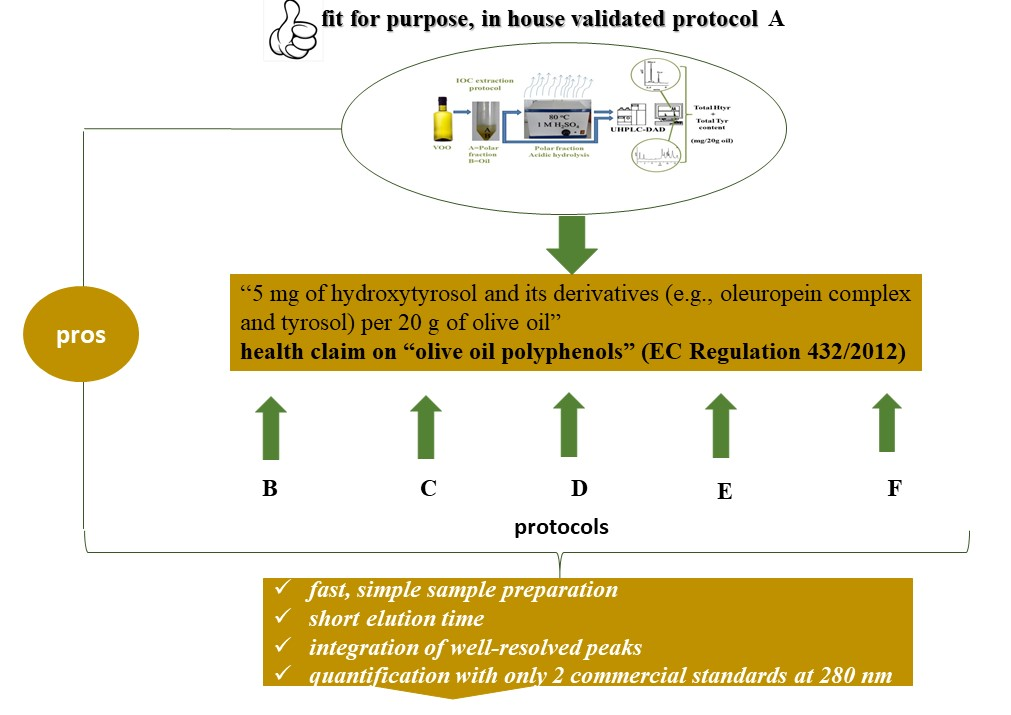
2.2.1. Protocols Involving Acidic Hydrolysis of the Polar Fraction (PF)
Protocols A and B involved determination of Htyr and Tyr in the polar fraction (PF) before and after acidic hydrolysis of all the Htyr and Tyr bound forms and were performed in Laboratory 1 by the same analysts. The target analytes were, thus, determined through integration of well-resolved peaks and with the aid of commercially available external standards at 280 nm. Extraction of the polar fraction under protocol A was as described in the IOC method (methanol:water, 80:20,
v/
v) without the addition of internal standard, whereas under protocol B, extraction was performed using methanol:water, 60:40,
v/
v, vortexing (2 min) and centrifugation (3500 rpm, 10 min). The results, shown in
Table 1 and
