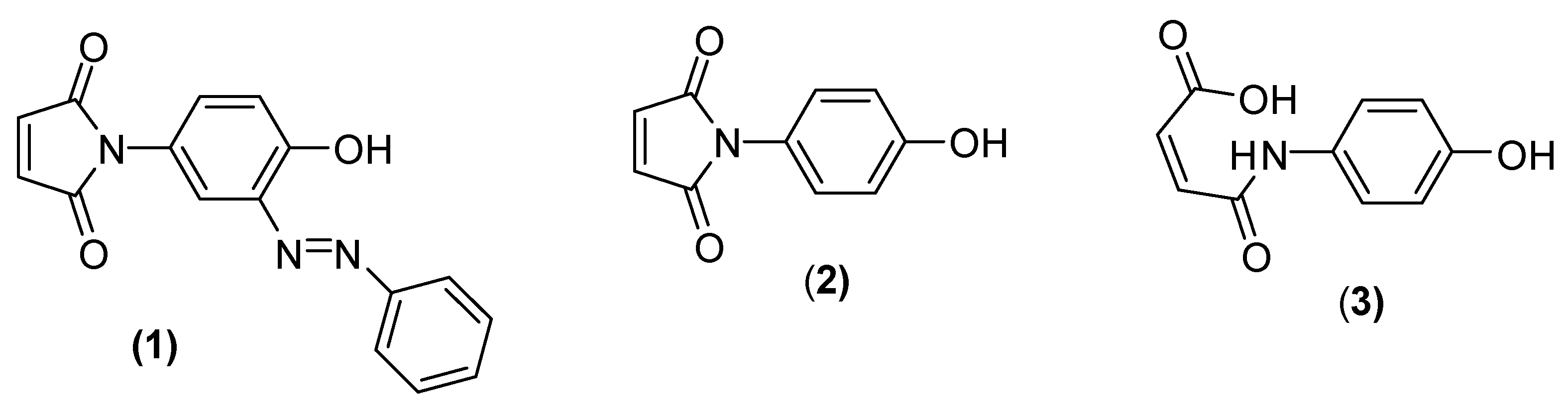
2. Results and Discussion
Reaction of
p-aminophenol with maleic anhydride, as described by Mohammed and Mustapha [
1], generated a yellow solid, albeit in a disappointingly poor yield, a result which is in accord with earlier observations. We believe that the
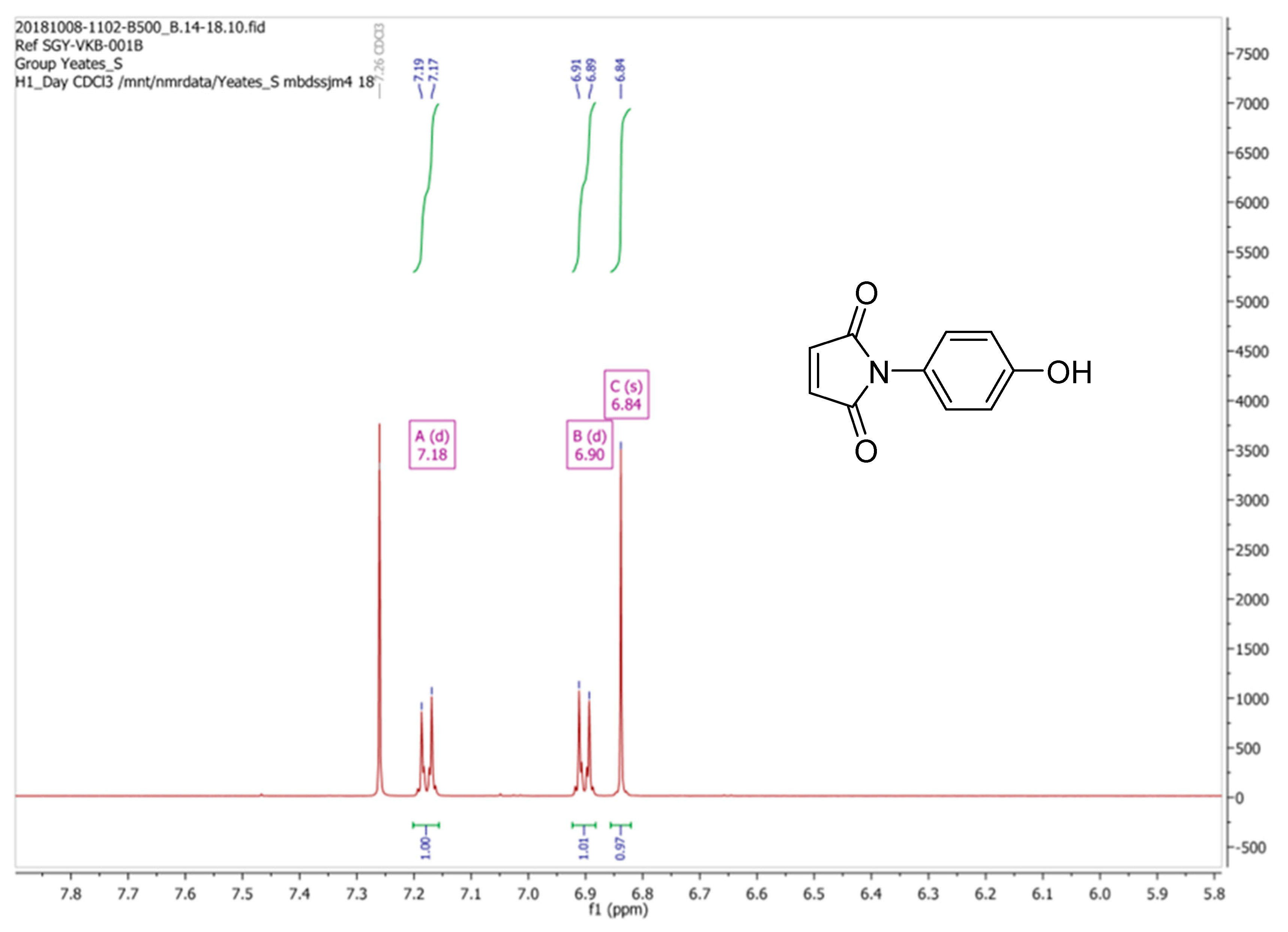
1H-NMR spectrum of (
2), when recorded in CDCl
3 (i.e., a 2H singlet at δ 6.83 ppm for the vinyl C-Hs of the maleimide moiety and an AB system, comprising of two, 2H doublets [δ 6.89 ppm and δ 7.17 ppm], for the aromatic hydrogens) as depicted in
Figure 2 to be in keeping with its supposed structure. However, our
1H-NMR data was found to be substantively different to the spectrum that was reported by Mohammed and Mustapha for the same compound which was recorded in CD
3OD. Critically these workers stated that the maleimide CHs appeared as two well-resolved doublets (centered at δ 6.79 ppm and δ 7.47 ppm) which is an assignment that is clearly improbable. In checking the literature, we note that the
