Importance of the Proximity and Orientation of Ligand-Linkage to the Design of Cinnamate-GW9662 Hybrid Compounds as Covalent PPARγ Agonists




Abstract
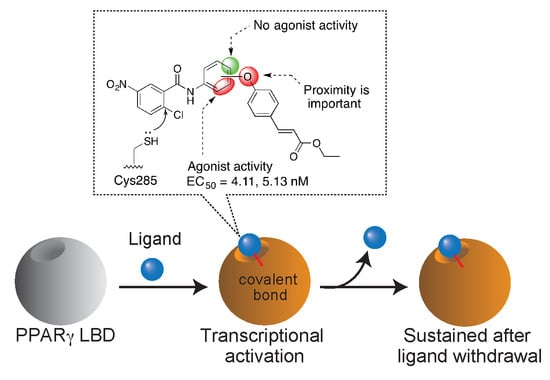
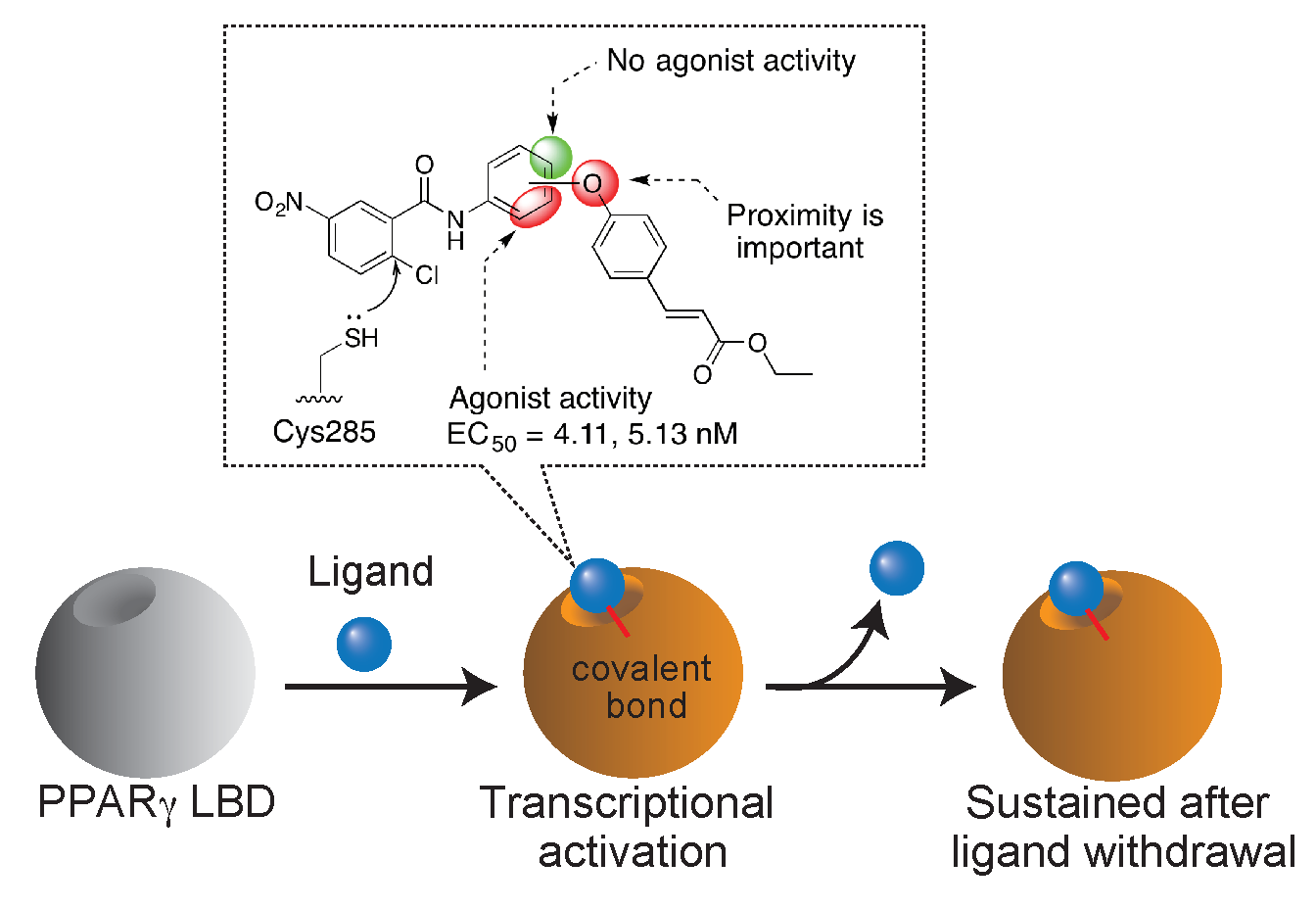
:
1. Introduction
2. Results and Discussion
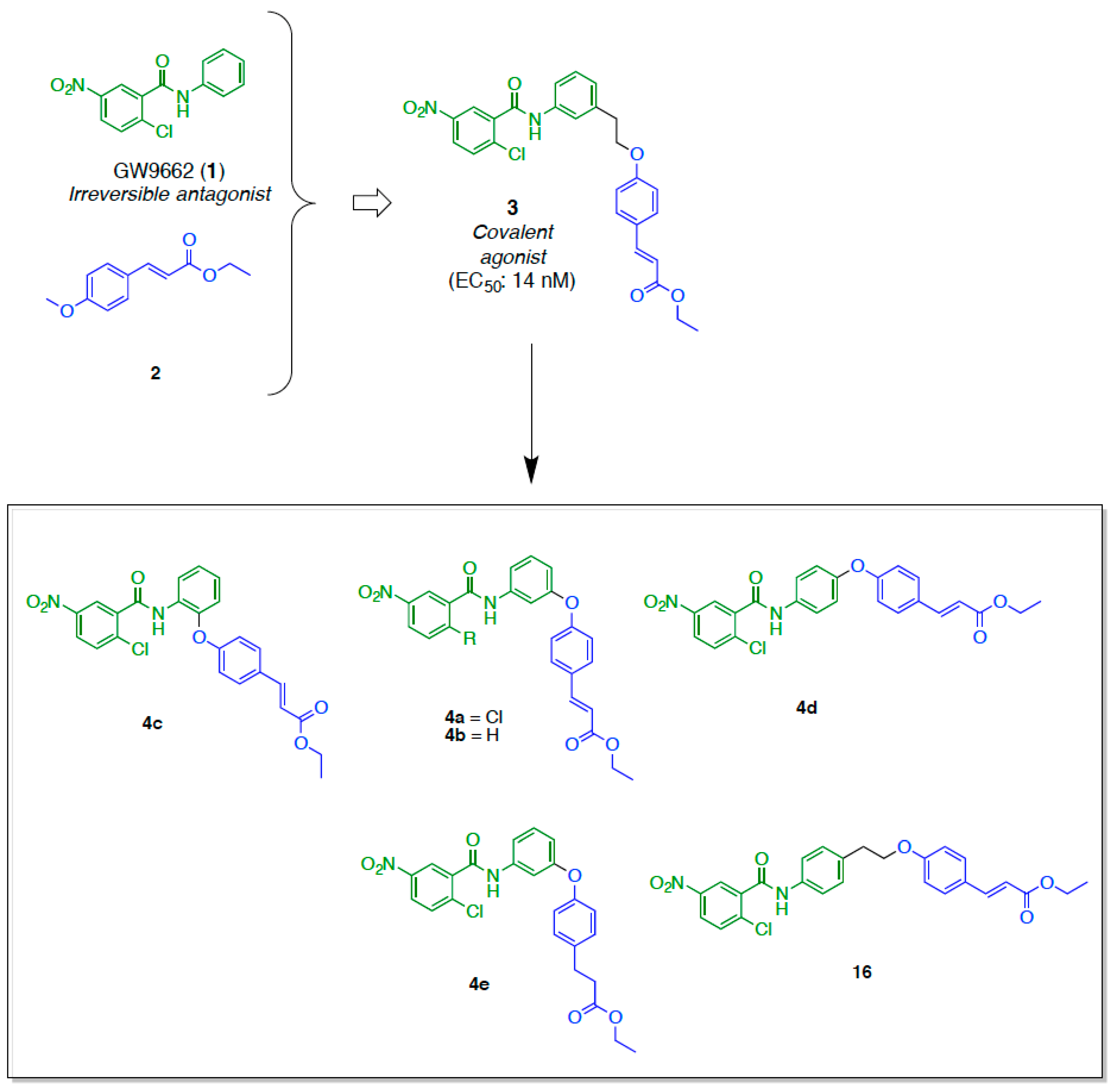
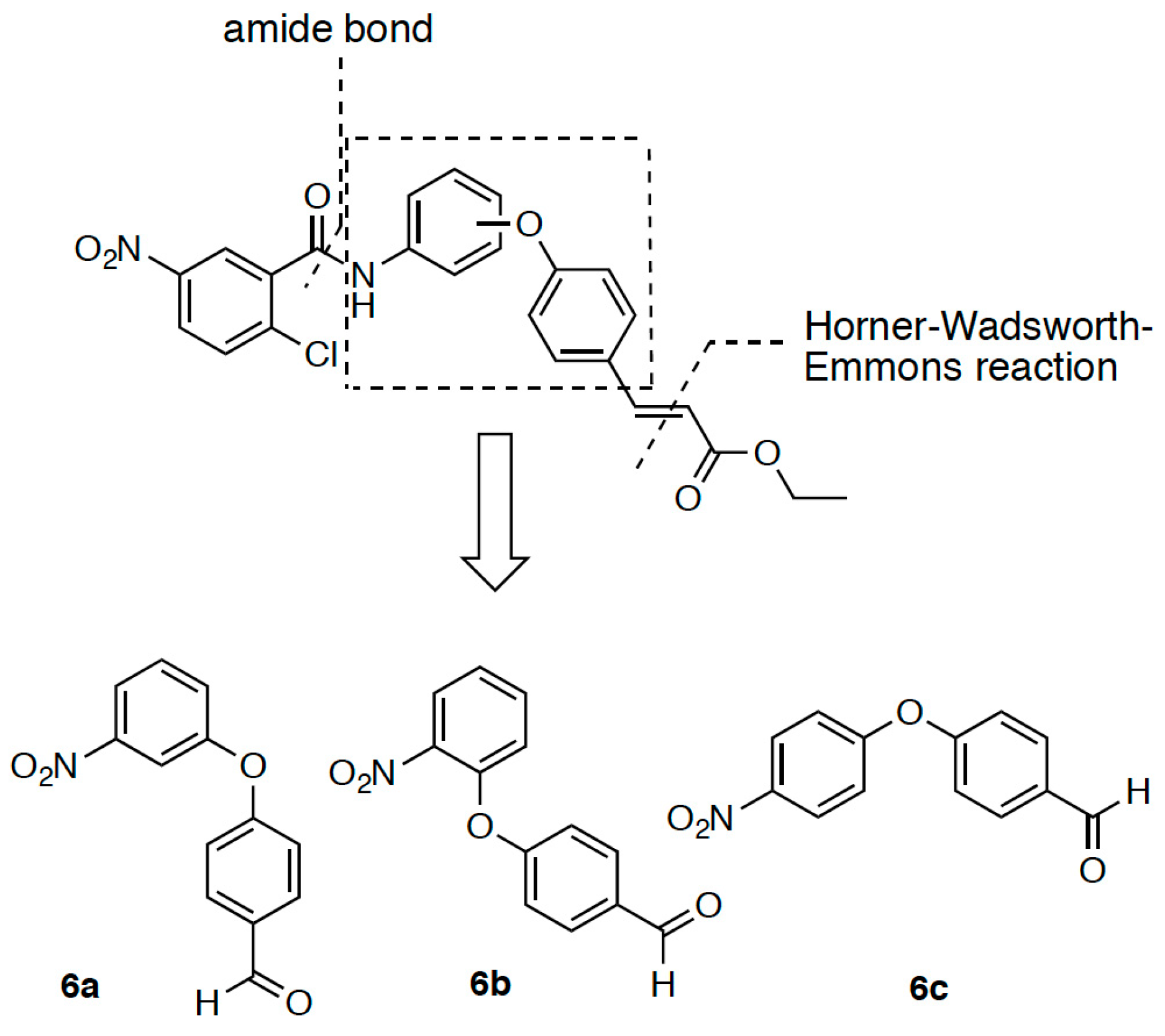
2.1. Retrosynthetic Analyses
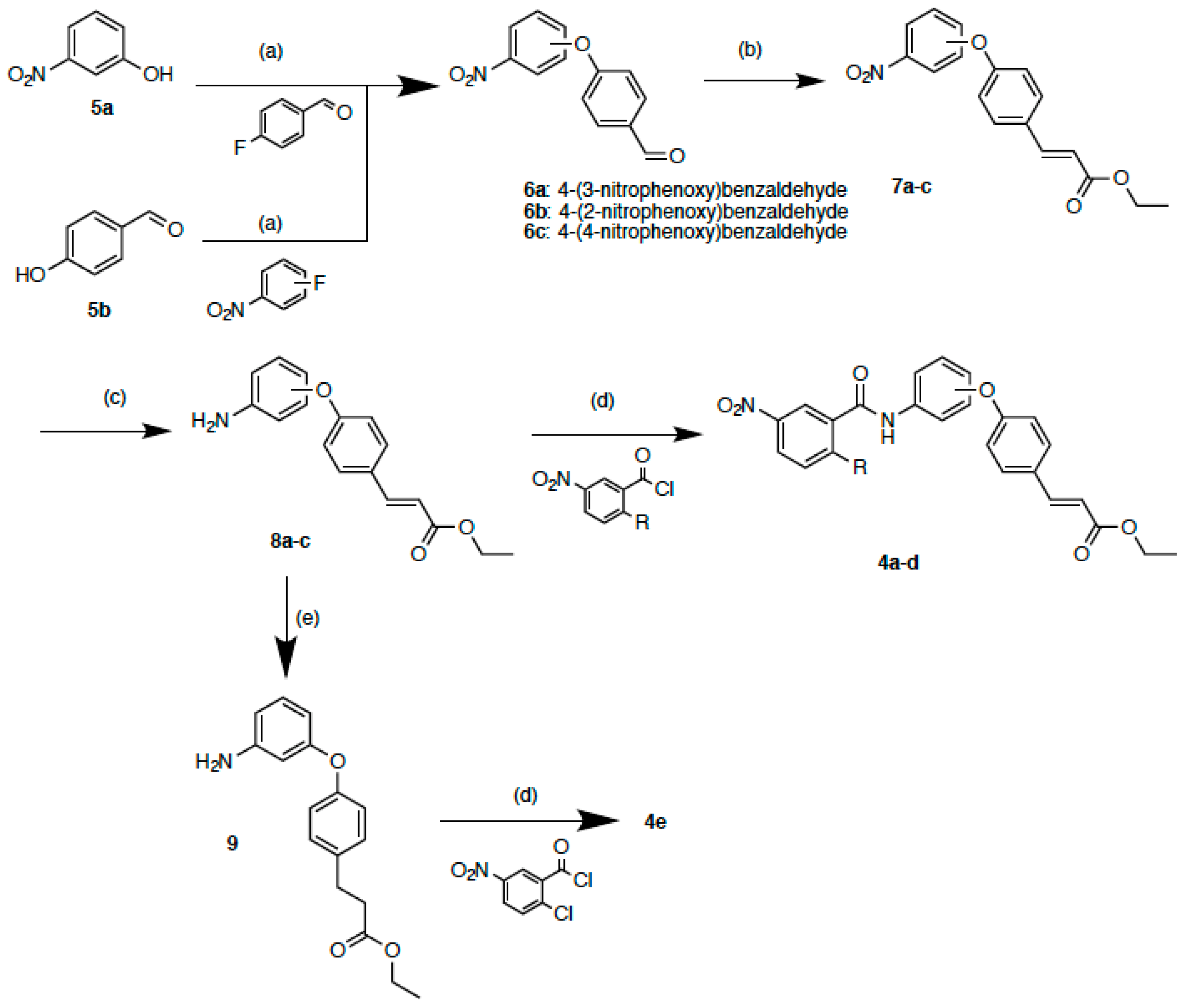
