Substitution Effect on the Charge Transfer Processes in Organo-Imido Lindqvist-Polyoxomolybdate

 , ,
, ,  , , and
, , and 
Abstract
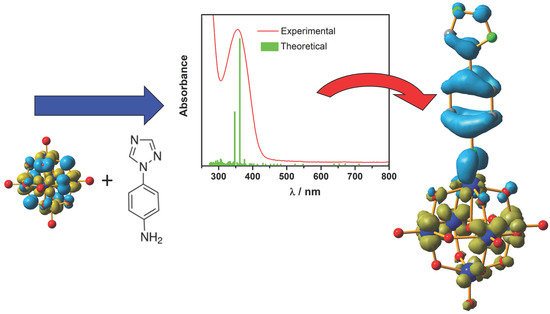
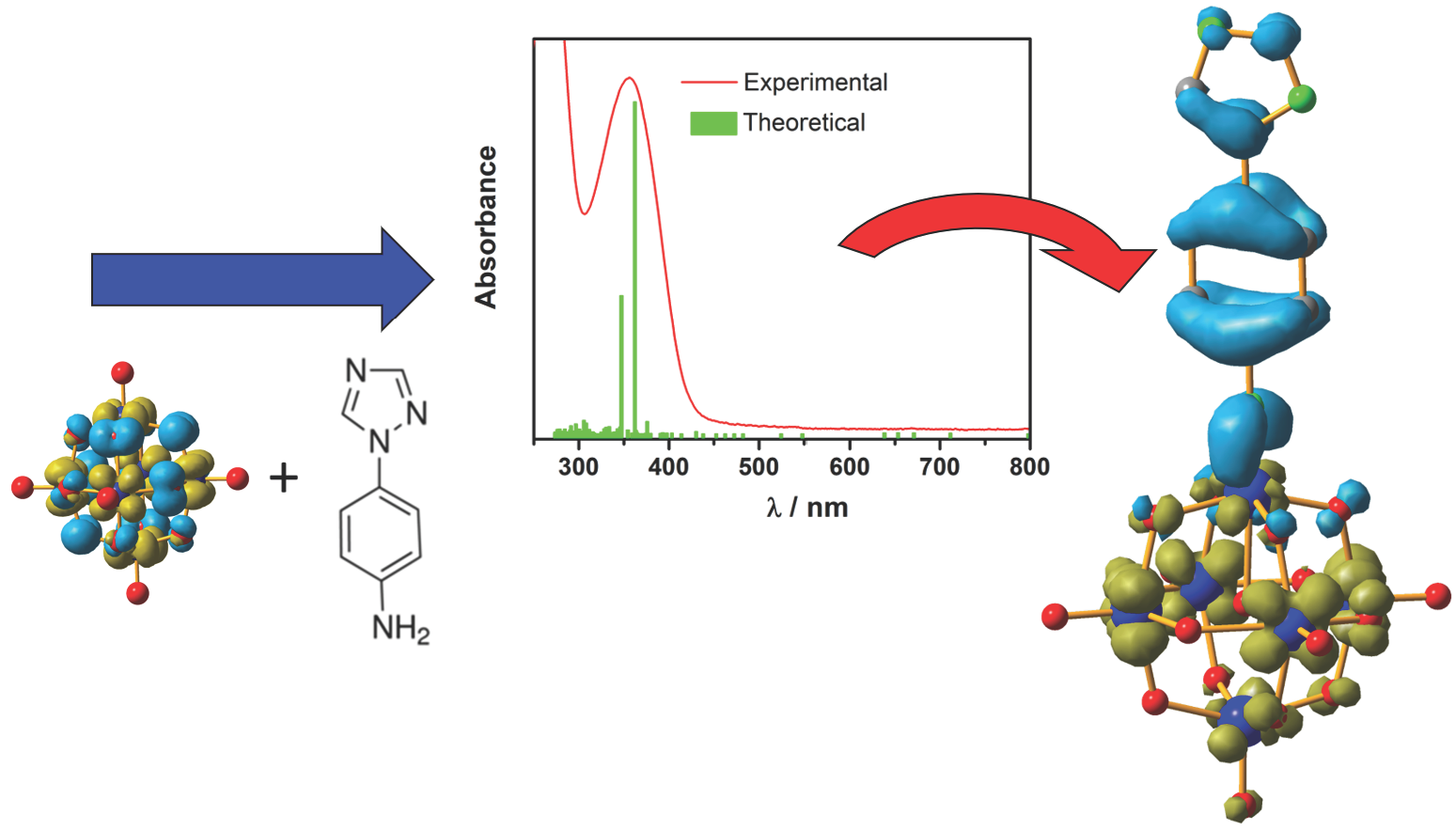
:
1. Introduction
2. Results and Discussions
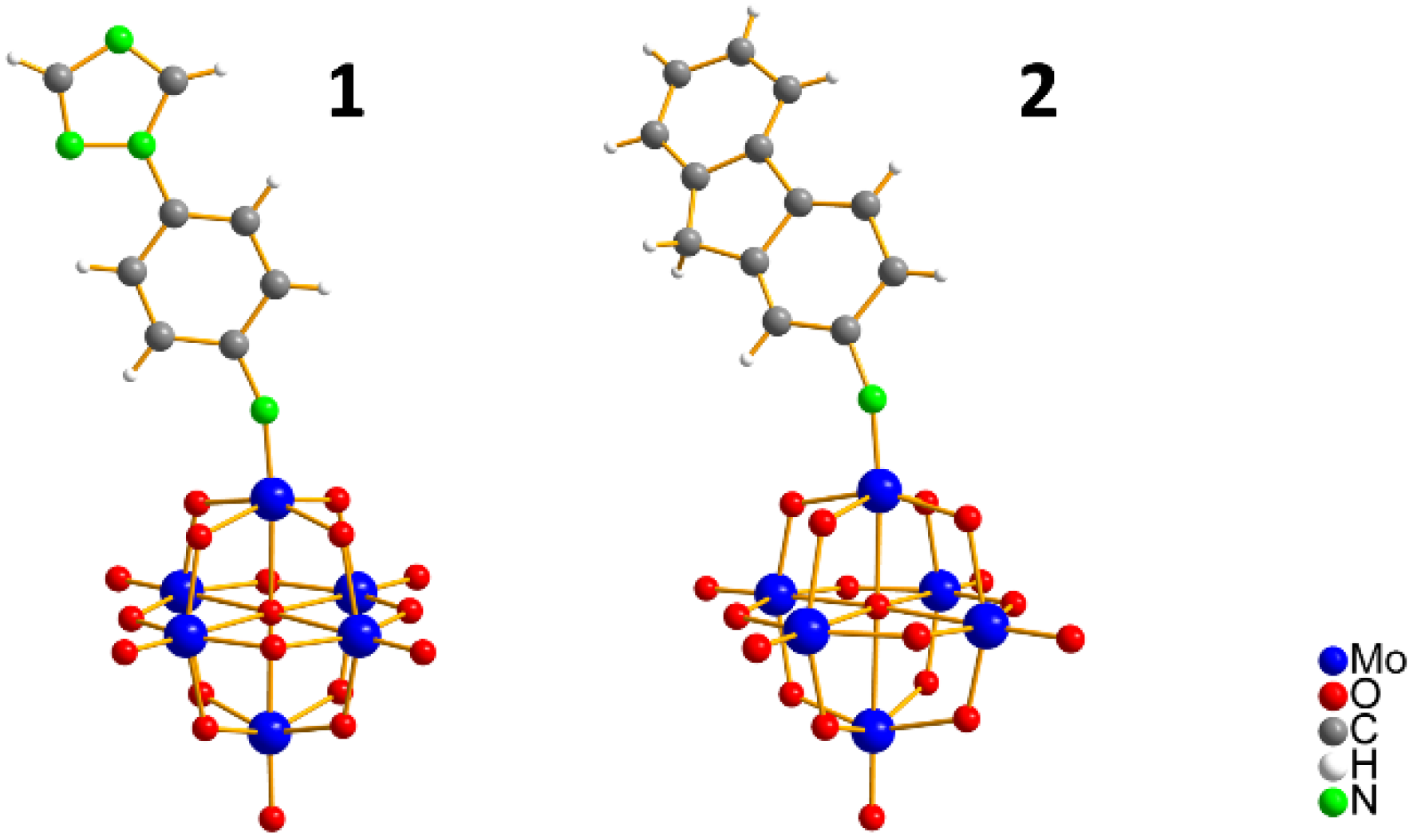
2.1. Synthesis and X-ray Structures
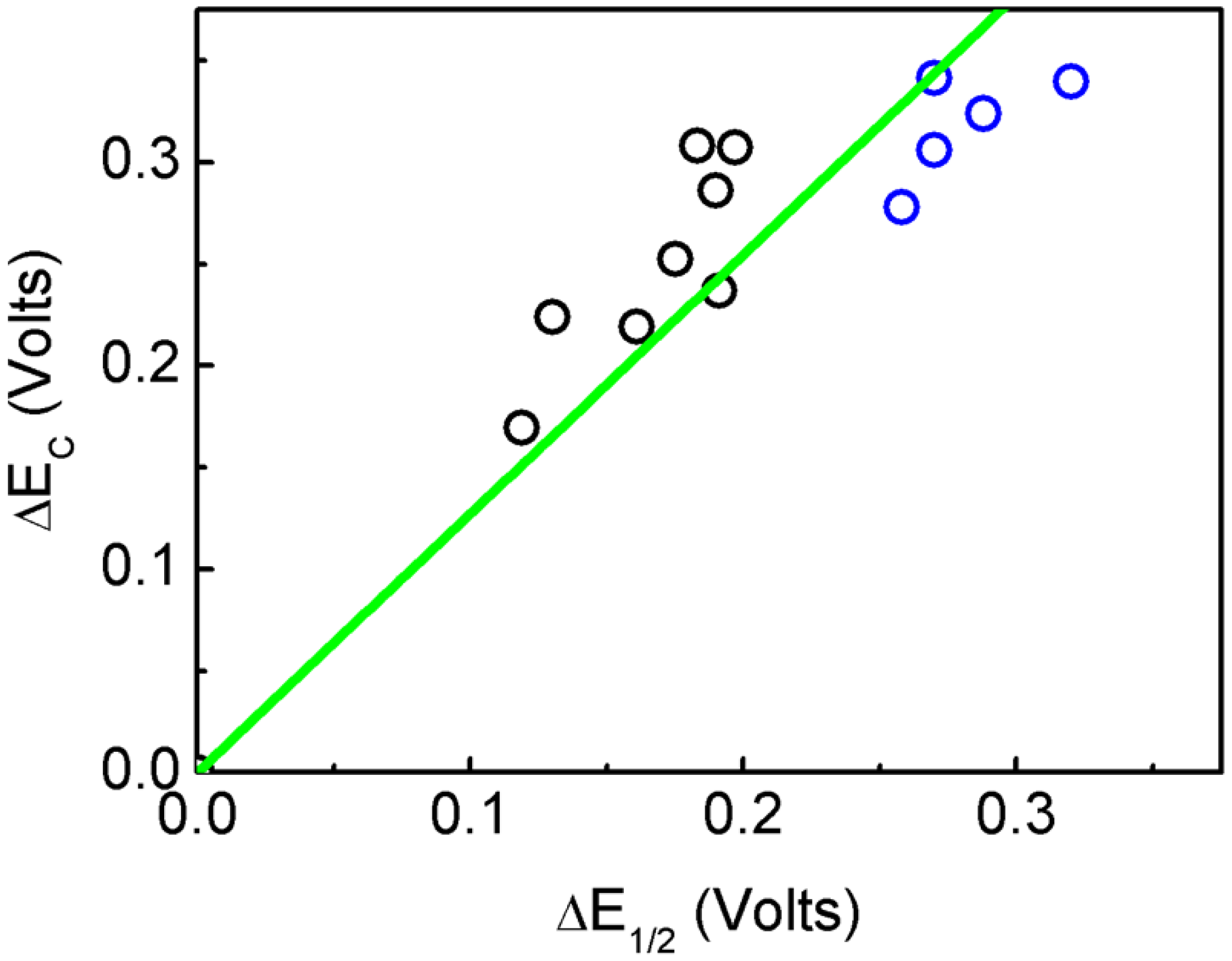
2.2. Electrochemical Properties
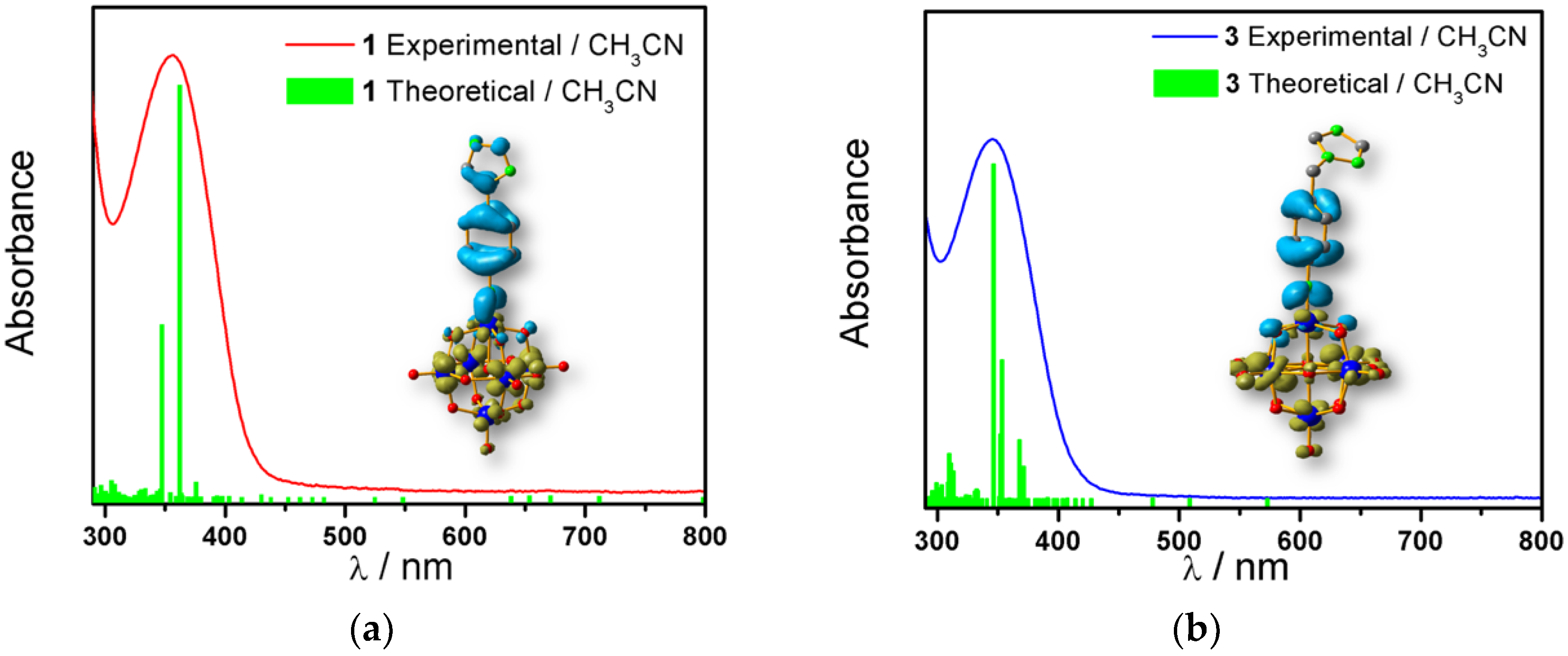
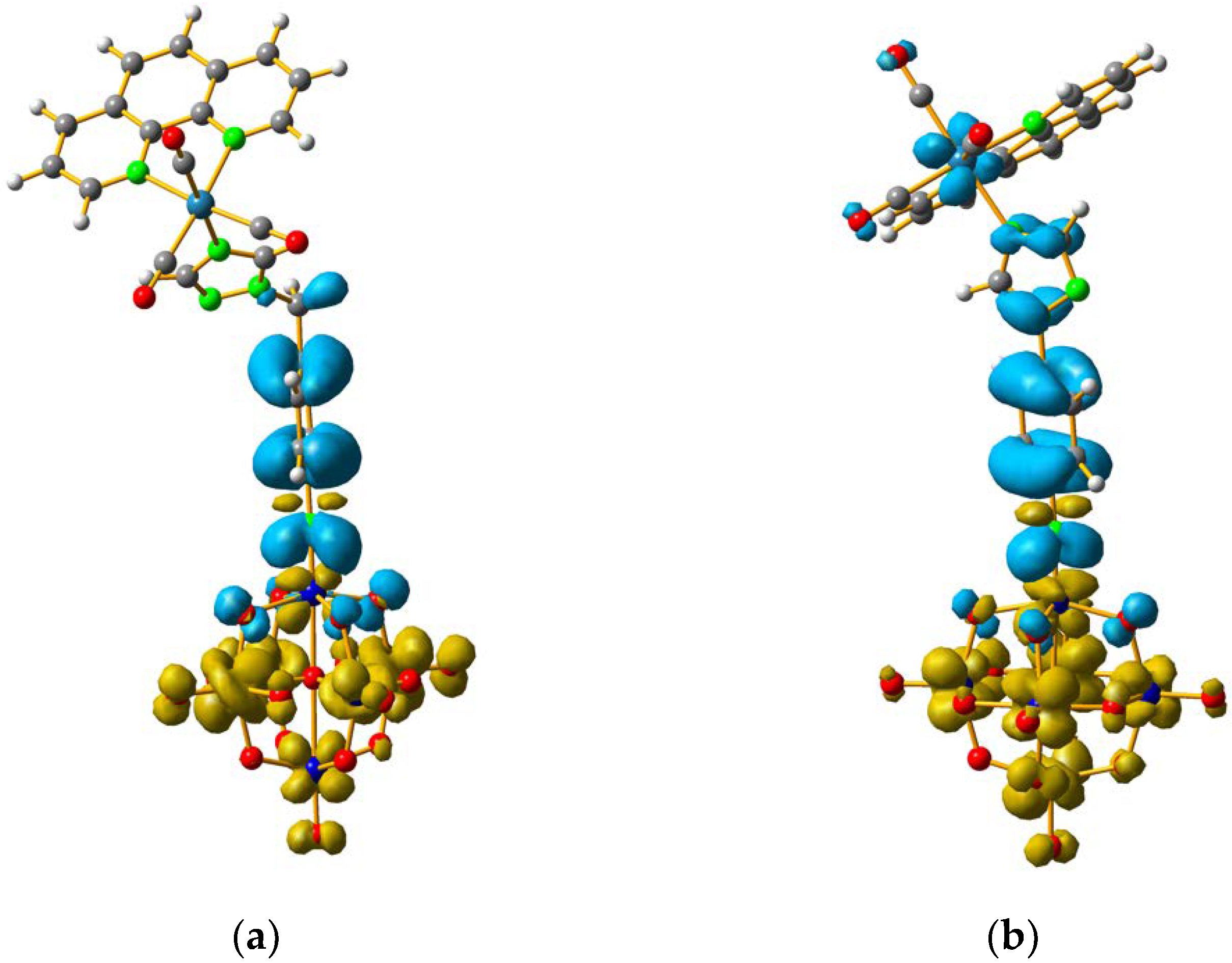
2.3. Spectroscopic Characterization
3. Materials and Methods
3.1. Reagents and Instruments
3.2. Synthesis of [n-Bu4N]2[Mo6O18NC6H4N3C2H2] (1)
3.3. Synthesis of [n-Bu4N]2[Mo6O18NC13H9] (2)
3.4. Crystal Structure Determination
3.5. Computational Details
3.5.1. Prediction of UV-Vis Spectra
3.5.2. Theoretical Redox Potentials
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Pope, M.T. Polyoxo anions: Synthesis and structure. In Comprehensive Coordination Chemistry II; Elsevier: New York, NY, USA, 2004; pp. 635–678. [Google Scholar]