1H-NMR spectrum for (
2) as reported by Tang et al. [
2], which was also recorded in CDCl
3, was identical to our data (
Figure 2). Being at a loss to explain these discrepancies the synthesis of (
2) was repeated a third time using the method reported by Jin et al. [
3]. Again the
1H-NMR spectrum (run in CDCl
3) for this new sample of (
2) was identical to the spectrum of the batch that we originally obtained using Mohammed and Mustapha’s procedure.
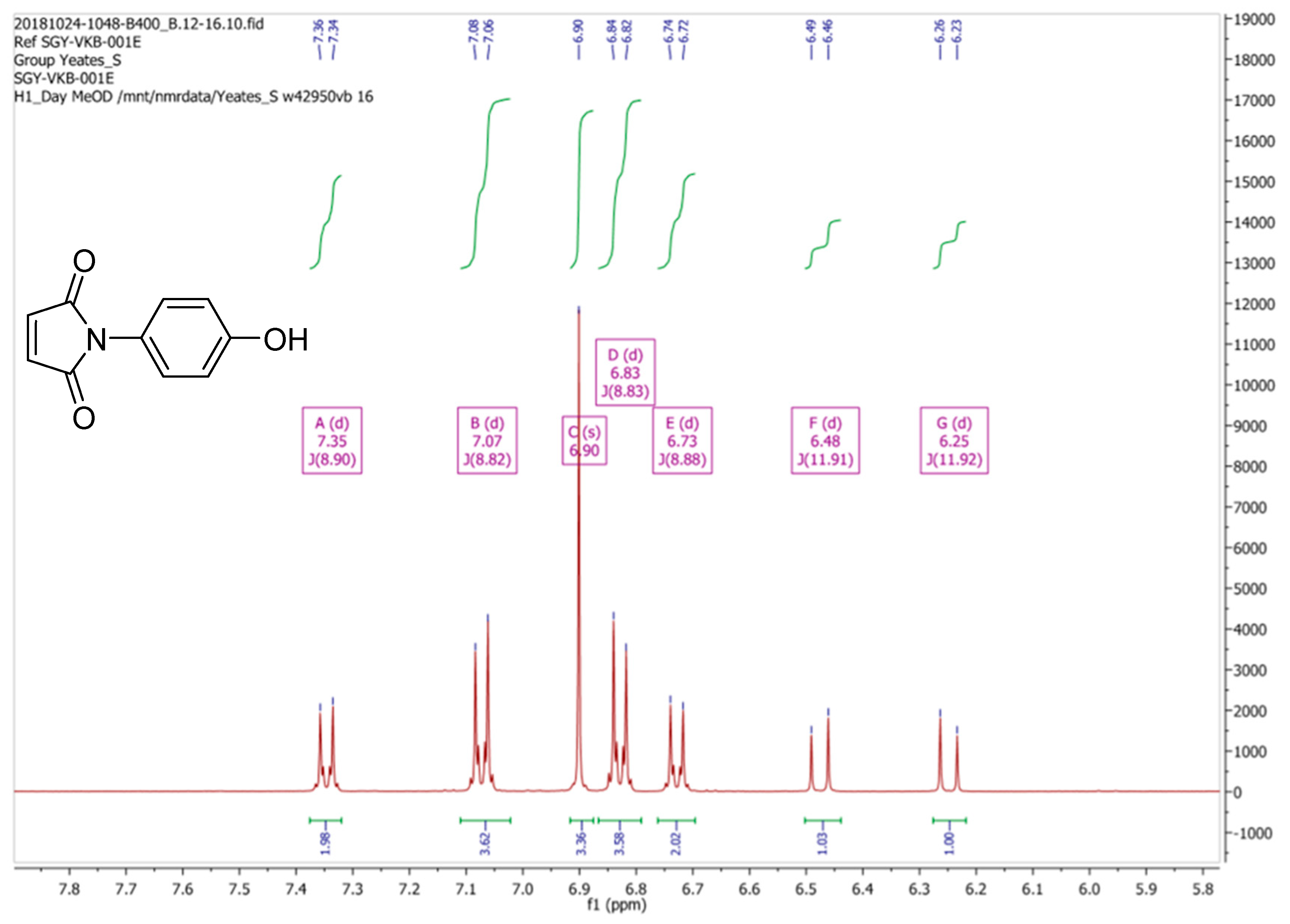
Due to these discrepancies the
1H-NMR spectrum of (
2) was then re-recorded in CD
3OD, as originally reported by Mohammed and Mustapha [
1]. Our spectral data for (
2) in CD
3OD is depicted in
Figure 3. Clearly this spectrum is substantially different, both in terms of signal multiplicities and chemical shifts, from the spectrum that was obtained in CDCl
