis. J. Surg. Res. 2010, 161, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Remsberg, C.M.; Yáñez, J.A.; Ohgami, Y.; Vega-Villa, K.R.; Rimando, A.M.; Davies, N.M. Pharmacometrics of pterostilbene: Preclinical pharmacokinetics and metabolism, anticancer, antiinflammatory, antioxidant and analgesic activity. Phytother. Res. 2008, 22, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Rimando, A.M.; Cuendet, M.; Desmarchelier, C.; Mehta, R.G.; Pezzuto, J.M.; Duke, S.O. Cancer chemopreventive and antioxidant activities of pterostilbene, a naturally occurring analogue of resveratrol. J. Agric. Food. Chem. 2002, 50, 3453–3457. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liu, S.; Wang, T.; Li, H.; Tang, S.; Wang, J.; Wang, Y.; Deng, X. Pterostilbene, a potential MCR-1 inhibitor that enhances the efficacy of polymyxin B. Antimicrob. Agents Chemother. 2018, 62, e02146-17. [Google Scholar] [CrossRef]
- Good, L.; Awasthi, S.K.; Dryselius, R.; Larsson, O.; Nielsen, P.E. Bactericidal antisense effects of peptide-PNA conjugates. Nat. Biotechnol. 2001, 19, 360–364. [Google Scholar] [CrossRef]
- Hatamoto, M.; Ohashi, A.; Imachi, H. Peptide nucleic acids (PNAs) antisense effect to bacterial growth and their application potentiality in biotechnology. Appl. Microbiol. Biotechnol. 2010, 86, 397–402. [Google Scholar] [CrossRef]
- Górska, A.; Markowska-Zagrajek, A.; Równicki, M.; Trylska, J. Scanning of 16S ribosomal RNA for peptide nucleic acid targets. J. Phys. Chem. B 2016, 120, 8369–8378. [Google Scholar] [CrossRef]
- Good, L.; Sandberg, R.; Larsson, O.; Nielsen, P.E.; Wahlestedt, C. Antisense PNA effects in Escherichia coli are limited by the outer-membrane LPS layer. Microbiology 2000, 146, 2665–2670. [Google Scholar] [CrossRef] [PubMed]
- Gryko, D.; Giedyk, M.; Jackowska, A.; Równicki, M.; Kolanowska, M.; Trylska, J. Vitamin B12 transports modified RNA into E. coli and S. typhimurium cells. Chem. Commun. 2018. [Google Scholar] [CrossRef]
- Forier, K.; Raemdonck, K.; De Smedt, S.C.; Demeester, J.; Coenye, T.; Braeckmans, K. Lipid and polymer nanoparticles for drug delivery to bacterial biofilms. J. Control. Release 2014, 190, 607–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abushahba, M.F.; Mohammad, H.; Thangamani, S.; Hussein, A.A.; Seleem, M.N. Impact of different cell penetrating peptides on the efficacy of antisense therapeutics for targeting intracellular pathogens. Sci. Rep. 2016, 6, 20832. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.Y.; Mao, X.G.; Li, Z.; Chen, Z.; Zhou, Y.; Hou, Z.; Li, M.K.; Meng, J.R.; Luo, X.X. A potent and selective antimicrobial poly(amidoamine) dendrimer conjugate with LED209 targeting QseC receptor to inhibit the virulence genes of Gram negative bacteria. Nanomed. Nanotechnol. 2015, 11, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Huschka, R.; Barhoumi, A.; Liu, Q.; Roth, J.A.; Ji, L.; Halas, N.J. Gene silencing by gold nanoshell-mediated delivery and laser-triggered release of antisense oligonucleotide and siRNA. ACS Nano 2012, 6, 7681–7691. [Google Scholar] [CrossRef] [PubMed]
- Daly, S.M.; Sturge, C.R.; Felder-Scott, C.F.; Geller, B.L.; Greenberg, D.E. MCR-1 inhibition with peptide-conjugated Phosphorodiamidate morpholino oligomers restores sensitivity to polymyxin in Escherichia coli. mBio 2017, 8, e01315-17. [Google Scholar] [CrossRef]
- Thomason, M.K.; Storz, G. Bacterial antisense RNAs: How many are there, and what are they doing? Annu. Rev. Genet. 2010, 44, 167–188. [Google Scholar] [CrossRef]