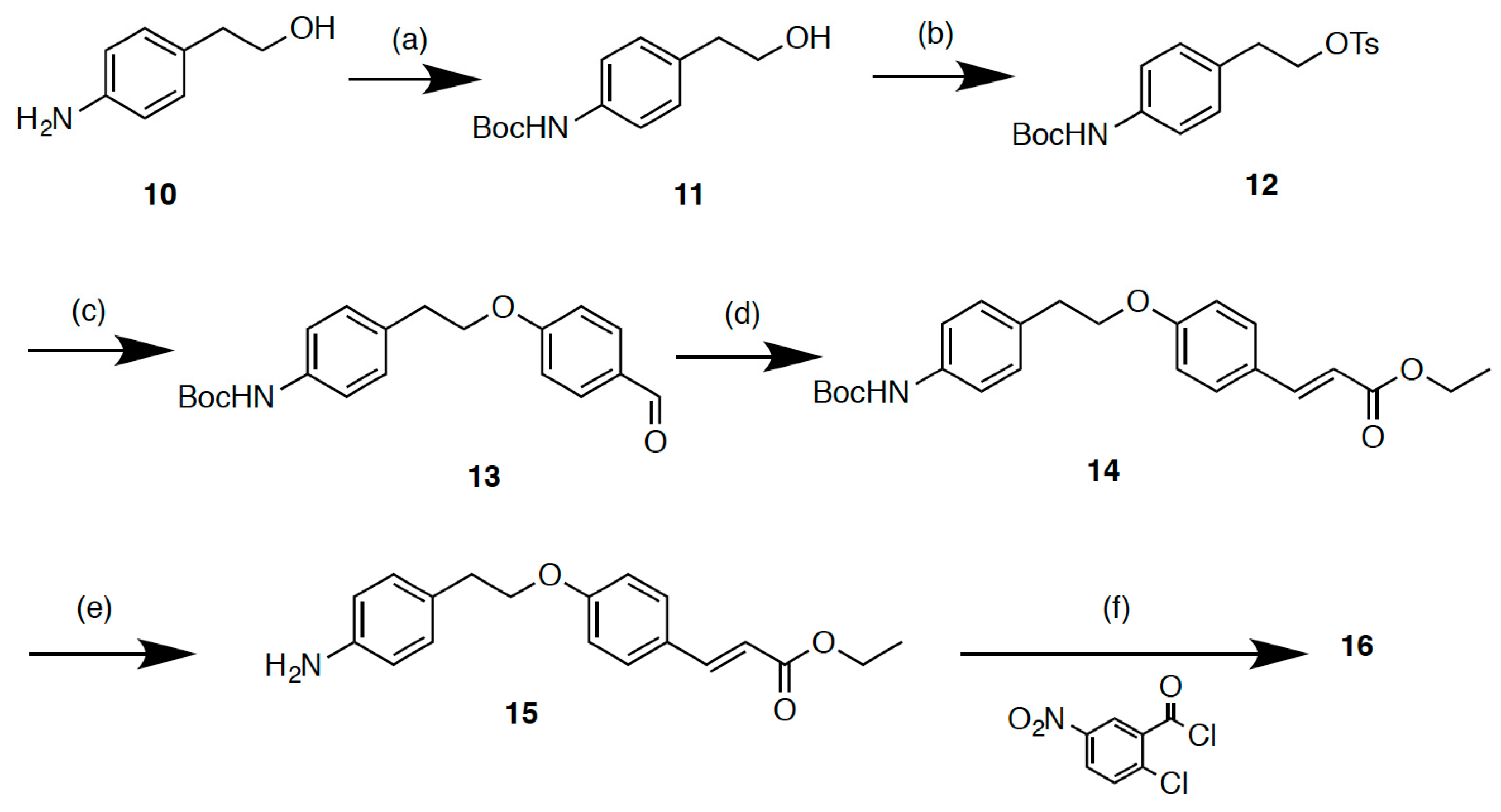
2.2. Synthesis
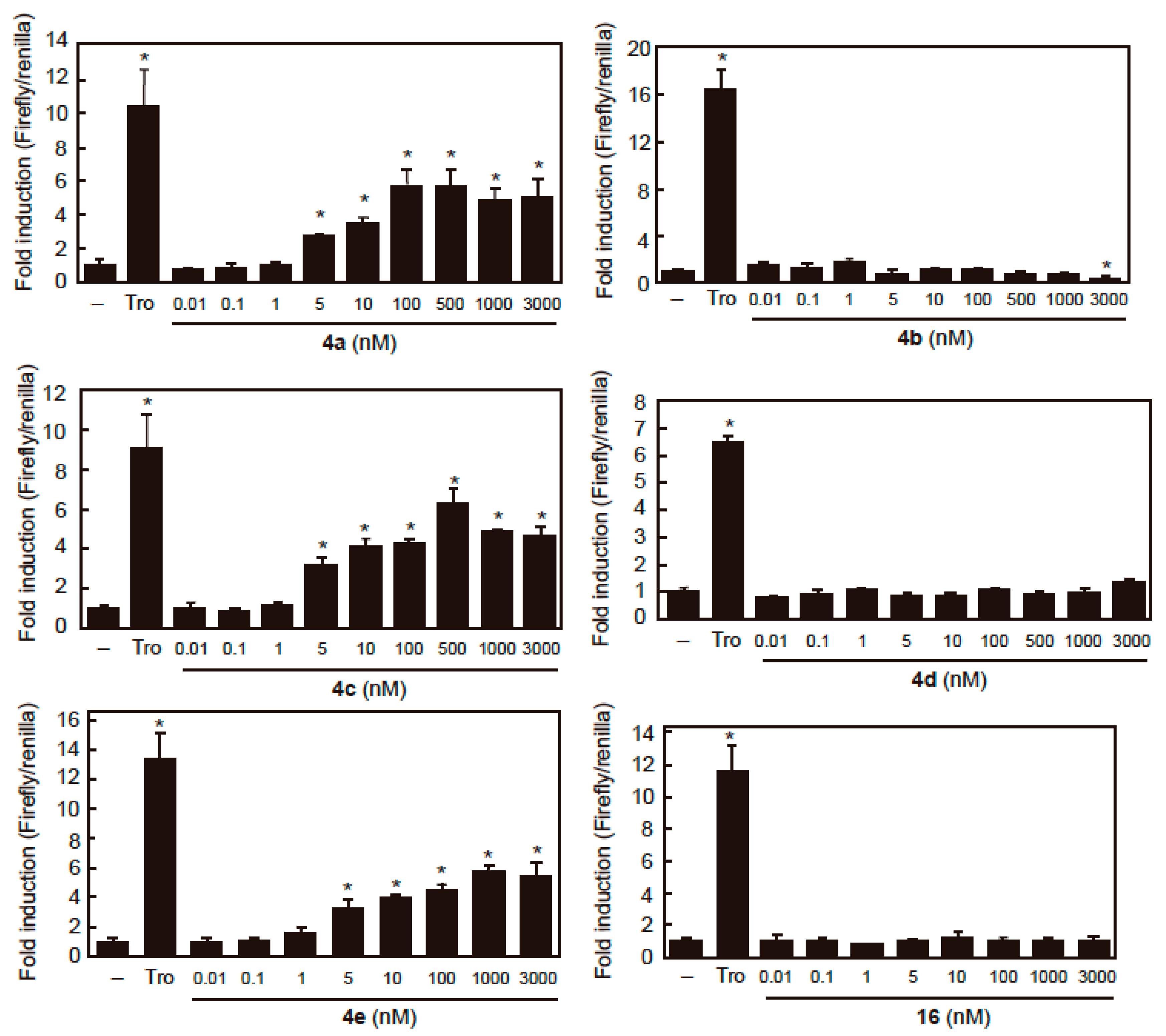
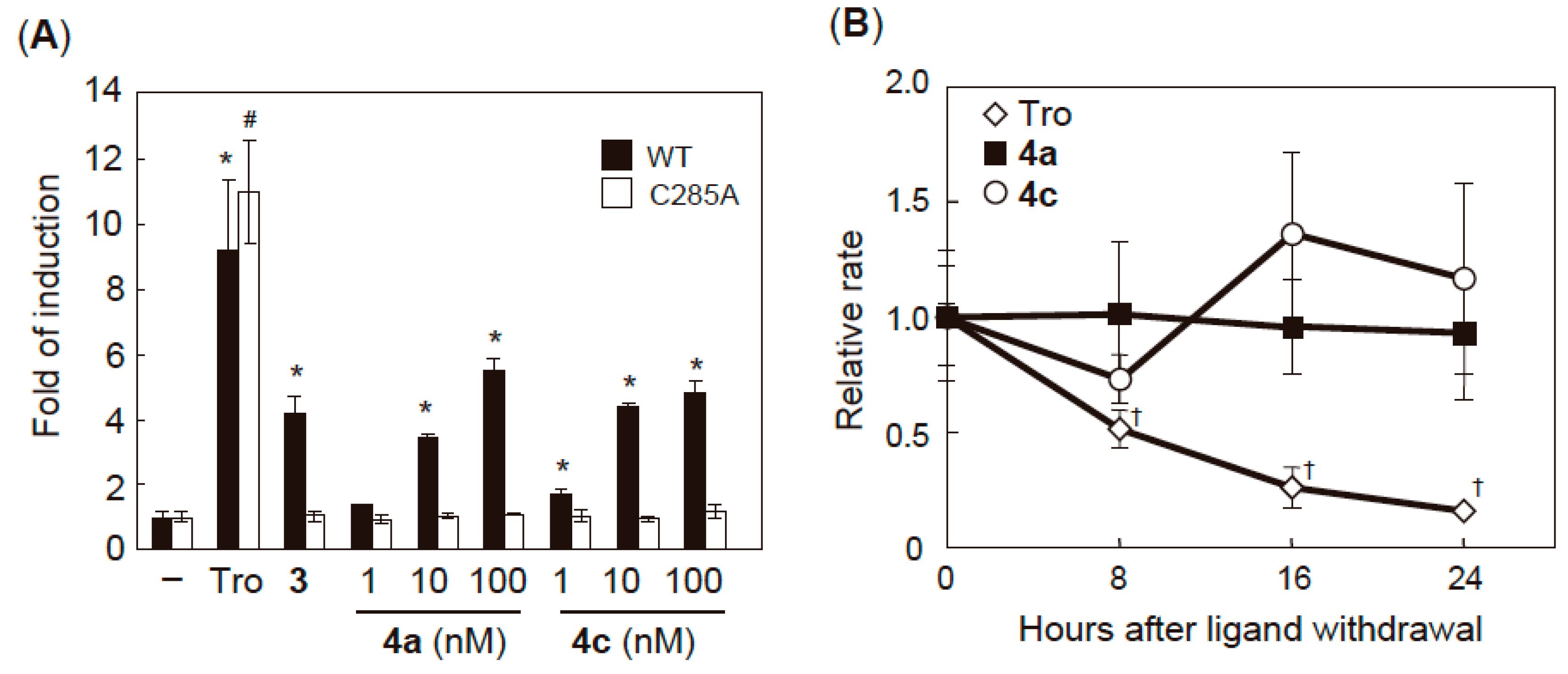
2.3. PPARγ Agonist Activity
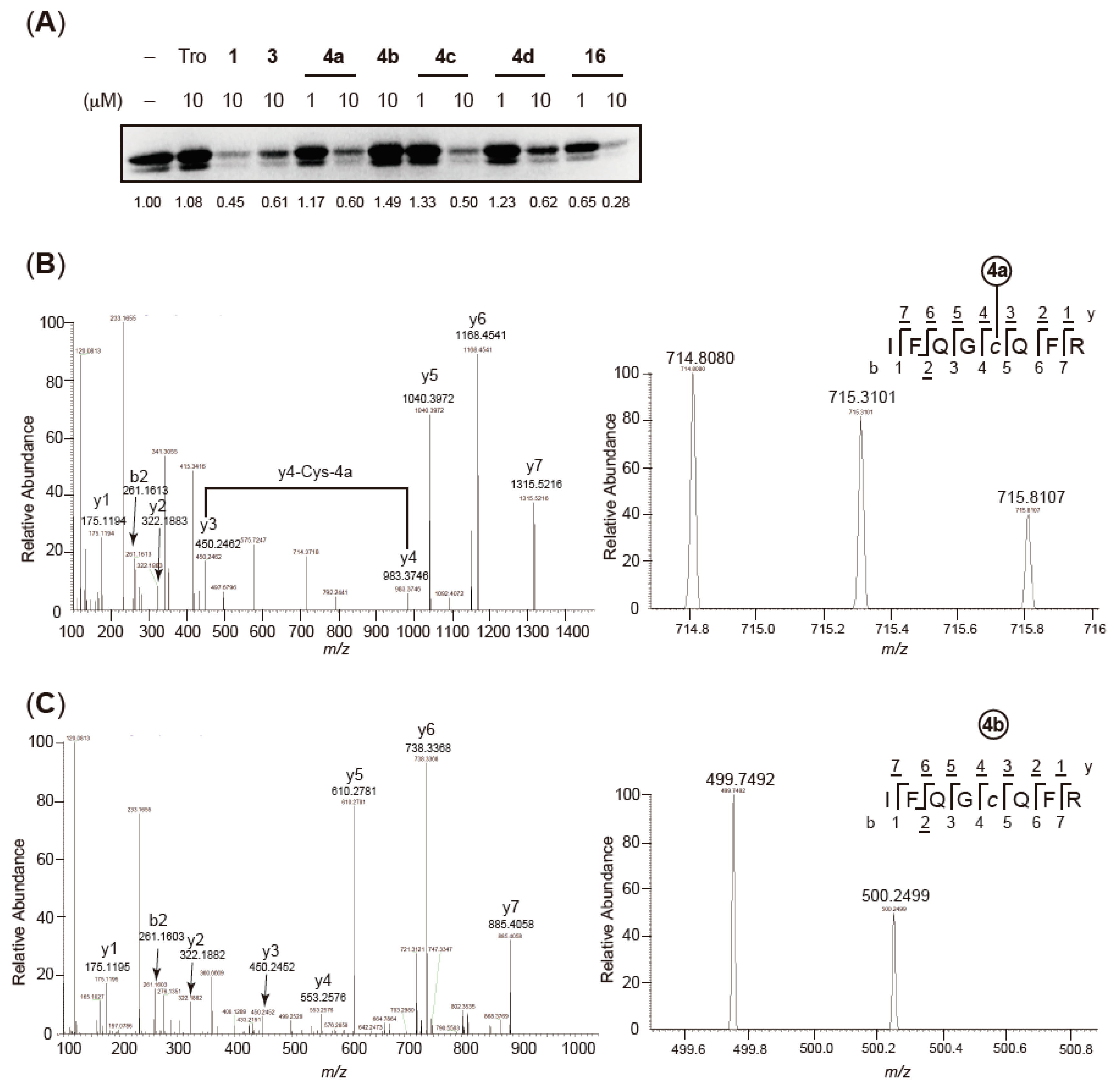
2.4. Validation of Covalent Bond Formation of the Synthesized Compounds with the Cys285 Residue
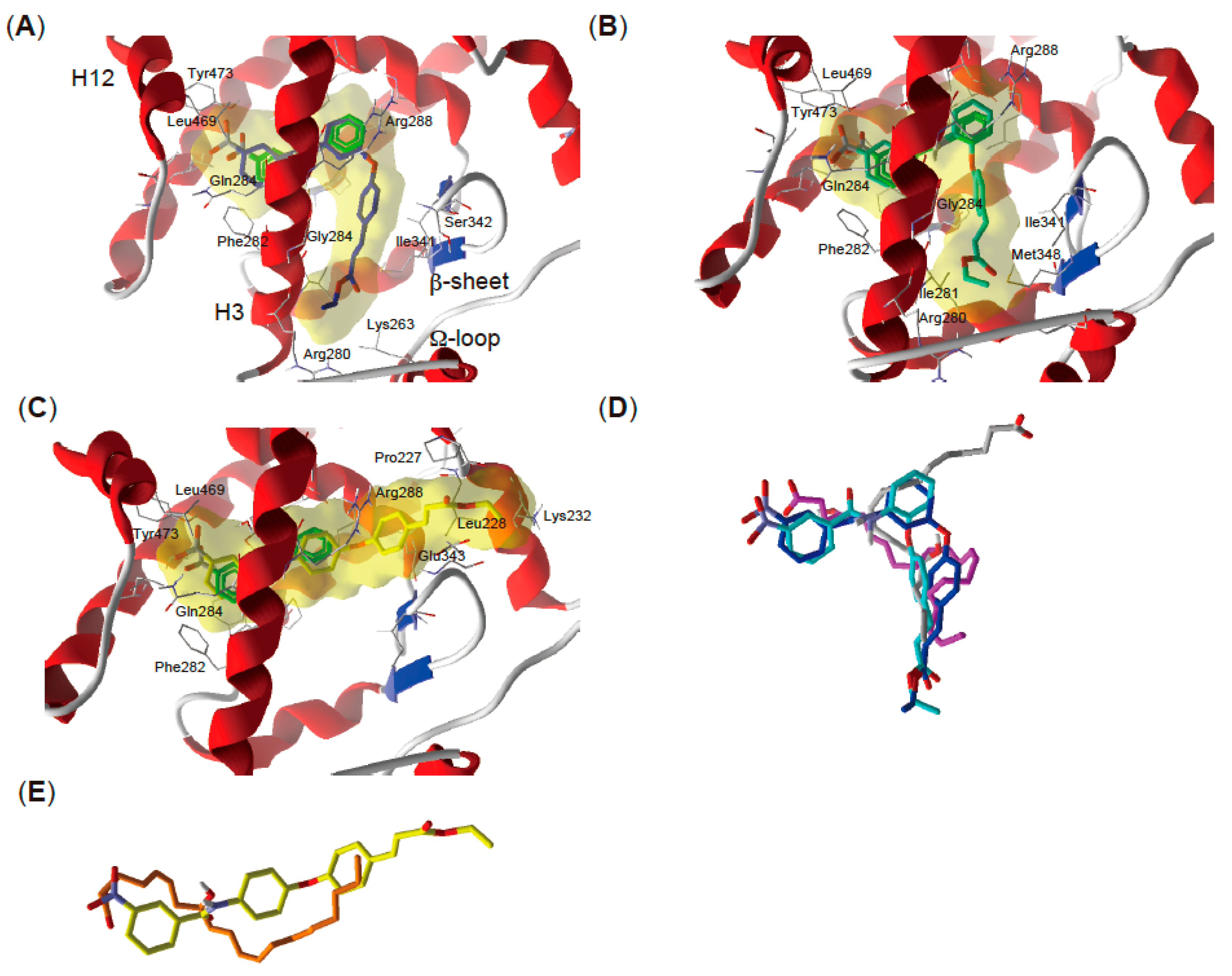
2.5. Docking Study
2.6. Characterization of Agonist-Type Covalent Ligands