3. Moreover, we observed that the
1H-NMR spectrum of (
2) in CD
3OD varied with time, a process which appeared to reach equilibrium after a period of one hour, resulting the
1H-NMR spectrum which is reported in
Figure 4.
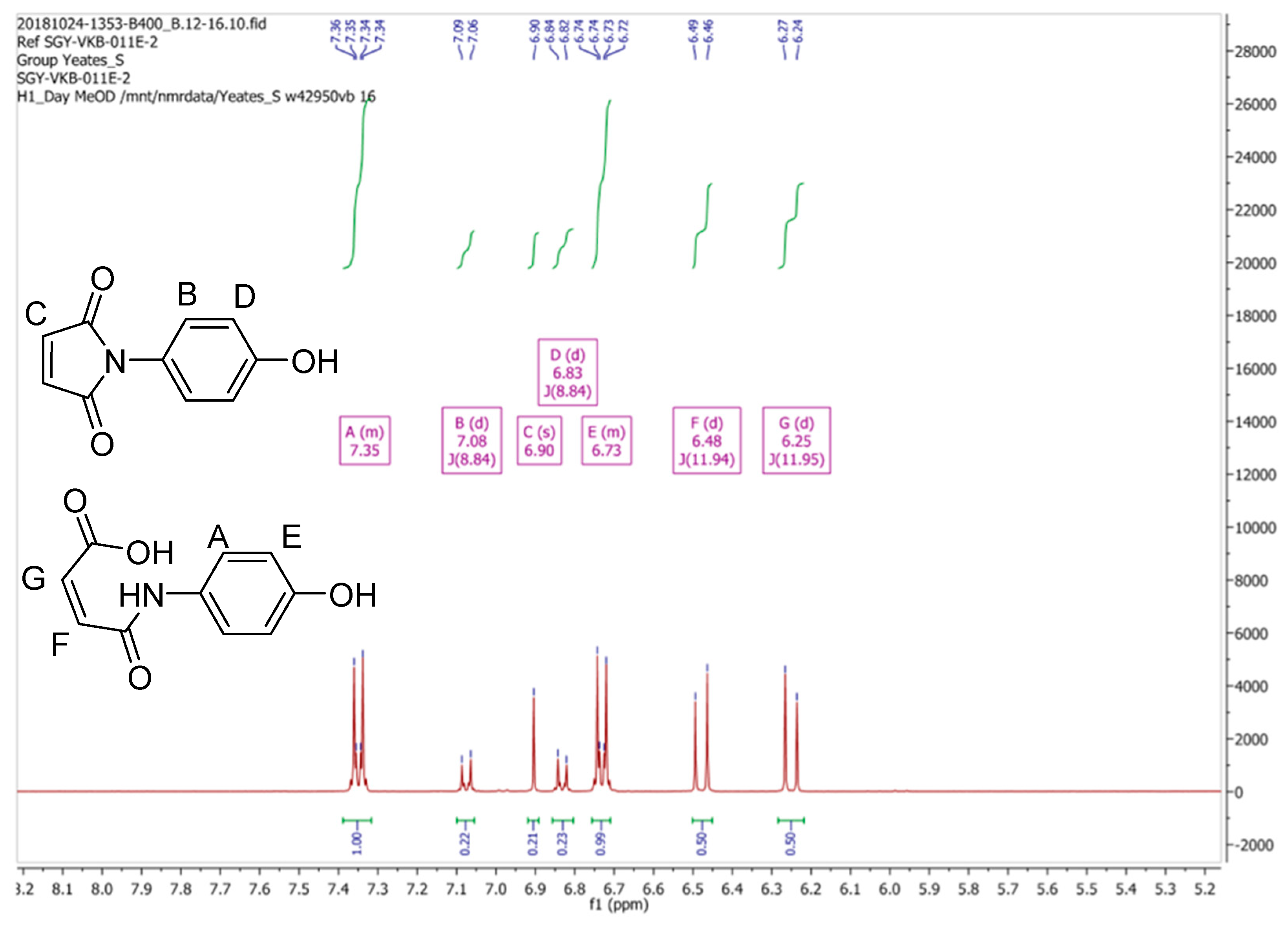
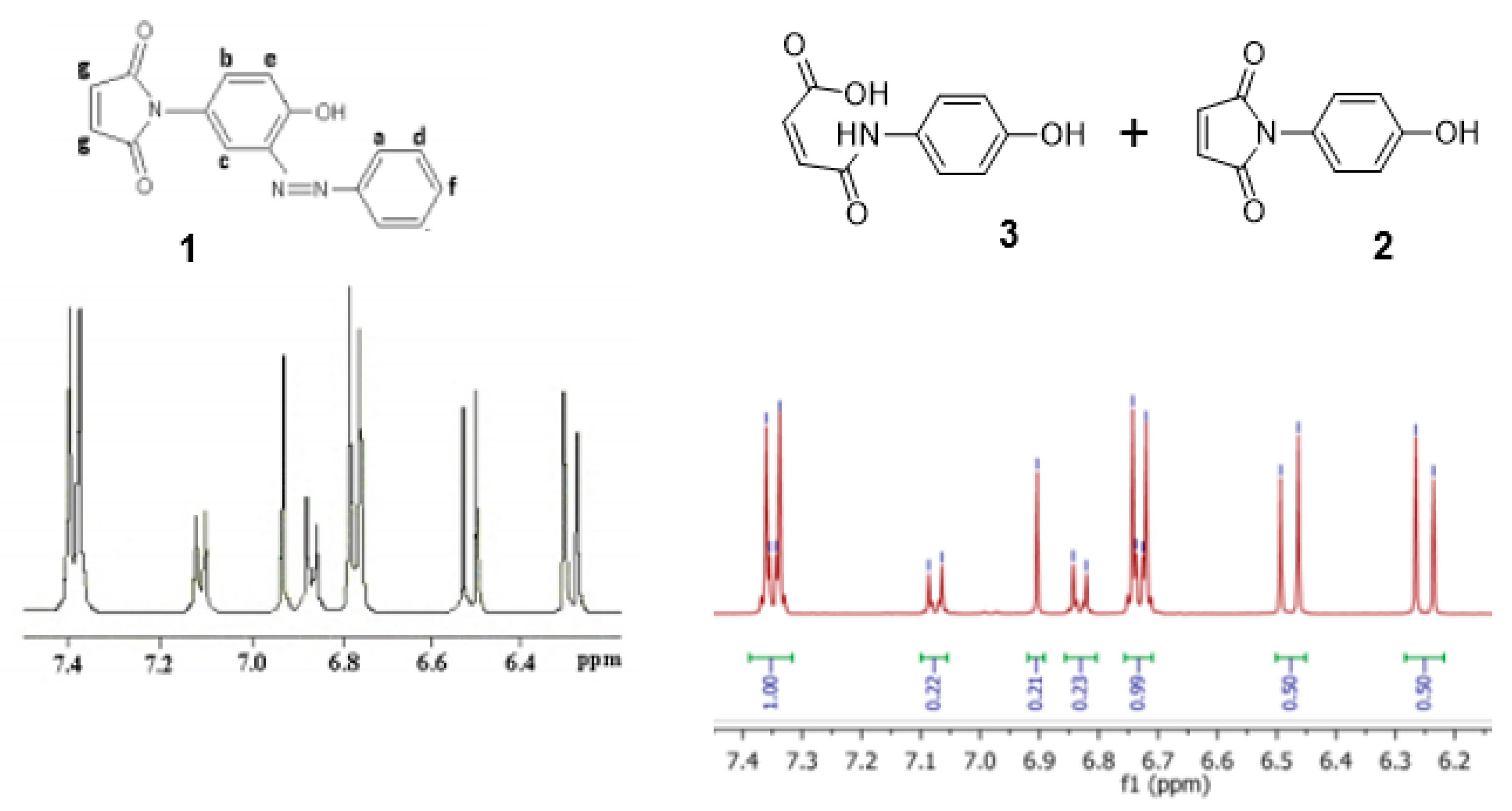
From the data presented in
Figure 3 and
Figure 4 we conclude that the resonances at δ 6.83, 6.90 and 7.08 ppm, which diminish in intensity with time, are associated with maleimide (
2). We also suggest that a second component present in this mixture, which is associated with the resonances at δ 6.25 (1H, d,
3J11.95 Hz), 6.48 (1H, d,
3J11.94 Hz), 6.73 (2H, m) and 7.35 (2H, m) ppm, which increase in intensity with time, can be assigned to the ring-opened, (
Z)-maleamic acid, (
3). Furthermore, a detailed analysis of the
1H spectrum of (
3), as a solution in DMSO-
d6, is available in the literature [