Table 2, were calculated in the same way taking into account correction for mass difference between free and bound forms and are expressed as the sum of total Htyr and Tyr rounded to the first integer. In
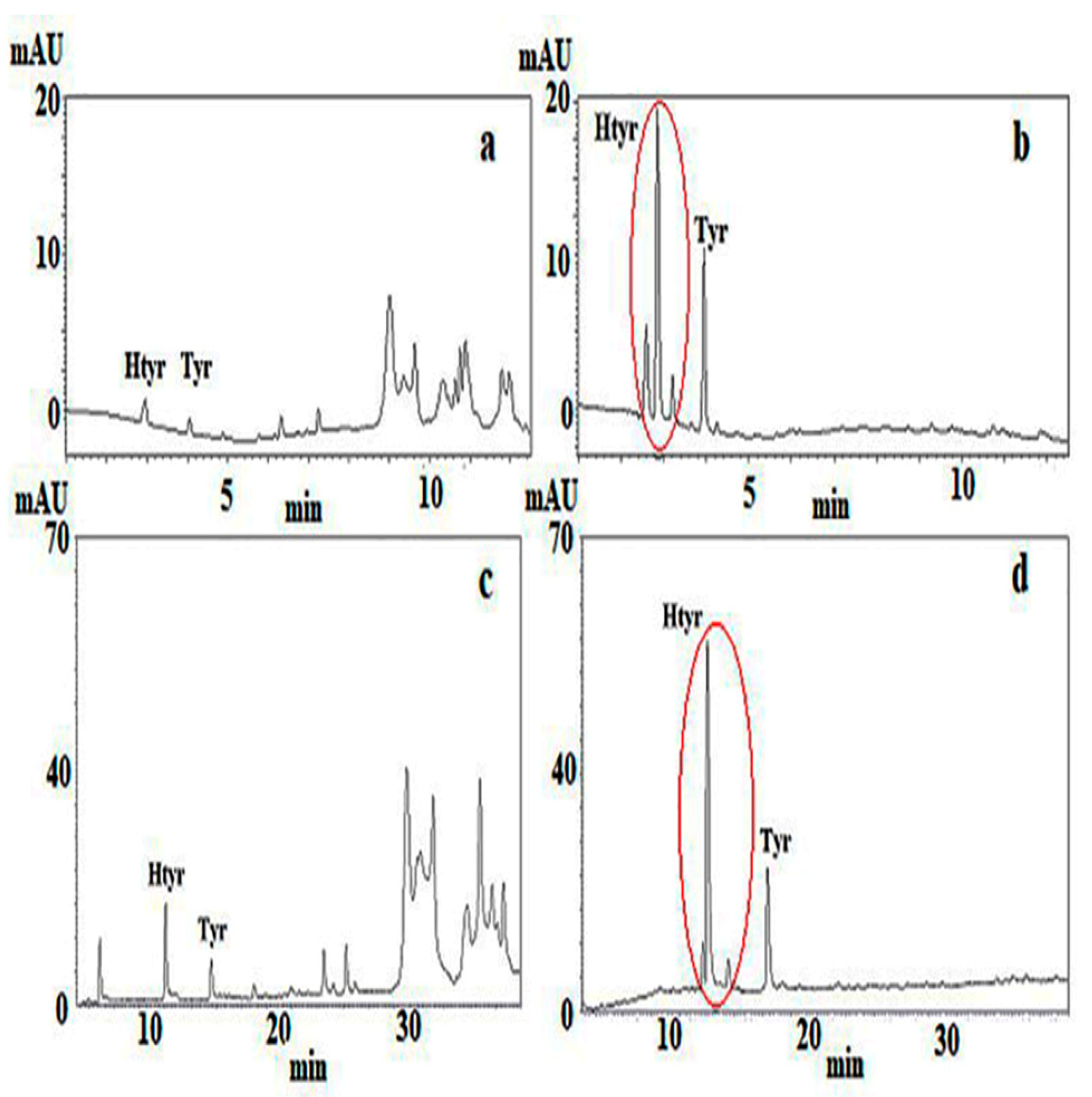
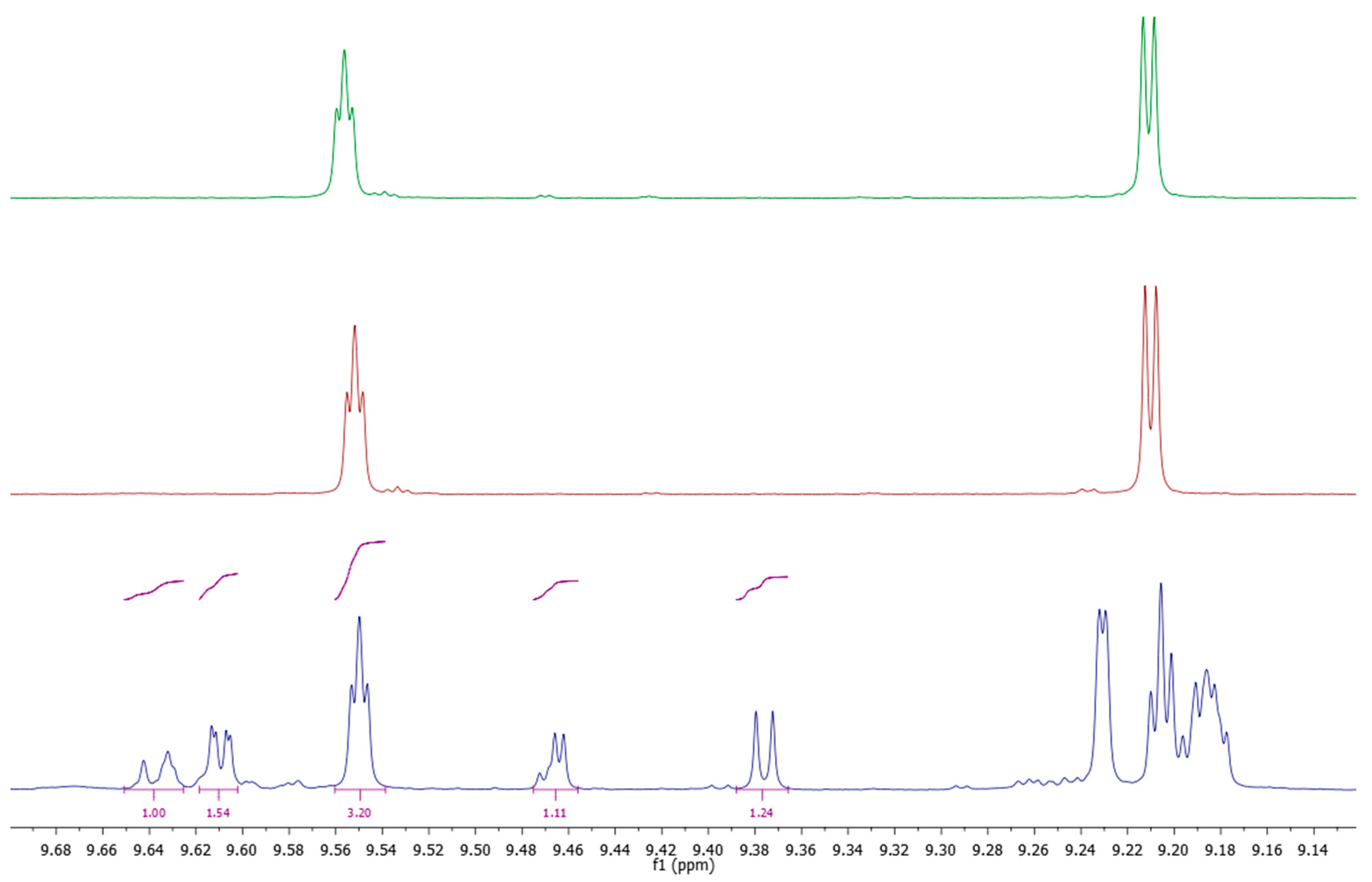
Figure 1a–d chromatograms before and after hydrolysis of the PF are also shown. Using protocol A the time needed for sample preparation involving isolation of the PF according to IOC [
9] and acidic hydrolysis as proposed by Mulinacci et al. [
24] was ~1.2-fold higher than that proposed by Mastralexi et al. [
10] (protocol B) but ~2.2-fold lower than that needed for the direct hydrolysis of phenols in the oil according to Romero and Brenes [
15]. Elution time using the UHPLC protocol was the ~1/3 of that needed using HPLC conditions (IOC elution protocol). Overall, except for the unequivocal assignment of peaks to target analytes, application of protocol A led to an increase in the number of samples analyzed per day and a significantly lower solvent consumption. The total Htyr and Tyr content ranged from 0 up to ~12 mg/20 g of oil. Frequency distribution of the rounded values revealed that 16 out of the 30 samples addressed the limit of ≥ 5 mg/20 g oil. It should be stated that if correction factors as proposed would not be taken into account the corresponding range would be 0–5 mg/20 g oil and only one sample, namely S-26 would satisfy the legal limit. Applying protocol B the same samples except for two (S-10, S-12) were found with a content ≥5 mg/20 g oil. It can be said that in case PF hydrolysis is applied the major pro of protocol A is the speed of analysis compared to HPLC conditions.