3. Materials and Methods
3.1. Chemistry
3.1.1. General Remarks
3.1.2. 4-(3-Nitrophenoxy)benzaldehyde (6a)
3.1.3. General Procedure of Preparation for Compounds 6b and c
3.1.4. 4-(2-Nitrophenoxy)benzaldehyde (6b)
3.1.5. 4-(4-Nitrophenoxy)benzaldehyde (6c)
3.1.6. General Procedure of Preparation for Compounds 7a–c
3.1.7. (E)-Ethyl 3-[4-(3-nitrophenoxy)phenyl]acrylate (7a)
3.1.8. (E)-Ethyl 3-[4-(2-nitrophenoxy)phenyl]acrylate (7b)
3.1.9. (E)-Ethyl 3-[4-(4-nitrophenoxy)phenyl]acrylate (7c)
3.1.10. General Procedure of Preparation for Compounds 8a–c
3.1.11. (E)-Ethyl 3-[4-(3-aminophenoxy)phenyl]acrylate (8a)
3.1.12. (E)-Ethyl 3-[4-(2-aminophenoxy)phenyl]acrylate (8b)
3.1.13. (E)-Ethyl 3-[4-(4-aminophenoxy)phenyl]acrylate (8c)
3.1.14. Ethyl 3-[4-(3-aminophenoxy)phenyl]propanoate (9)
3.1.15. General Procedure of Preparation for Compounds 4a–e
