
Electronic Structure and Lithium Diffusion in LiAl2(OH)6Cl Studied by First Principle Calculations




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Atomic and Electronic Structure of LiAl2(OH)6Cl
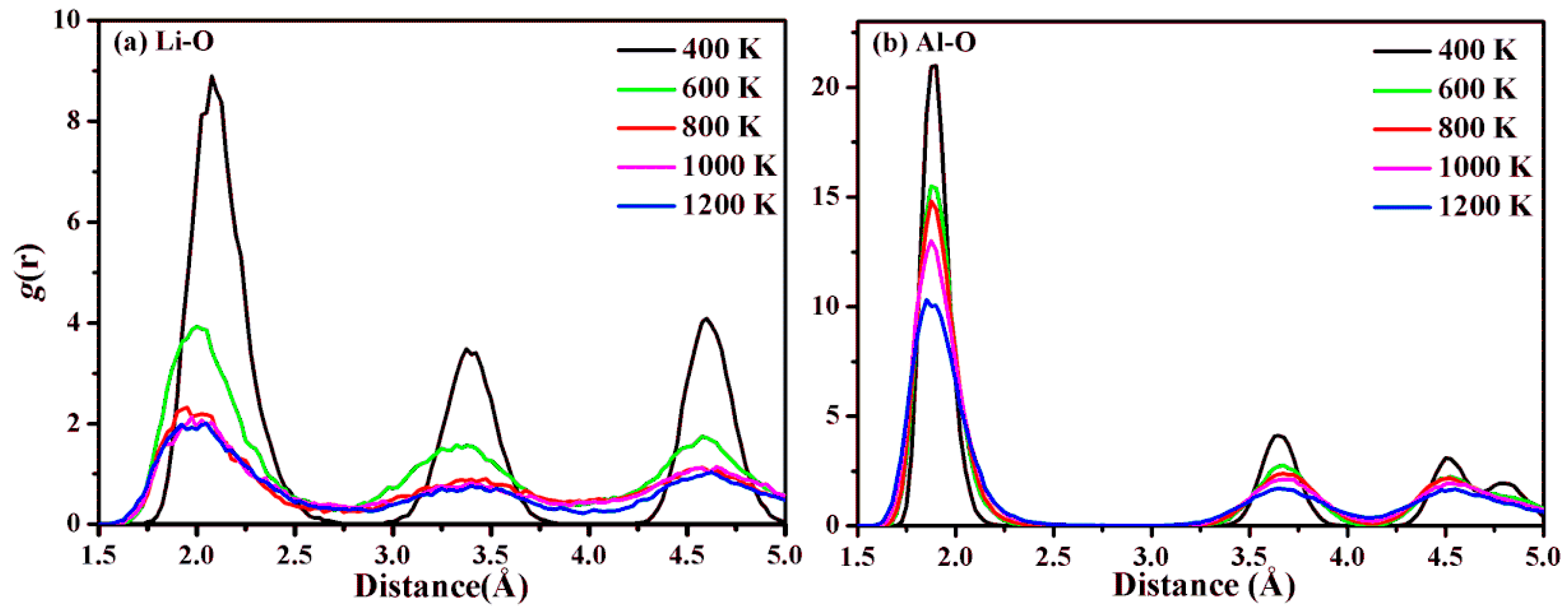
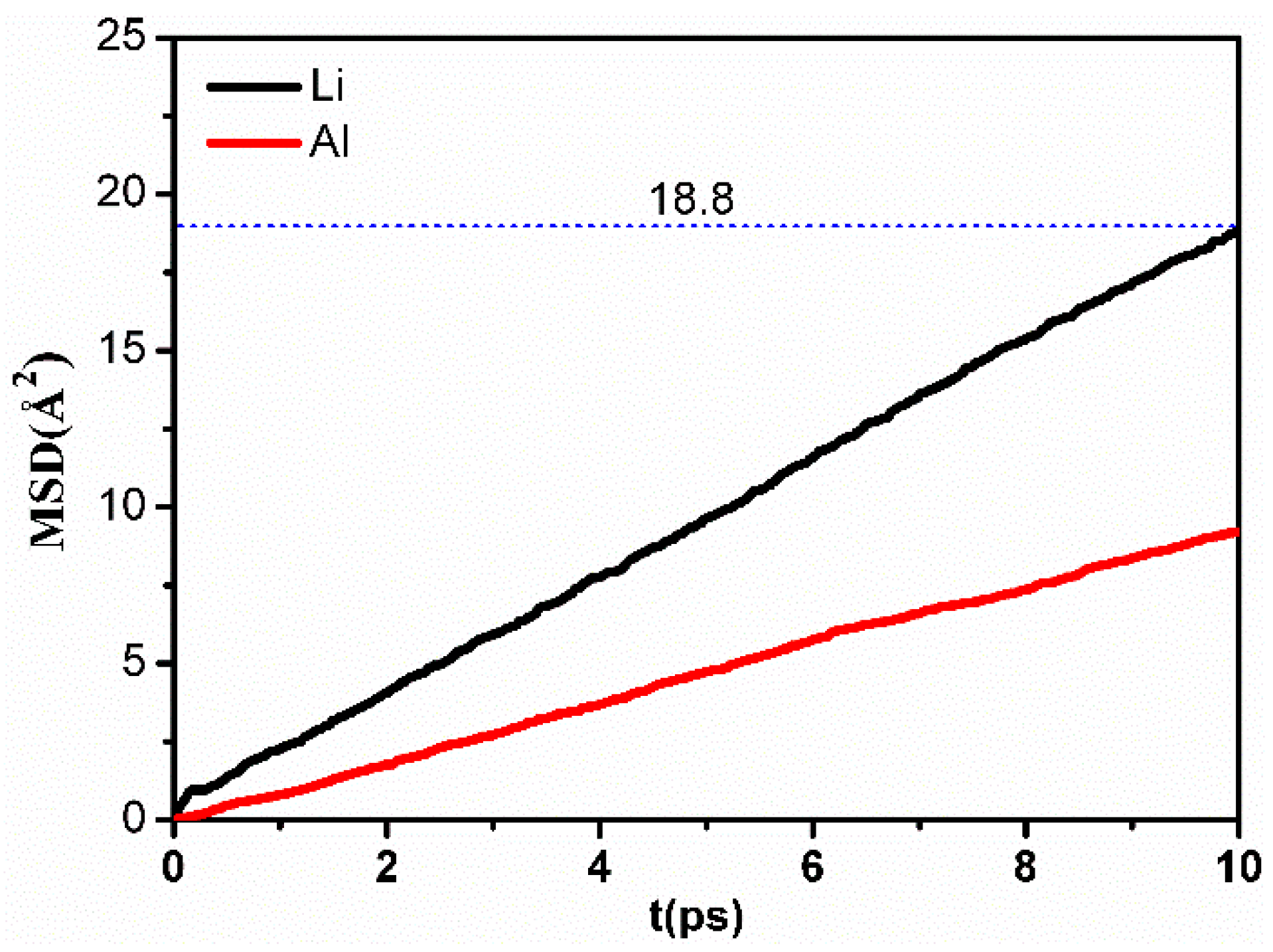
2.2. Lithium Diffusion in LiAl2(OH)6Cl during Heating
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mohapatra, L.; Parida, K. A review on the recent progress, challenges and perspective of layered double hydroxides as promising photocatalysts. J. Mater. Chem. A 2016, 4, 10744–10766. [Google Scholar] [CrossRef]
- Feng, J.T.; He, Y.F.; Liu, Y.N.; Du, Y.Y.; Li, D.Q. Supported catalysts based on layered double hydroxides for catalytic oxidation and hydrogenation: General functionality and promising application prospects. Chem. Soc. Rev. 2015, 44, 5291–5319. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.B.; Zhang, S.T.; Pan, T.; Xu, S.M.; Zhou, A.W.; Pu, M.; Yan, H.; Han, J.B.; Wei, M.; Evans, D.G.; et al. TiO2 @ layered double hydroxide core-shell nanospheres with largely enhanced photocatalytic activity toward O2 generation. Adv. Funct. Mater. 2015, 25, 2243–2249. [Google Scholar] [CrossRef]
- Abello, S.; Bolshak, E.; Montane, D. Ni-Fe catalysts derived from hydrotalcite-like precursors for hydrogen production by ethanol steam reforming. Appl. Catal. a-Gen. 2013, 450, 261–274. [Google Scholar] [CrossRef]
- Kuthati, Y.; Kankala, R.K.; Lee, C.H. Layered double hydroxide nanoparticles for biomedical applications: Current status and recent prospects. Appl. Clay. Sci. 2015, 112, 100–116. [Google Scholar] [CrossRef]
- Choy, J.H.; Kwak, S.Y.; Park, J.S.; Jeong, Y.J.; Portier, J. Intercalative nanohybrids of nucleoside monophosphates and DNA in layered metal hydroxide. J. Am. Chem. Soc. 1999, 121, 1399–1400. [Google Scholar] [CrossRef]
- Wang, Y.L.; Ji, H.Q.; Peng, W.; Liu, L.; Gao, F.; Li, M.G. Gold nanoparticle-coated Ni/Al layered double hydroxides on glassy carbon electrode for enhanced methanol electro-oxidation. Int. J. Hydrogen Energ. 2012, 37, 9324–9329. [Google Scholar] [CrossRef]
- Chen, X.B.; Li, C.; Gratzel, M.; Kostecki, R.; Mao, S.S. Nanomaterials for renewable energy production and storage. Chem. Soc. Rev. 2012, 41, 7909–7937. [Google Scholar] [CrossRef]
