CDFT-Based Reactivity Descriptors as a Useful MEDT Chemoinformatics Tool for the Study of the Virotoxin Family of Fungal Peptides




Abstract
:
1. Introduction
2. Computational Methodology
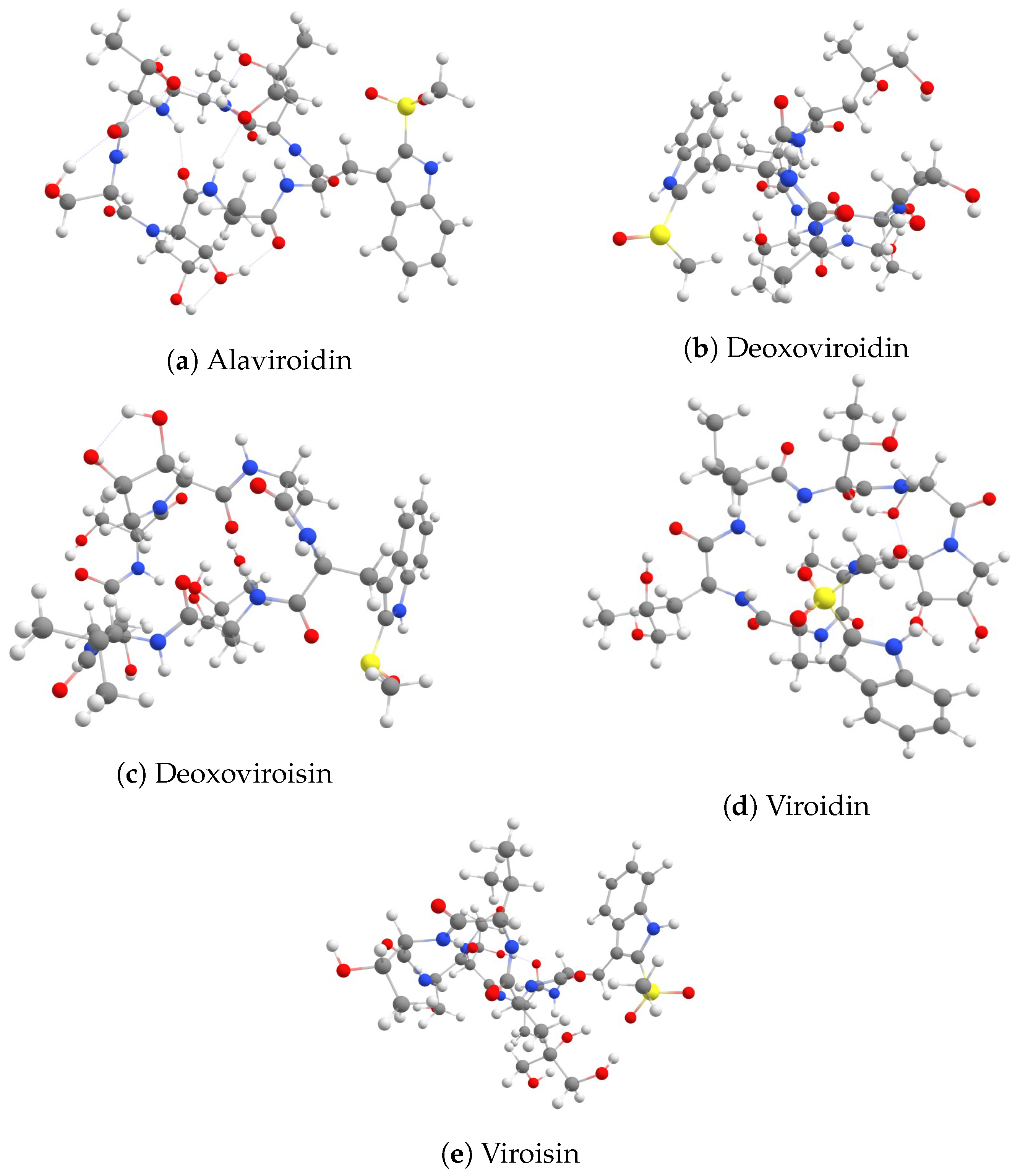
3. Results and Discussion
3.1. Calculation of Global Reactivity Descriptors
3.2. Calculation of the pKas of the Five Fungal Peptides of the Virotoxin Family
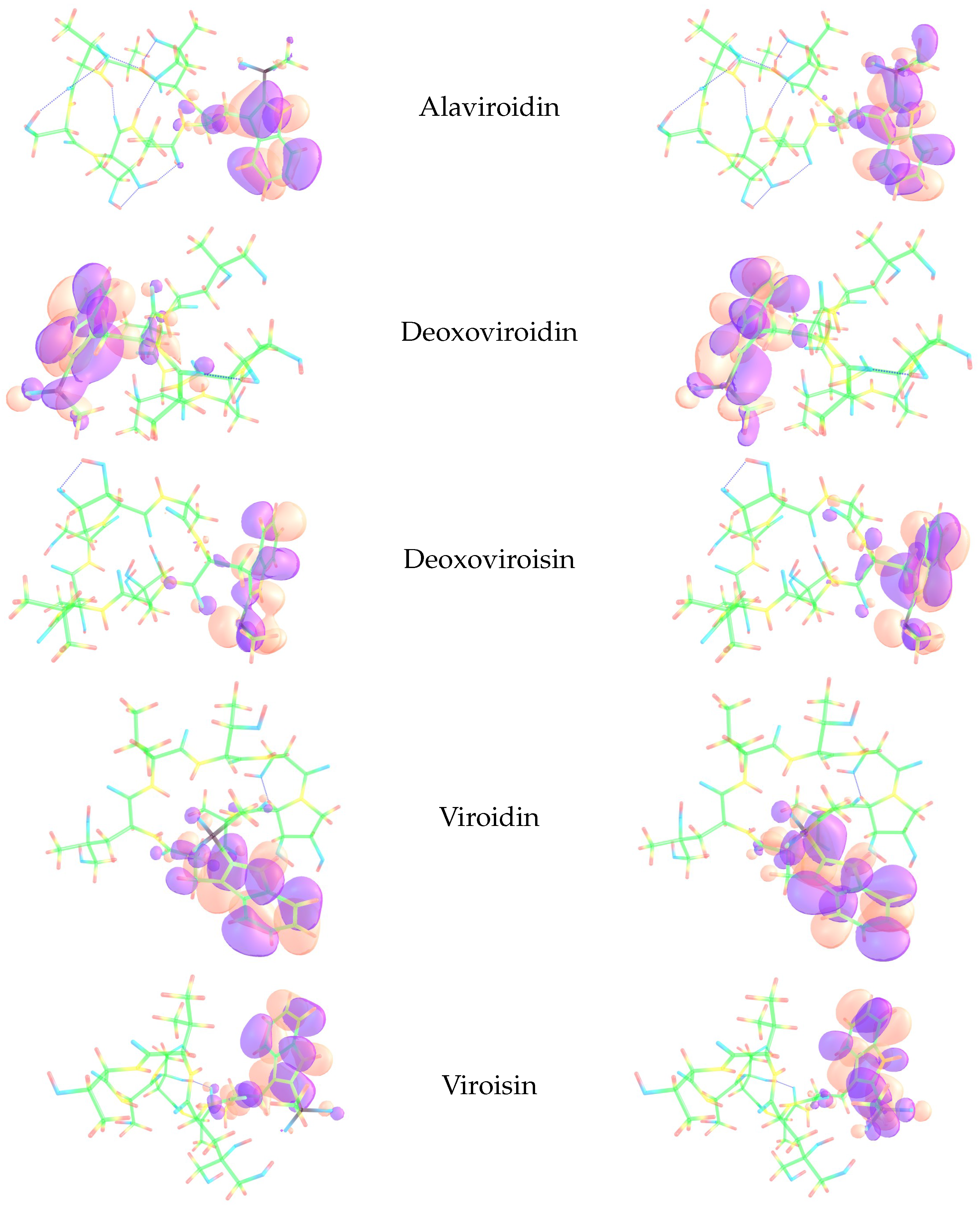
3.3. Local Reactivity Descriptors Calculation
| Nucleophilic Parr Function | ||
| Electrophilic Parr Function |
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhou, P. (Ed.) Computational Peptidology; Humana Press: New York, NY, USA, 2015. [Google Scholar]
- Guha, R.; Bender, A. (Eds.) Computational Approaches in Cheminformatics and Bioinformatics; Wiley: Hoboken, NJ, USA, 2012. [Google Scholar]
- Wieland, T.; Bodanszky, M. The World of Peptides: A Brief History of Peptide Chemistry; Springer: Berlin/Heidelberg, Germany, 1991. [Google Scholar]
- Gilbert, J.; Senyuva, H.Z. (Eds.) Bioactive Compounds in Foods; Blackwell Pub: Oxford, UK, 2008. [Google Scholar]
- Vetter, J. Toxins of Amanita phalloides. Toxicon 1998, 36, 13–24. [Google Scholar] [CrossRef]
- Karlson-Stiber, C.; Persson, H. Cytotoxic Fungi—An Overview. Toxicon 2003, 42, 339–349. [Google Scholar] [CrossRef]
- Wei, J.; Wu, J.; Chen, J.; Wu, B.; He, Z.; Zhang, P.; Li, H.; Sun, C.; Liu, C.; Chen, Z.; et al. Determination of Cyclopeptide Toxins in Amanita subpallidorosea and Amanita virosa by High-Performance Liquid Chromatography Coupled with High-Resolution Mass Spectrometry. Toxicon 2017, 133, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Walton, J. The Cyclic Peptide Toxins of Amanita and Other Poisonous Mushrooms; Springer: Cham, Switzerland, 2018. [Google Scholar]