3.1.16. (E)-Ethyl 3-{4-[3-(2-chloro-5-nitrobenzamido)phenoxy]phenyl}acrylate (4a)
3.1.17. (E)-Ethyl 3-{4-[3-(3-nitrobenzamido)phenoxy]phenyl}acrylate (4b)
3.1.18. (E)-Ethyl 3-{4-[2-(2-chloro-5-nitrobenzamido)phenoxy]phenyl}acrylate (4c)
3.1.19. (E)-Ethyl 3-{4-[4-(2-chloro-5-nitrobenzamido)phenoxy]phenyl}acrylate (4d)
3.1.20. Ethyl 3-{4-[3-(2-chloro-5-nitrobenzamido)phenoxy]phenyl}propanoate (4e)
3.1.21. tert-Butyl N-(4-{2-[(4-methylbenzenesulfonyl)oxy]ethyl}phenyl)carbamate (12)
3.1.22. tert-Butyl N-{4-[2-(4-formylphenoxy)ethyl]phenyl}carbamate (13)
3.1.23. (E)-Ethyl 3-{4-[2-(4-{[(tert-butoxy)carbonyl]amino}phenyl)ethoxy]phenyl}acrylate (14)
3.1.24. (E)-Ethyl 3-{4-[2-(4-aminophenyl)ethoxy]phenyl}acrylate (15)
3.1.25. (E)-Ethyl 3-(4-{2-[4-(2-chloro-5-nitrobenzamido)phenyl]ethoxy}phenyl)acrylate (16)