4], which is in accord with our analysis. We assume that adventitious water present in CD
3OD is responsible for this ring opening reaction [
5].
At this point we note that the reporting of the NMR data for (
2) by Mohammed and Mustapha is less than ideal. The
1H-NMR spectrum purported to be associated with (
2) as reproduced in
Figure 5 (doublets/multiplets at δ 6.32. 6.58, 6.79 and 7.47ppm) is at odds with that which is reported for
2 in the experimental section of the same paper [
1]:
“1H-NMR (CD3OD): 7.20–7.43 (aromatic), 6.93–7.25 (HC=CH of maleimide), 6.26–6.52 (HC=CH) ppm.”
The proposed ring opening from (
2) to (
3) seems to come to an equilibrium and we have not seen it progress to full conversion. Given this and the fact that we appear to have clean material that does not show this ring-opening behavior in chloroform we decided to continue with the synthetic route reported to prepare compound (
1). Unfortunately, despite numerous attempts this failed as the procedure detailed by Mohammed and Mustapha [
1] merely afforded dark red/brown intractable solids with greater than stoichiometric yields. The most promising material resulting from these investigations afforded the proton NMR displayed in
Figure 6, below. Unfortunately, GC-MS analysis of this material, which again was reported by Mohammed and Mustapha to generate a MS in accord with it structure (more on this later), proved to be less than conclusive. Indeed, we were unable to find any trace of this product by GC-MS, with the only identifiable component being due to residual (
2) which eluted at 4.5 minutes at approximately 62 °C. We assume that other components of the mixture, as evidenced from its
1H-NMR spectrum, were either too volatile or in-volatile for GC-MS analysis despite allowing a long run on the instrument holding at 300 °C for 20 minutes. Again, our results appear to be at odds with those described by Mohammed and Mustapha [
1].