- Mingos, D.M.P. Bonding and Charge Distribution in Polyoxometalates: A Bond Valence Approach; Springer: Berlin, Germany, 1999. [Google Scholar]
- Hill, C.L. Polyoxometalates: Reactivity. In Comprehensive Coordination Chemistry II: Transition Metal Groups 3–6; Wedd, A.G., Ed.; Elsevier: New York, NY, USA, 2004; Volume 4. [Google Scholar]
- Nagano, O.; Sasaki, Y. Structure of the hydrated potassium hexamolybdate complex of hexaoxacyclooctadecane (18-crown-6). Acta Cryst. 1979, B35, 2387–2389. [Google Scholar] [CrossRef]
- Hutin, M.; Rosnes, M.H.; Long, D.; Cronin, L. Polyoxometalates: Synthesis and Structure-From Building Blocks to Emergent Materials; Elsevier: New York, NY, USA, 2013. [Google Scholar]
- Gumerova, N.I.; Rompel, A. Synthesis, structures and applications of electron-rich polyoxometalates. Nat. Rev. Chem. 2018, 2, 0112. [Google Scholar] [CrossRef]
- Strong, J.B.; Ostrander, R.; Rheingold, A.L.; Maatta, E.A. Ensheathing a Polyoxometalate: Convenient Systematic Introduction of Organoimido Ligands at Terminal Oxo Sites in [Mo6O19]2−. J. Am. Chem. Soc. 1994, 116, 3601–3602. [Google Scholar] [CrossRef]
- Sima, G.; Li, Q.; Zhu, Y.; Lv, C.; Khan, R.N.N.; Hao, J.; Zhang, J.; Wei, Y. Organoimido-derivatized hexamolybdates with a remote carboxyl group: Syntheses and structural characterizations. Inorg. Chem. 2013, 52, 6551–6558. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhang, J.; Hao, J.; Wei, Y. A general and highly regioselective synthesis approach to multi-functionalized organoimido derivatives of Polyoxometalates. Sci. Rep. 2016, 6, 24759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Yasari, A.; Van Steerteghem, N.; Kearns, H.; El Moll, H.; Faulds, K.; Wright, J.A.; Brunschwig, B.S.; Clays, K.; Fielden, J. Organoimido-polyoxometalate