2.2.2. Protocols Not Involving Acidic Hydrolysis of the PF
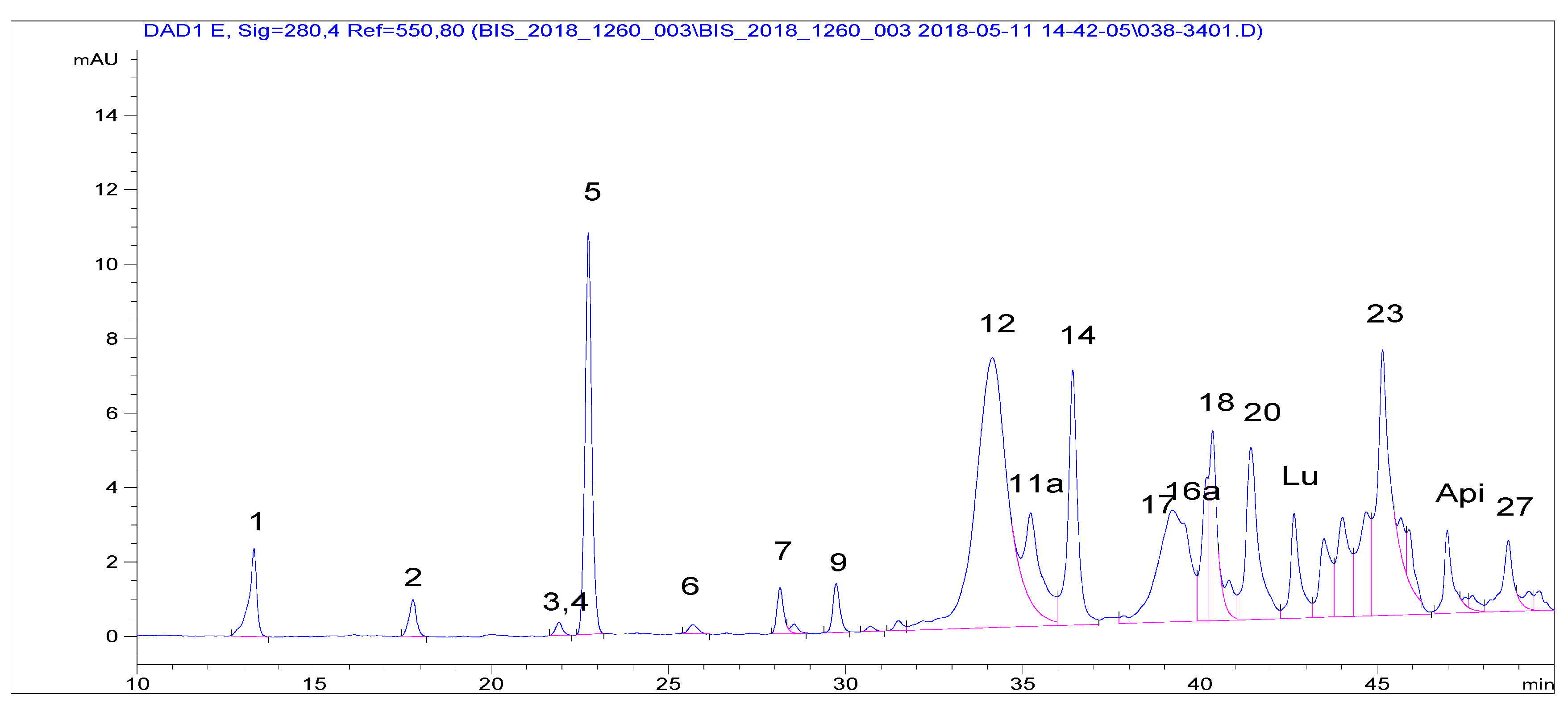
Laboratories 2–4 extracted VOO PFs by the IOC recommended procedure. The accredited Laboratory 2 applied the IOC elution conditions (protocol C), therefore, the corresponding run was three-fold longer than that of protocol A (
Figure 2). Peak resolution was better than that according to the IOC HPLC conditions (
Figure 1c) that can be assigned to a certain extent to the fact that a more advanced packing material than the recommended one by IOC was used. This material had the same pore size (80 Å) and a surface area two-fold higher and 19% carbon load instead of 11.5. The compounds had been identified at the setup of the method by LC–MS and then upon application in terms of relative retention time to that of the internal standard, syringic acid. Only the peaks assigned to Htyr, Tyr and their bound forms, including four oxidized ones, were integrated. Using a second detection means (LC–MS (TQd)) some additional peaks were assigned as Htyr and Tyr bound forms. It is pointed out that quantification assumes that all compounds present the same response toward the internal standard. Results are shown in
Table 3.
Using protocol C, only half of the samples that satisfied the limit using protocol A were found to contain ≥5 mg/20 g oil. These samples (S-1, S-3, S-4, S-15, S-17, S-24, S-26), also satisfied the required limit with protocol B. Inclusion of additional peaks in the calculation affected marginally the number of samples satisfying the minimum required amount (
Table S1). The pros of protocols A and B involving PF hydrolysis over the IOC procedure is obvious and in line with the decision of this body to search for a fit for the purpose method. Except of speed, protocols A and B provide results without the need of MS detection even at the step of method introduction in a laboratory.
Protocol D (Laboratory 3) involved a gradient elution system of 73 min (~2.4-fold longer than that of protocol A) of a binary mixture (acidified water and methanol) instead of the ternary one (acidified H