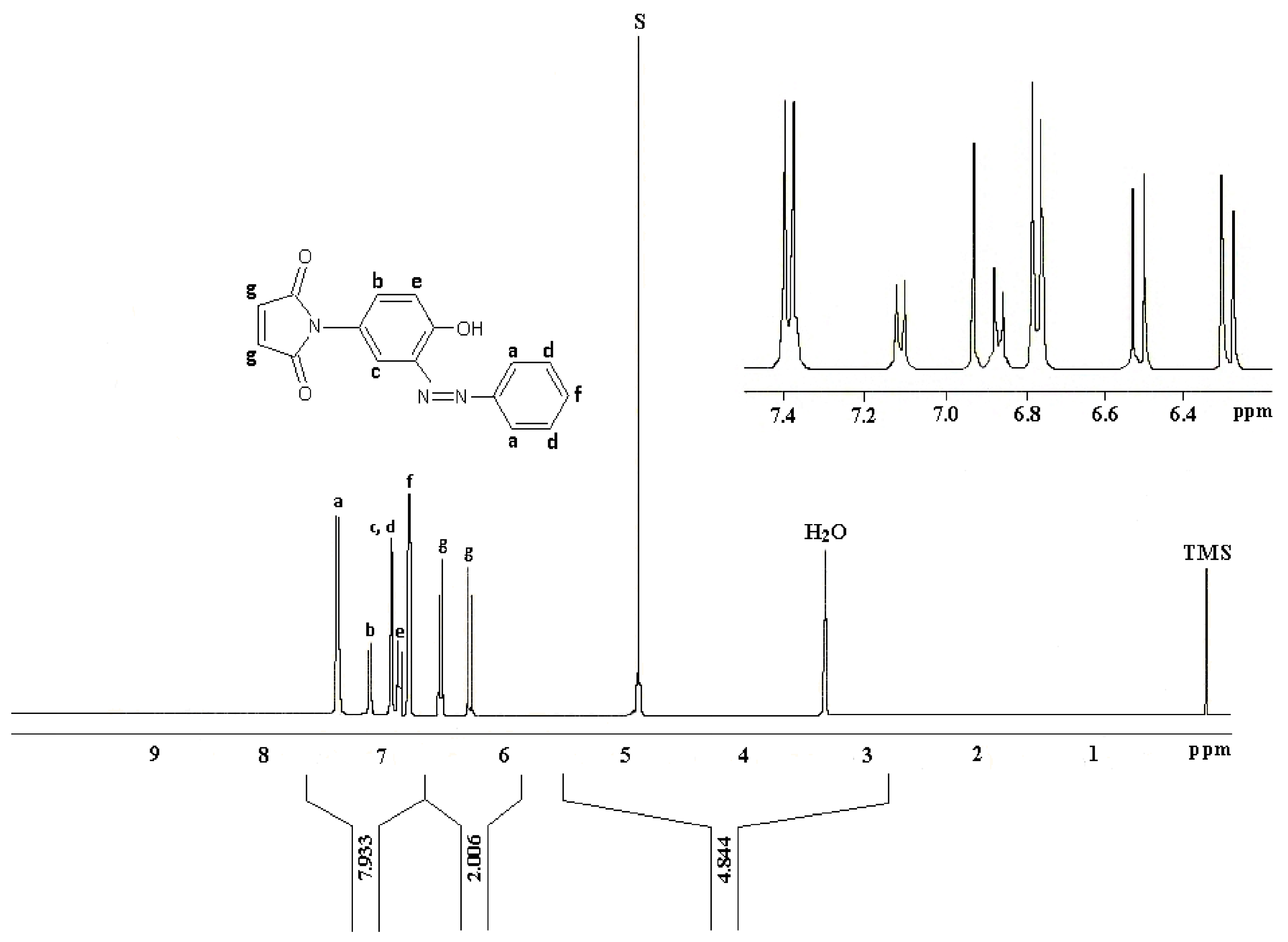
The
1H-NMR data reported by Mohammed and Mustapha for (
1) is reproduced in
Figure 7. Here again this analysis was a matter of some concern especially with regards to the assignment of the maleimide CHs (“g”,
Figure 7) which were reported as two doublets centered at δ 6.3 ppm and δ 6.5 ppm.
At this juncture we noticed a remarkable similarity between the
1H-NMR spectrum reported by Mohammed and Mustapha’s for azo-compound (
1) and our
1H-NMR spectrum of the equilibrating mixture of (
2) and (
2)
Figure 8. We propose that these two spectra depict the same mixture, though perhaps in different relative proportions.