- Parr, R.; Yang, W. Density—Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Chemical Reactivity Theory—A Density Functional View; Chattaraj, P. (Ed.) CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2009. [Google Scholar]
- Lewars, E. Computational Chemistry—Introduction to the Theory and Applications of Molecular and Quantum Mechanics; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2003. [Google Scholar]
- Young, D. Computational Chemistry—A Practical Guide for Applying Techniques to Real-World Problems; John Wiley & Sons: New York, NY, USA, 2001. [Google Scholar]
- Jensen, F. Introduction to Computational Chemistry, 2nd ed.; John Wiley & Sons: Chichester, UK, 2007. [Google Scholar]
- Cramer, C. Essentials of Computational Chemistry—Theories and Models, 2nd ed.; John Wiley & Sons: Chichester, UK, 2004. [Google Scholar]
- Domingo, L.R. Molecular Electron Density Theory: A Modern View of Reactivity in Organic Chemistry. Molecules 2016, 21, 1319. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [PubMed]
- Frau, J.; Flores-Holguín, N.; Glossman-Mitnik, D. Chemical Reactivity Properties, pKa Values, AGEs Inhibitor Abilities and Bioactivity Scores of the Mirabamides A–H Peptides of Marine Origin Studied by Means of Conceptual DFT. Mar. Drugs 2018, 16, 302. [Google Scholar] [CrossRef] [PubMed]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. An Alternative Approach to Computational Peptidology Based on Conceptual DFT and Empirical Bioactivity Scores. Med. Chem. 2019, 9, 27–30. [Google Scholar]