2O, methanol, acetonitrile) proposed in the IOC protocol. The column packing material used (ODS-1) though of the same pore size and surface area had a carbon load of 6.2% instead of the 11.5% of the ODS-2 recommended one. This issue, plus the fact of artifact formation due to the increased amount of methanol in the elution system may partially explain the poorer resolution pattern of the chromatogram of
Figure 3 in relation to that of
Figure 2 or even of
Figure 1c for the same VOO sample. The latter was obtained using an end-capped Nucleosil ODS with a 1.6-fold higher surface than that of ODS-1 and a carbon load of 15%. According to protocol D calculation of total Htyr and Tyr content was based on the quantification of six compounds using external standards for five of them, which were either commercially available or laboratory isolated and the response factor of oleocanthal for ligstroside aglycone. In this case no need for mass correction is needed, the mg of the six compounds were summed up to give the final content of all Htyr and Tyr forms.
As evidenced, peaks corresponding to bound forms of Htyr and Tyr are not well resolved making difficult their integration and consequently accurate quantification for the purpose of the health claim. Formation of artifacts due to interaction with methanol might add difficulty to quantification of compounds 3 and 4. Data for the total Htyr and Tyr content delivered according to Laboratory 3 expertise are given in
Table 4.
This protocol produced higher values than those using protocol A for 23 samples, so that 21 out of the 30 fulfilled the desirable amount of 5 mg/20g oil. From these samples, five were found of the same content as with protocol A, nine (S-4, S-5, S-10, S-12, S-16, S-19, S-28, S-29, S-30) were found to contain slightly higher content by 1 mg/20 g oil, whereas in 14 (S-1, S-3, S-6–S-9, S-11, S-14, S-15, S-17, S-18, S-24–S-26) the difference was in the range of 2 to 7 mg/20 g oil. Findings need further investigation taking into account that only six compounds were determined. Protocol A seemed to retain the pros of speed and less sophistication in standards used (two external curves instead of five ones and a response factor).