3.2. Cell Line and Cell Culture
3.3. Plasmids and Recombinant Protein
3.4. PPARγ Reporter Assay
3.5. Rhodamine-Maleimide Assay.
3.6. LC-MS/MS Detection of the Hybrid Ligand Modification of PPARγ
3.7. Docking Studies
3.8. Statistics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chawla, A.; Repa, J.J.; Evans, R.M.; Mangelsdorf, D.J. Nuclear receptors and lipid physiology: Opening the X-files. Science 2001, 294, 1866–1870. [Google Scholar] [CrossRef]
- Berger, J.; Moller, D.E. The mechanisms of action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef]
- Nolte, R.T.; Wisely, G.B.; Westin, S.; Cobb, J.E.; Lambert, M.H.; Kurokawa, R.; Rosenfeld, M.G.; Willson, T.M.; Glass, C.K.; Milburn, M.V. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 1998, 395, 137–143. [Google Scholar] [CrossRef]
- Nagy, L.; Schwabe, J.W. Mechanism of the nuclear receptor molecular switch. Trends Biochem. Sci. 2004, 29, 317–324. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Sundseth, S.S.; Jones, S.A.; Brown, P.J.; Wisely, G.B.; Koble, C.S.; Devchand, P.; Wahli, W.; Willson, T.M.; Lenhard, J.M.; et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc. Natl. Acad. Sci. USA 1997, 94, 4318–4323. [Google Scholar] [CrossRef]
- Tontonoz, P.; Spiegelman, B.M. Fat and beyond: The diverse biology of PPARgamma. Annu. Rev. Biochem. 2008, 77, 289–312. [Google Scholar] [CrossRef]
- Ricote, M.; Valledor, A.F.; Glass, C.K. Decoding transcriptional programs regulated by PPARs and LXRs in the macrophage: Effects on lipid homeostasis, inflammation, and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 230–239. [Google Scholar] [CrossRef]
- Guan, Y.; Hao, C.; Cha, D.R.; Rao, R.; Lu, W.; Kohan, D.E.; Magnuson, M.A.; Redha, R.; Zhang, Y.; Breyer, M.D. Thiazolidinediones expand body fluid volume through PPARgamma stimulation of ENaC-mediated renal salt absorption. Nat. Med. 2005, 11, 861–866. [Google Scholar] [CrossRef]
- Home, P.D.; Pocock, S.J.; Beck-Nielsen, H.; Curtis, P.S.; Gomis, R.; Hanefeld, M.; Jones, N.P.; Komajda, M.; McMurray, J.J. Rosiglitazone evaluated for cardiovascular outcomes in oral agent combination therapy for type 2 diabetes (RECORD): A multicentre, randomised, open-label trial. Lancet 2009, 373, 2125–2135. [Google Scholar] [CrossRef]
- Wright, M.B.; Bortolini, M.; Tadayyon, M.; Bopst, M. Minireview: Challenges and opportunities in development of PPAR agonists. Mol. Endocrinol. 2014, 28, 1756–1768. [Google Scholar] [CrossRef]
- Tagami, T.; Yamamoto, H.; Moriyama, K.; Sawai, K.; Usui, T.; Shimatsu, A.; Naruse, M. A selective peroxisome proliferator-activated receptor-gamma modulator, telmisartan, binds to the receptor in a different fashion from thiazolidinediones. Endocrinology 2009, 150, 862–870. [Google Scholar] [CrossRef]
- Fujimura, T.; Sakuma, H.; Konishi, S.; Oe, T.; Hosogai, N.; Kimura, C.; Aramori, I.; Mutoh, S. FK614, a novel peroxisome proliferator-activated receptor gamma modulator, induces differential transactivation through a unique ligand-specific interaction with transcriptional coactivators. J. Pharmacol. Sci. 2005, 99, 342–352. [Google Scholar] [CrossRef]
- Chang, C.H.; McNamara, L.A.; Wu, M.S.; Muise, E.S.; Tan, Y.; Wood, H.B.; Meinke, P.T.; Thompson, J.R.; Doebber, T.W.; Berger, J.P.; et al. A novel selective peroxisome proliferator-activator receptor-gamma modulator-SPPARgammaM5 improves insulin sensitivity with diminished adverse cardiovascular effects. Eur. J. Pharmacol. 2008, 584, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Ohtera, A.; Miyamae, Y.; Yoshida, K.; Maejima, K.; Akita, T.; Kakizuka, A.; Irie, K.; Masuda, S.; Kambe, T.; Nagao, M. Identification of a New Type of Covalent PPARgamma Agonist using a Ligand-Linking Strategy. ACS Chem. Biol. 2015, 10, 2794–2804. [Google Scholar] [CrossRef]
- Waku, T.; Shiraki, T.; Oyama, T.; Maebara, K.; Nakamori, R.; Morikawa, K. The nuclear receptor PPARgamma individually responds to serotonin- and fatty acid-metabolites. EMBO J. 2010, 29, 3395–3407. [Google Scholar] [CrossRef]
- Leesnitzer, L.M.; Parks, D.J.; Bledsoe, R.K.; Cobb, J.E.; Collins, J.L.; Consler, T.G.; Davis, R.G.; Hull-Ryde, E.A.; Lenhard, J.M.; Patel, L.; et al. Functional consequences of cysteine modification in the ligand binding sites of peroxisome proliferator activated receptors by GW9662. Biochemistry 2002, 41, 6640–6650. [Google Scholar] [CrossRef] [PubMed]