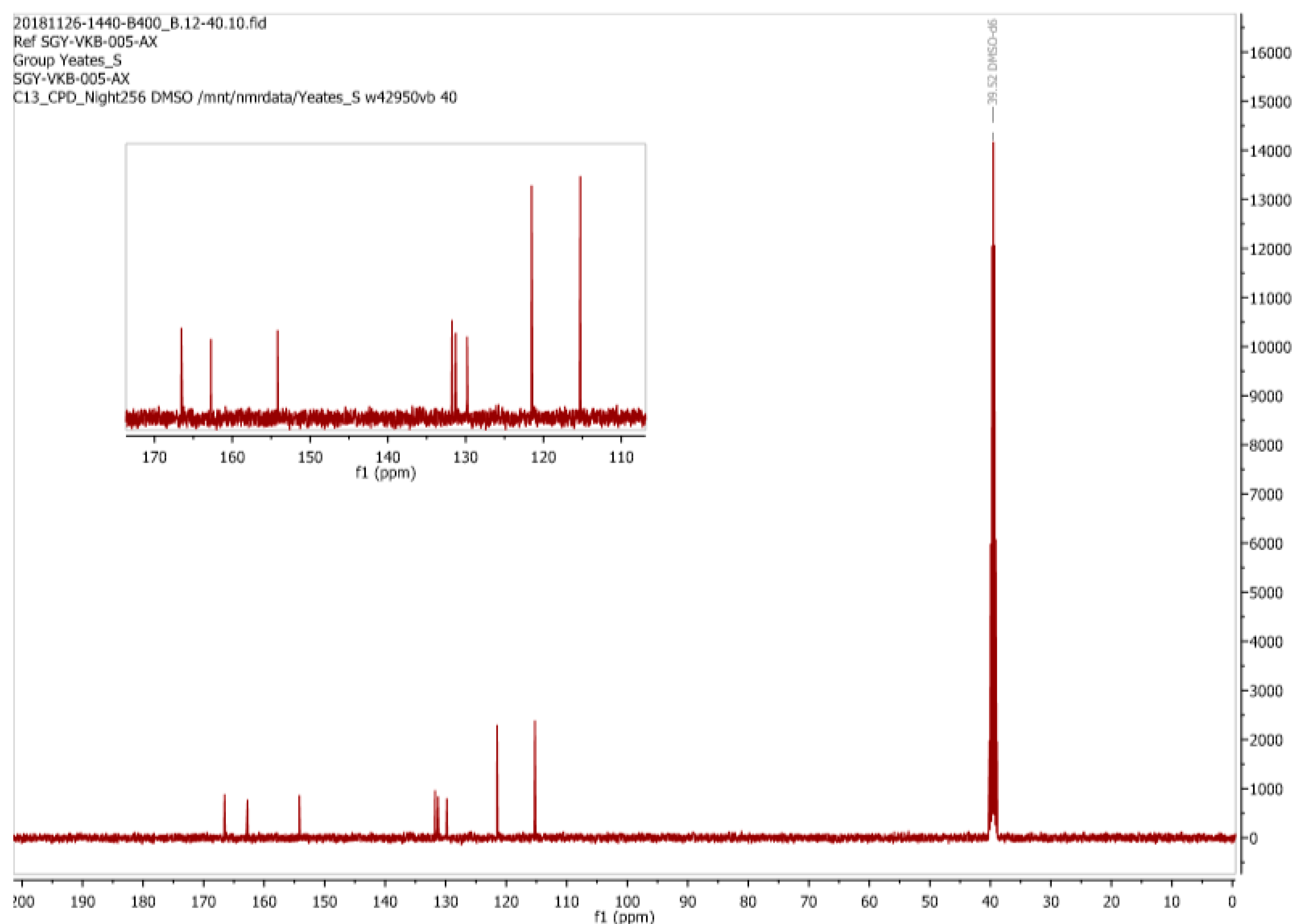
We next turned our attention to the
13C-NMR data for (
1) (recorded in CD
3OD) that was reported by Mohammed and Mustapha [
1],
Figure 9. Here we find the assignment of what appears to be only 8 resonances for (
1) to be erroneous. Significantly a resonance at around δ 164 ppm was simply ignored while there is an apparent accidental equivalence of three carbon signals at δ 122 ppm This is highly unusual for such a densely functionalized, low molecular weight, molecule of this type.
As reported above we prepared an authentic sample of (
3) by the method reported by Jin et al. [
3] so that its NMR spectra could be recorded in DMSO-
d6. In this solvent the exchangeable protons of the carboxylic acid (δ 13.71 ppm), amide (δ 10.3 ppm) and phenol (δ 9.35 ppm) residues were clearly apparent. The
13C-NMR spectrum of our “authentic” (
3) in DMSO-
d6 (
Figure 10) is also identical to that reported by Trujillo-Ferrara [
4]. While we should be cognizant of potential solvent effects, we again observe a remarkable similarity between the
13C-NMR spectrum for (
1) in CD
3OD as reported by Mohammed and Mustapha and the spectrum that we observed for an authentic sample of (
3) which was run in DMSO-
d6. We contend therefore that Mohammed and Mustapha isolated (
3) rather (
1) from their attempted diazonium coupling reaction, a realization that is emphatically supported by the appearance of only 8 carbon signals in their reported
13C-NMR spectrum (
Figure 9). It is now obvious that the resonances at δ 163 and δ 166 ppm reported by Mohammed and Mustapha for (
1) are in reality associated with the two carbonyl groups (i.e., the carboxylic acid and amide functionalities) of (
3). Awkwardly there is also a mismatch between the assignments as depicted in
Figure 9 and that reported in the experimental of Mohammed and Mustapha’s original paper [
1] which unexplainably does list two carbonyls but only 6 signals in total.