nonlinear optical chromophores: A structural, spectroscopic, and computational study. Inorg. Chem. 2017, 56, 10181–10194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xiao, F.; Hao, J.; Wei, Y. The chemistry of organoimido derivatives of polyoxometalates. Dalton Trans. 2012, 41, 3599–3615. [Google Scholar] [CrossRef]
- Hermosilla-Ibáñez, P.; Wrighton-Araneda, K.; Prado, G.; Paredes-García, V.; Pizarro, N.; Vega, A.; Venegas-Yazigi, D. The first ReI organometallic complex with an organoimido-polyoxometalate ligand. Dalton Trans. 2017, 46, 8611–8620. [Google Scholar] [CrossRef]
- Ravelli, D.; Dondi, D.; Fagnoni, M.; Albini, A.; Bagno, A. Predicting the UV spectrum of polyoxometalates by TD-DFT. J. Comput. Chem. 2011, 32, 2983–2987. [Google Scholar] [CrossRef]
- López, X.; Carbó, J.J.; Bo, C.; Poblet, J.M. Structure, properties and reactivity of polyoxometalates: A theoretical perspective. Chem. Soc. Rev. 2012, 41, 7537–7571. [Google Scholar] [CrossRef]
- Winget, P.; Weber, E.J.; Cramer, C.J.; Truhlar, D.G. Computational electrochemistry: Aqueous one-electron oxidation potentials for substituted anilines. Phys. Chem. Chem. Phys. 2000, 2, 1231–1239. [Google Scholar] [CrossRef]
- Stark, J.L.; Young, G., Jr.; Maatta, E.A. A functionalized polyoxometalate bearing a ferrocenylimido ligand: Preparation and structure of [(FcN)Mo6O18]2−. Angew. Chem. Int. Ed. Engl. 1995, 34, 2547–2548. [Google Scholar] [CrossRef]
- Kang, J.; Nelson, J.A.; Lu, M.; Xie, B.; Peng, Z.; Powell, D.R. Charge-transfer hybrids containing covalently bonded polyoxometalates and ferrocenyl units. Inorg. Chem. 2004, 43, 6408–6413. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Xu, B.; Peng, Z.; Zhu, X.; Wei, Y.; Powell, D.R. Molecular and polymeric hybrids based on covalently linked polyoxometalates and transition-metal complexes. Angew. Chem. Int. Ed. 2005, 44, 6902–6905. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Peng, Z.; Wei, Y.; Powell, D.R. Polyoxometalates covalently bonded with terpyridine ligandsl. Chem. Commun. 2003, 375, 2562–2563. [Google Scholar] [CrossRef]
- Zhu, Y.; Yin, P.; Xiao, F.; Li, D.; Bitterlich, E.; Xiao, Z.; Zhang, J.; Hao, J.; Liu, T.; Wang, Y.; et al. Bottom-up construction of POM-based macrostructures: Coordination assembled paddle-wheel macroclusters and their vesicle-like supramolecular aggregation in solution. J. Am. Chem. Soc. 2013, 135, 17155–17160. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, L.; Yin, P.; Wei, Y.; Hao, J.; Zhu, Y.; Zhu, L.; Yuan, G. Convenient syntheses and structural characterizations of mono-substituted alkylimido hexamolybdates: [Mo6O18(NR)]2− (R = Me, Et, n-Pr, i-Pr, n-Bu, t-Bu, Cy, Hex, Ode). Dalton Trans. 2009, 18, 1172–1179. [Google Scholar] [CrossRef]
- Li, Q.; Wei, Y.; Guo, H.; Zhan, C.G. Syntheses, structural characterizations and electronic absorption spectra simulation of three phenylimido substituted hexamolybdates incorporating a remote chloro group. Inorg. Chim. Acta 2008, 361, 2305–2313. [Google Scholar] [CrossRef]
- Proust, A.; Thouvenot, R.; Chaussade, M.; Robert, F.; Gouzerh, P. Phenylimido derivatives of [Mo6O19]2−: syntheses, X-ray structures, vibrational, electrochemical, 95Mo and 14N NMR studies. Inorg. Chim. Acta 1994, 224, 81–95. [Google Scholar] [CrossRef]
- Le Bahers, T.; Adamo, C.; Ciofini, I. A qualitative index of spatial extent in charge-transfer excitations. J. Chem. Theory Comput. 2011, 7, 2498–2506. [Google Scholar] [CrossRef]
- Hur, N.H.; Klemperer, W.G.; Wang, R.C. Inorganic Syntheses; John Wiley & Sons: Hoboken, NJ, USA, 1990; Volume 27, pp. 77–79. [Google Scholar]
- SAINTPLUS Version 6.02; Brucker AXS: Madison, WI, USA, 1999.