- Itoh, T.; Fairall, L.; Amin, K.; Inaba, Y.; Szanto, A.; Balint, B.L.; Nagy, L.; Yamamoto, K.; Schwabe, J.W.R. Structural basis for the activation of PPARγ by oxidized fatty acids. Nat. Struct.Mol. Biol. 2008, 15, 924–931. [Google Scholar] [CrossRef]
- Schopfer, F.J.; Cole, M.P.; Groeger, A.L.; Chen, C.S.; Khoo, N.K.; Woodcock, S.R.; Golin-Bisello, F.; Motanya, U.N.; Li, Y.; Zhang, J.; et al. Covalent peroxisome proliferator-activated receptor gamma adduction by nitro-fatty acids: Selective ligand activity and anti-diabetic signaling actions. J. Biol. Chem. 2010, 285, 12321–12333. [Google Scholar] [CrossRef]
- Wei, T.; Furgal, J.C.; Scott, T.F. In situ deprotection and dynamic covalent assembly using a dual role catalyst. Chem. Commun. 2017, 53, 3874–3877. [Google Scholar] [CrossRef]
- Ohtera, A.; Miyamae, Y.; Nakai, N.; Kawachi, A.; Kawada, K.; Han, J.; Isoda, H.; Neffati, M.; Akita, T.; Maejima, K.; et al. Identification of 6-octadecynoic acid from a methanol extract of Marrubium vulgare L. as a peroxisome proliferator-activated receptor gamma agonist. Biochem. Biophys. Res. Commun. 2013, 440, 204–209. [Google Scholar] [CrossRef]
- Shiraki, T.; Kamiya, N.; Shiki, S.; Kodama, T.S.; Kakizuka, A.; Jingami, H. Alpha,beta-unsaturated ketone is a core moiety of natural ligands for covalent binding to peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 2005, 280, 14145–14153. [Google Scholar] [CrossRef]
- de Groot, J.C.; Weidner, C.; Krausze, J.; Kawamoto, K.; Schroeder, F.C.; Sauer, S.; Bussow, K. Structural characterization of amorfrutins bound to the peroxisome proliferator-activated receptor gamma. J. Med. Chem. 2013, 56, 1535–1543. [Google Scholar] [CrossRef] [PubMed]
- Amato, A.A.; Rajagopalan, S.; Lin, J.Z.; Carvalho, B.M.; Figueira, A.C.; Lu, J.; Ayers, S.D.; Mottin, M.; Silveira, R.L.; Souza, P.C.; et al. GQ-16, a novel peroxisome proliferator-activated receptor gamma (PPARgamma) ligand, promotes insulin sensitization without weight gain. J. Biol. Chem. 2012, 287, 28169–28179. [Google Scholar] [CrossRef]
- Hughes, T.S.; Giri, P.K.; de Vera, I.M.S.; Marciano, D.P.; Kuruvilla, D.S.; Shin, Y.; Blayo, A.-L.; Kamenecka, T.M.; Burris, T.P.; Griffin, P.R.; et al. An alternate binding site for PPARγ ligands. Nat. Commun. 2014, 5, 3571. [Google Scholar] [CrossRef] [PubMed]
- Waku, T.; Shiraki, T.; Oyama, T.; Fujimoto, Y.; Maebara, K.; Kamiya, N.; Jingami, H.; Morikawa, K. Structural insight into PPARgamma activation through covalent modification with endogenous fatty acids. J. Mol. Biol. 2009, 385, 188–199. [Google Scholar] [CrossRef]
- Bauer, R.A. Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today 2015, 20, 1061–1073. [Google Scholar] [CrossRef]
- Baillie, T.A. Targeted Covalent Inhibitors for Drug Design. Angew. Chem. Int. Ed. 2016, 55, 13408–13421. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 4a, 4b, 4c, 4d, 4e, and 16, as well as the docking data, are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | EC50 (nM) |
|---|---|
| 4a | 5.13 ± 0.45 |
| 4b | N.D. |
| 4c | 4.11 ± 0.13 |
| 4d | N.D. |
| 4e | 2.54 ± 1.30 |
| 16 | N.D. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Utsugi, Y.; Kobuchi, H.; Kawamura, Y.; Atito, A.S.A.; Nagao, M.; Isoda, H.; Miyamae, Y. Importance of the Proximity and Orientation of Ligand-Linkage to the Design of Cinnamate-GW9662 Hybrid Compounds as Covalent PPARγ Agonists. Molecules 2019, 24, 2019. https://doi.org/10.3390/molecules24102019
Utsugi Y, Kobuchi H, Kawamura Y, Atito ASA, Nagao M, Isoda H, Miyamae Y. Importance of the Proximity and Orientation of Ligand-Linkage to the Design of Cinnamate-GW9662 Hybrid Compounds as Covalent PPARγ Agonists. Molecules. 2019; 24(10):2019. https://doi.org/10.3390/molecules24102019
Chicago/Turabian StyleUtsugi, Yuki, Hirona Kobuchi, Yukio Kawamura, Ahmed Salahelden Aboelhamd Atito, Masaya Nagao, Hiroko Isoda, and Yusaku Miyamae. 2019. "Importance of the Proximity and Orientation of Ligand-Linkage to the Design of Cinnamate-GW9662 Hybrid Compounds as Covalent PPARγ Agonists" Molecules 24, no. 10: 2019. https://doi.org/10.3390/molecules24102019
APA StyleUtsugi, Y., Kobuchi, H., Kawamura, Y., Atito, A. S. A., Nagao, M., Isoda, H., & Miyamae, Y. (2019). Importance of the Proximity and Orientation of Ligand-Linkage to the Design of Cinnamate-GW9662 Hybrid Compounds as Covalent PPARγ Agonists. Molecules, 24(10), 2019. https://doi.org/10.3390/molecules24102019