We also re-examined the mass spectral data for compound (
1) as presented in
Figure 3 of the original paper [
1]. Although the spectrum does look authentic the molecular ion would be expected to have a mass of 293 and not of 279 as presented. It is difficult to account for how a difference of 14 can be related as a mass loss from the expected molecular ion. As reported earlier, our attempts at finding even a trace of the desired compound (
1) by GC-MS failed but that we did find some of the starting material at
m/
z 189. This may also be the case for the published spectra with the possibly an impurity / column artefact at
m/
z 279. This is further supported by the observation that the published spectral data also displays a peak at
m/
z 189.
As we were only interested in synthesizing (
1) we did not attempt to replicate any further work in the contested paper [
1]. The apparent ring opening in methanol is intriguing but not what we sought [
5]. The reported characterization for the other materials synthesized in [
1] are somewhat difficult to follow as the proton NMR’s have no integration reported and few assignments. Most worryingly the maleimde CHs of all the products are as quoted as “multiplets”. We believe this to be erroneous. On a quick check over the number of expected
13C signals vs reported signals we also find far fewer reported than expected in all the diazo products reported in [
1]. However, without access to the original spectroscopic data it is difficult to establish how well interpreted the written experimental for products other than (
1) might be.
Beyond NMR interpretation all six of the azo compounds prepared in [
1] are reported as having sharp melting points and in all cases excellent microanalysis results are claimed. In the case of microanalysis the greatest deviation between “found” and “expected” values is a difference of 0.18% on carbon for the phenylazo-3-
N-(4-hydroxyphenyl)maleimide product. Unfortunately, these results must be in error as all of the calculated molecular formulas are incorrect. In the case of (
1) the results looked very good however rather than having 23 hydrogens, (
1) has 11. Obviously, this discrepancy of 12 H cannot possibly represent the true structure of (
1) so we compared the results to
3 which is the best candidate from the spectroscopy presented for (
1),
Table 1.
Evidently (
3) is not a good candidate structure for the reported figures so we looked at all of the other elemental analysis published in [
1]. Strangely we found that in all six of the newly reported azo compounds the same 12 hydrogen excess features in all of their published formula. We postulate that the authors may have wrongly drawn the structures as containing fully reduced, cyclohexyl, rather than aryl rings. As an example, (
4) has an excellent match to the reported microanalysis of (
1) and this structural alteration and its excellent fit is entirely consistent throughout the whole series of six compounds reported (
Figure 11).














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
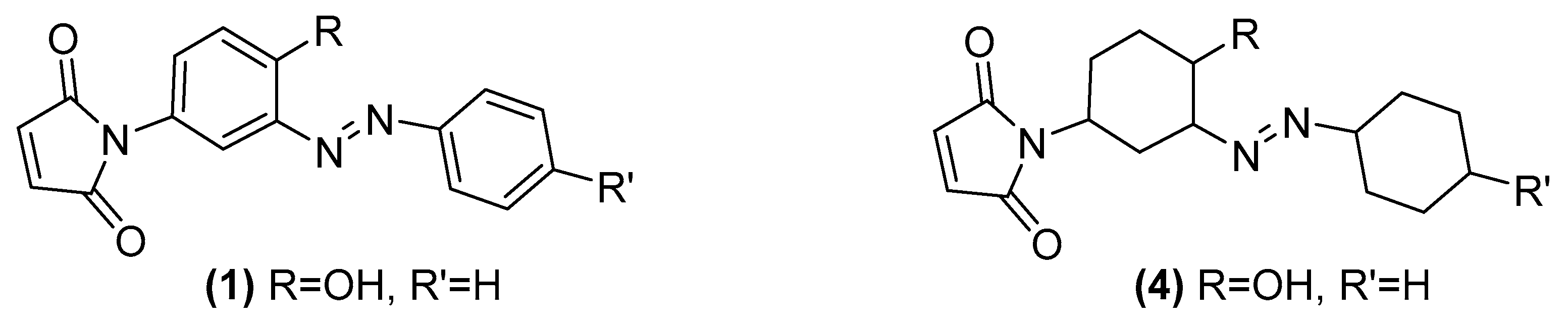{kind=link}