- Cavani, F.; Trifiro, F.; Vaccari, A. Hydrotalcite-type anionic clays: Preparation, properties and applications. Catal. Today 1991, 11, 173–301. [Google Scholar] [CrossRef]
- Newman, S.P.; Greenwell, H.C.; Coveney, P.V.; Jones, W. Layered Double Hydroxides: Present Andfuture; Nova Science Publishers, Inc.: New York, NY, USA, 2001. [Google Scholar]
- Duan, X.; Evans, D.G. Layered Double Hydroxides; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Dutta, P.K.; Puri, M. Anion-exchange in lithium aluminate hydroxides. J. Phys. Chem. 1989, 93, 376–381. [Google Scholar] [CrossRef]
- Intissar, M.; Jumas, J.C.; Besse, J.P.; Leroux, F. Reinvestigation of the layered double hydroxide containing tetravalent cations: Unambiguous response provided by XAS and Mossbauer spectroscopies. Chem. Mater. 2003, 15, 4625–4632. [Google Scholar] [CrossRef]
- Velu, S.; Suzuki, K.; Okazaki, M.; Osaki, T.; Tomura, S.; Ohashi, F. Synthesis of new Sn-incorporated layered double hydroxides and their thermal evolution to mixed oxides. Chem. Mater. 1999, 11, 2163–2172. [Google Scholar] [CrossRef]
- Besserguenev, A.V.; Fogg, A.M.; Francis, R.J.; Price, S.J.; OHare, D.; Isupov, V.P.; Tolochko, B.P. Synthesis and structure of the gibbsite intercalation compounds [LiAl2(OH)6]X {X=Cl, Br, NO3} and [LiAl2(OH)6]Cl H2O using synchrotron X-ray and neutron powder diffraction. Chem. Mater. 1997, 9, 241–247. [Google Scholar] [CrossRef]
- Fogg, A.M.; O’Hare, D. Study of the intercalation of lithium salt in gibbsite using time-resolved in situ X-ray diffraction. Chem. Mater. 1999, 11, 1771–1775. [Google Scholar] [CrossRef]
- Lei, L.X.; Vijayan, R.P.; O’Hare, D. Preferential anion exchange intercalation of pyridinecarboxylate and toluate isomers in the layered double hydroxide [LiAl2(OH)6]Cl H2O. J. Mater. Chem. 2001, 11, 3276–3280. [Google Scholar] [CrossRef]
- Lei, L.X.; Millange, F.; Walton, R.I.; O’Hare, D. Efficient separation of pyridinedicarboxylates by preferential anion exchange intercalation in [LiAl2(OH)6]Cl H2O. J. Mater. Chem. 2000, 10, 1881–1886. [Google Scholar] [CrossRef]
- Hou, X.Q.; Bish, D.L.; Wang, S.L.; Johnston, C.T.; Kirkpatrick, R.J. Hydration, expansion, structure, and dynamics of layered double hydroxides. Am. Mineral 2003, 88, 167–179. [Google Scholar] [CrossRef]
- Fogg, A.M.; Rohl, A.L.; Parkinson, G.M.; O’Hare, D. Predicting guest orientations in layered double hydroxide intercalates. Chem. Mater. 1999, 11, 1194–1200. [Google Scholar] [CrossRef]