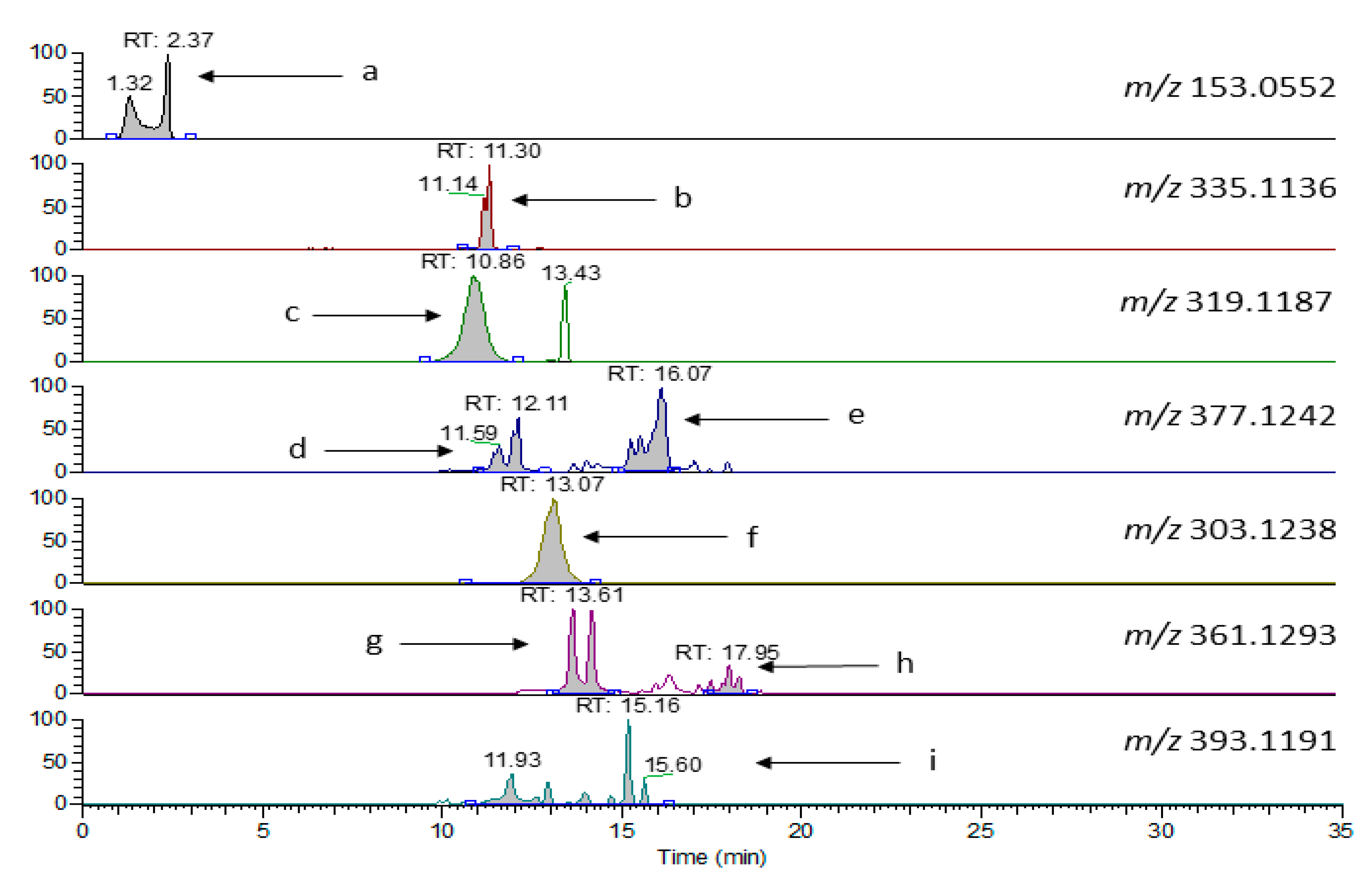
Laboratory 4 (protocol E) exploited the ability of MS to determine specific analytes with no influence from overlapping signals under UV detection mode. A short (10 cm) fused core column, performing in between of UHPLC and conventional HPLC capability, was used. Focus was on major Htyr and Tyr bound forms, their isomers and their oxidation products (
Figure 4). As evidenced the total run lasts 35 min, close to that for PF analysis before hydrolysis using protocol A.