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M.S. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Brandenburg, K. Diamond, Version 3.2k; Crystal Impact GbR: Bonn, Germany, 2014. [Google Scholar]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew--Burke--Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. GAUSSIAN 09 (Revision D.2); Gaussian, Inc.: Pittsburgh, PA, USA, 2009. [Google Scholar]
- Cameron, J.M.; Fujimoto, S.; Kastner, K.; Wei, R.J.; Robinson, D.; Sans, V.; Newton, G.N.; Oshio, H.H. Orbital engineering: Photoactivation of an organofunctionalized polyoxotungstate. Chem. A Eur. J. 2017, 23, 47–50. [Google Scholar] [CrossRef]
- Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. Energy-adjusted abinitio pseudopotentials for the first row transition elements. J. Chem. Phys. 1987, 86, 866–872. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1 and 2 are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mo6O18N-R | Mo–N–C Angle (°) | Ref. |
|---|---|---|
 1 | 159.8(5) | This work |
 2 | 158.1(2) | This work |
 3 | 163.5(2)/164.1(1) | [12] |
| –CH2CH3 | 178.1(1) | [21] |
| –CH2CH2CH3 | 177.9(5) | [21] |
| –CH2CH2CH2CH3 | 178.3(8) | [21] |
| –CH2CH2CH2CH2CH2CH3 | 176.7(1) | [21] |
| Mo6O18N–R | ΔE1/2 (V) | ΔEC (V) | E1/2 (V) | E1/2-Teo (V) | Ref. |
|---|---|---|---|---|---|
 1 | 0.190 | 0.286 | −1.026 | -0.973 | This work |
 2 | 0.183 | 0.308 | −1.019 | −0.995 | This work |
 3 | 0.175 | 0.252 | −1.011 | −0.939 | [12] |
 4 | 0.130 | 0.224 | −0.966 | −0.910 | [12] |
| –CH3 | 0.270 | 0.341 | −1.028 | [21] | |
| –CH2CH3 | 0.320 | 0.340 | −1.020 | [21] | |
| –CH2CH2CH3 | 0.258 | 0.278 | −0.964 | [21] | |
| –CH2CH2CH2CH3 | 0.288 | 0.324 | −1.011 | [21] | |
| –CH2CH2CH2CH2CH2CH3 | 0.270 | 0.306 | −0.993 | [21] | |
 | 0.190 | 0.286 | −0.973 | [23] | |
 | 0.191 | 0.237 | −0.924 | [22] | |
 | 0.197 | 0.307 | −0.994 | [22] | |
 | 0.161 | 0.219 | −0.906 | [10] | |
 | 0.119 | 0.169 | −0.856 | [10] |
| Mo6O18N–R | λ O.F. → Mo (nm) | f | References |
|---|---|---|---|
 1 | 375 | 0.2951 | This work |
 2 | 389 | 0.5770 | This work |
 3 | 346 | 0.3772 | [12] |
 4 | 345 | 0.3976 | [12] |
 5 | 366 | 0.5993 | This work |
 6 | 328 | 0.0100 | [21] |
 7 | 328 | 0.0119 | [21] |
 8 | 342 | 0.3901 | [23] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hermosilla-Ibáñez, P.; Wrighton-Araneda, K.; Cañón-Mancisidor, W.; Gutiérrez-Cutiño, M.; Paredes-García, V.; Venegas-Yazigi, D. Substitution Effect on the Charge Transfer Processes in Organo-Imido Lindqvist-Polyoxomolybdate. Molecules 2019, 24, 44. https://doi.org/10.3390/molecules24010044
Hermosilla-Ibáñez P, Wrighton-Araneda K, Cañón-Mancisidor W, Gutiérrez-Cutiño M, Paredes-García V, Venegas-Yazigi D. Substitution Effect on the Charge Transfer Processes in Organo-Imido Lindqvist-Polyoxomolybdate. Molecules. 2019; 24(1):44. https://doi.org/10.3390/molecules24010044
Chicago/Turabian StyleHermosilla-Ibáñez, Patricio, Kerry Wrighton-Araneda, Walter Cañón-Mancisidor, Marlen Gutiérrez-Cutiño, Verónica Paredes-García, and Diego Venegas-Yazigi. 2019. "Substitution Effect on the Charge Transfer Processes in Organo-Imido Lindqvist-Polyoxomolybdate" Molecules 24, no. 1: 44. https://doi.org/10.3390/molecules24010044
APA StyleHermosilla-Ibáñez, P., Wrighton-Araneda, K., Cañón-Mancisidor, W., Gutiérrez-Cutiño, M., Paredes-García, V., & Venegas-Yazigi, D. (2019). Substitution Effect on the Charge Transfer Processes in Organo-Imido Lindqvist-Polyoxomolybdate. Molecules, 24(1), 44. https://doi.org/10.3390/molecules24010044