- Hou, X.J.; Li, H.Q.; He, P.; Sun, Z.H.; Li, S.P. Structural and electronic analysis of Li/Al layered double hydroxides and their adsorption for CO2. Appl. Surf. Sci. 2017, 416, 411–423. [Google Scholar] [CrossRef]
- Hou, X.Q.; Kalinichev, A.G.; Kirkpatrick, R.J. Interlayer structure and dynamics of Cl--LiAl2-layered double hydroxide: Cl-35 NMR observations and molecular dynamics modeling. Chem. Mater. 2002, 14, 2078–2085. [Google Scholar] [CrossRef]
- Hou, X.Q.; Kirkpatrick, R.J. Thermal evolution of the Cl--LiAl2 layered double hydroxide: A multinuclear MAS NMR and XRD perspective. Inorg. Chem. 2001, 40, 6397–6404. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cheng, K.; Jia, G.Z. The molecular dynamics simulation of hydrogen bonding in supercritical water. Physica A 2019, 516, 365–375. [Google Scholar] [CrossRef]
- Han, X.J.; Li, J.G.; Schober, H.R. High temperature breakdown of the Stokes-Einstein relation in a computer simulated Cu-Zr melt. J. Chem. Phys. 2016, 144. [Google Scholar] [CrossRef] [PubMed]
- Sansone, R.; Ebner, C.; Probst, M. Quantum chemical and molecular dynamics study on the hydration of cyanide and thiocyanate anions. J. Mol. Liq. 2000, 88, 129–150. [Google Scholar] [CrossRef]
- Yu, C.Y.; Hui, X.D.; Chen, X.H.; Liu, X.J.; Lin, D.Y.; Liu, Z.K.; Chen, G.L. Ab initio molecular dynamics simulation of the atom packing and density of Al-Ni amorphous alloys. Sci. China Technol. Sc. 2010, 53, 3175–3182. [Google Scholar] [CrossRef]
- Shokri, S.; Naderi, O.; Moradi, K.; Sadeghi, R.; Ebrahimi, S. A combined molecular dynamic simulation and experimental study of thermo-physical properties of the new synthesized amino acid-based ionic liquids. J. Mol. Liq. 2019, 277, 290–301. [Google Scholar] [CrossRef]
- Zhu, Z.Y.; Chu, I.H.; Ong, S.P. Li3Y(PS4)2 and Li5PS4Cl2: New lithium superionic conductors predicted from silver thiophosphates using efficiently tiered ab initio molecular dynamics simulations. Chem. Mater. 2017, 29, 2474–2484. [Google Scholar] [CrossRef]
- Chu, I.H.; Nguyen, H.; Hy, S.; Lin, Y.C.; Wang, Z.B.; Xu, Z.H.; Deng, Z.; Meng, Y.S.; Ong, S.P. Insights into the performance limits of the Li7P3S11 superionic conductor: A combined first-principles and experimental study. Acs Appl. Mater. Inter. 2016, 8, 7843–7853. [Google Scholar] [CrossRef]