- Ryszard, K.; Krainer, A.R.; Sidney, A. RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug. Discov. 2012, 11, 125–140. [Google Scholar]
- Bennett, C.F.; Swayze, E.E. RNA targeting therapeutics: Molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293. [Google Scholar] [CrossRef]
- Geller, B.L.; Deere, J.D.; Stein, D.A.; Kroeker, A.D.; Moulton, H.M.; Iversen, P.L. Inhibition of gene expression in Escherichia coli by antisense phosphorodiamidate morpholino oligomers. Antimicrob. Agents Chemother. 2003, 47, 3233–3239. [Google Scholar] [CrossRef] [PubMed]
- Mellbye, B.L.; Puckett SETilley, L.D. Variations in amino acid composition of antisense peptide-phosphorodiamidate morpholino oligomer affect potency against Escherichia coli in vitro and in vivo. Antimicrob. Agents Chemother. 2009, 53, 525–530. [Google Scholar] [CrossRef]
- Mellbye, B.L.; Weller, D.D.; Hassinger, J.N.; Reeves, M.D.; Lovejoy, C.E.; Iversen, P.L.; Geller, B.L. Cationic phosphorodiamidate morpholino oligomers efficiently prevent growth of Escherichia coli in vitro and in vivo. J. Antimicrob. Chemother. 2010, 65, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.J.; Sturge, C.R.; Moustafa, D.A.; Daly, S.M.; Marshallbatty, K.R.; Felder, C.F.; Zamora, D.; Yabegill, M.; Labandeirarey, M.; Bailey, S.M. Inhibition of Pseudomonas aeruginosa by peptide-conjugated phosphorodiamidate morpholino oligomers. Antimicrob. Agents Chemother. 2017, 61, e01938-16. [Google Scholar] [CrossRef]
- Sully, E.K.; Geller, B.L.; Li, L.; Moody, C.M.; Bailey, S.M.; Moore, A.L.; Wong, M.; Nordmann, P.; Daly, S.M.; Sturge, C.R. Peptide-conjugated phosphorodiamidate morpholino oligomer (PPMO) restores carbapenem susceptibility to NDM-1-positive pathogens in vitro and in vivo. J Antimicrob Chemother 2017, 72, 782. [Google Scholar] [PubMed]
- Ji, Y.; Marra, A.; Rosenberg, M.; Woodnutt, G. Regulated antisense RNA eliminates alpha-toxin virulence in Staphylococcus aureus infection. J. Bacteriol. 1999, 181, 6585–6590. [Google Scholar] [PubMed]
- Chi, N.T.; Giangrossi, M.; Prosseda, G.; Brandi, A.; Martino, M.L.D.; Colonna, B.; Falconi, M. A multifactor regulatory circuit involving H-NS, VirF and an antisense RNA modulates transcription of the virulence gene icsA of Shigella flexneri. Nucleic Acids Res. 2011, 39, 8122–8134. [Google Scholar]
- Eun-Jin, L.; Groisman, E.A. An antisense RNA that governs the expression kinetics of a multifunctional virulence gene. Mol. Microbiol. 2010, 76, 1020–1033. [Google Scholar] [Green Version]
- Mara, G.; Gianni, P.; Nhan, T.C.; Anna, B.; Bianca, C.; Maurizio, F. A novel antisense RNA regulates at transcriptional level the virulence gene icsA of Shigella flexneri. Nucleic Acids Res. 2010, 38, 3362–3375. [Google Scholar] [Green Version]
- Panthee, S.; Paudel, A.; Hamamoto, H.; Sekimizu, K. Advantages of the silkworm as an animal model for developing novel antimicrobial agents. Front. Microbiol. 2017, 8, 373. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Li, R.; Xiao, X.; Wang, Z. Molecules that Inhibit Bacterial Resistance Enzymes. Molecules 2019, 24, 43. https://doi.org/10.3390/molecules24010043
Liu Y, Li R, Xiao X, Wang Z. Molecules that Inhibit Bacterial Resistance Enzymes. Molecules. 2019; 24(1):43. https://doi.org/10.3390/molecules24010043
Chicago/Turabian StyleLiu, Yuan, Ruichao Li, Xia Xiao, and Zhiqiang Wang. 2019. "Molecules that Inhibit Bacterial Resistance Enzymes" Molecules 24, no. 1: 43. https://doi.org/10.3390/molecules24010043
APA StyleLiu, Y., Li, R., Xiao, X., & Wang, Z. (2019). Molecules that Inhibit Bacterial Resistance Enzymes. Molecules, 24(1), 43. https://doi.org/10.3390/molecules24010043