Quantification was then based on the assumption that the response factor for all compounds relative to
o-coumaric acid used as the internal standard was 1, a compromise that introduces some bias in the quantified levels considering the findings of Mateos et al. [
25] on olive oil phenol response factors relatively to used internal standards (ISs) under LC–MS conditions of analysis. Additionally, the concentration determined for the aldehyde derivatives should be lower than expected due to artifact formation with methanol and water, used as solvents for the elution in HPLC [
26], since these artifacts would have a different molecular ion than those depicted in
Figure 4. The quantitative results are provided in
Table 5.
As evidenced, 12 samples were found with a content of total Htyr and Tyr ≥ 5 mg/20 g oil of which the 11 were those found with protocols A and B. Samples S-14 and S-18 were found to contain 5 mg instead for 4 mg using protocol A. The pro of protocol A is cost effectiveness, including detection means requirement, and simplicity in approach.
2.2.3. Data Comparison
Regarding the content of total Htyr and Tyr (free and bound forms) obtained for the same samples using the A-E liquid separation protocols (
Table 1,
Table 2,
Table 3,
Table 4 and
Table 5), it is clear that there are differences in absolute values but there may be a good agreement in terms of statistical comparisons. Pearson correlation analysis is a typical statistical approach to compare different methods. However, in literature there is disagreement whether such correlation provides accurate information for method comparison. Researchers argue that it cannot show whether there is a constant or proportional difference between methods. In addition, it is sensitive to data distribution and outliers and assumes no error in the measurement [
27]. Therefore, alternative approaches were sought as more appropriate. For example, comparison using regression lines is recommended when methods are to be compared over a range of different analyte concentrations [
28]. Passing-Bablok regression analysis is appropriate for such a task and is widely used in the field of clinical chemistry. This treatment allows estimation of method agreement and detection of possible bias between them. It is considered as robust, non-parametric and non-sensitive to the distribution of errors and data outliers. The regression equation via intercept and slope values provides information on constant and proportional measurement error and in combination with calculated confidence limits conclusion on agreement of measurements can be made [
27]. The derived data showed that there was equality of measurements only between protocols A and B because the criteria set for slope and intercept according to the calculated confidence limits were met (
Table S2). Bland Altman analysis is a more recent statistical tool. It examines the agreement between two analytical methods by studying the mean difference in measurements for a set of samples and constructing limits of agreement. The ‘difference plot’ permits to evaluate distance of measurements from zero (bias) and its variability (error). Interpretation of the calculated limits of agreement relies on scientists, who accept them or not [
29]. Examination of ‘difference plots’ (
Figures S1–S5) of protocol A values compared pairwise with the values of the other chromatographic protocols provided the following mean difference (bias) values (mg/20 g oil): −0.67 (B), −2.07 (C), −1.33 (C’), 1.93 (D) and −1.0 (E). Thus, the closer values were those obtained by protocol B. The negative sign indicated that protocol A afforded higher values than the other protocols with the exception of protocol D. The calculated mean difference values indicated that the smallest one was between protocols A and B. Such a value was rather low, justifying, thus, the equality between the two protocols found with Passing-Bablok regression analysis. On the other hand, the mean difference value in the range −1.0 to 1.93 (mg/20 g oil) explained the conclusion drawn from the Passing-Bablok regression analysis that protocols C (C’), D and E differed in performance from A.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}