- Qi, X.F.; Li, H.Y.; Zhao, Y.; Yan, N. Comparison of the structural and physical properties of nitrocellulose plasticized by N-butyl-N-(2-nitroxy-ethyl) nitramine and nitroglycerin: Computational simulation and experimental studies. J. Hazard. Mater. 2019, 362, 303–310. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for brillouin-zone integrations. Phys. Rev. B. 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Dronskowski, R.; Blochl, P.E. Crystal Orbital Hamilton Populations (COHP)-energy-resolved visualization of chemical bonding in solids based on density-functional calculations. J. Phys. Chem. 1993, 97, 8617–8624. [Google Scholar] [CrossRef]
- Deringer, V.L.; Tchougreeff, A.L.; Dronskowski, R. Crystal Orbital Hamilton Population (COHP) analysis as projected from plane-wave basis sets. J. Phys. Chem. A 2011, 115, 5461–5466. [Google Scholar] [CrossRef] [PubMed]
- Maintz, S.; Deringer, V.L.; Tchougreeff, A.L.; Dronskowski, R. Analytic projection from plane-wave and PAW wavefunctions and application to chemical-bonding analysis in solids. J. Comput. Chem. 2013, 34, 2557–2567. [Google Scholar] [CrossRef] [PubMed]
- Maintz, S.; Deringer, V.L.; Tchougreeff, A.L.; Dronskowski, R. LOBSTER: A tool to extract chemical bonding from plane-wave based DFT. J. Comput. Chem. 2016, 37, 1030–1035. [Google Scholar] [CrossRef]
Sample Availability: Not available. |






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Cheng, X.; Wu, C.; Köhler, J.; Deng, S. Electronic Structure and Lithium Diffusion in LiAl2(OH)6Cl Studied by First Principle Calculations. Molecules 2019, 24, 2667. https://doi.org/10.3390/molecules24142667
Zhang Y, Cheng X, Wu C, Köhler J, Deng S. Electronic Structure and Lithium Diffusion in LiAl2(OH)6Cl Studied by First Principle Calculations. Molecules. 2019; 24(14):2667. https://doi.org/10.3390/molecules24142667
Chicago/Turabian StyleZhang, Yueping, Xiyue Cheng, Chen Wu, Jürgen Köhler, and Shuiquan Deng. 2019. "Electronic Structure and Lithium Diffusion in LiAl2(OH)6Cl Studied by First Principle Calculations" Molecules 24, no. 14: 2667. https://doi.org/10.3390/molecules24142667
APA StyleZhang, Y., Cheng, X., Wu, C., Köhler, J., & Deng, S. (2019). Electronic Structure and Lithium Diffusion in LiAl2(OH)6Cl Studied by First Principle Calculations. Molecules, 24(14), 2667. https://doi.org/10.3390/molecules24142667