- Frau, J.; Flores-Holguín, N.; Glossman-Mitnik, D. Conceptual Density Functional Theory Study of the Chemical Reactivity Properties and Bioactivity Scores of the Leu-Enkephalin Opioid Peptide Neutrotransmitter. Comput. Mol. Biosci. 2019, 9, 16–23. [Google Scholar] [CrossRef]
- Frau, J.; Flores-Holguín, N.; Glossman-Mitnik, D. Chemical Reactivity Theory and Empirical Bioactivity Scores as Computational Peptidology Alternative Tools for the Study of Two Anticancer Peptides of Marine Origin. Molecules 2019, 24, 1115. [Google Scholar] [CrossRef] [PubMed]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. A Comparison of Conceptual DFT and Molecular Electron Density Theory (MEDT) Descriptors of Local Chemical Reactivity Properties: Oxytocin and Vasopressin Peptide Hormones as Test Cases. MOJ Bioorganic Org. Chem. 2019, 2, 45–49. [Google Scholar]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Computational Prediction of Bioactivity Scores and Chemical Reactivity Properties of the Parasin I Therapeutic Peptide of Marine Origin through the Calculation of Global and Local Conceptual DFT Descriptors. Theor. Chem. Acc. 2019, 138, 78. [Google Scholar] [CrossRef]
- Frau, J.; Glossman-Mitnik, D. Molecular Reactivity and Absorption Properties of Melanoidin Blue-G1 through Conceptual DFT. Molecules 2018, 23, 559. [Google Scholar] [CrossRef] [PubMed]
- Frau, J.; Glossman-Mitnik, D. Conceptual DFT Study of the Local Chemical Reactivity of the Dilysyldipyrrolones A and B Intermediate Melanoidins. Theor. Chem. Acc. 2018, 137, 1210. [Google Scholar] [CrossRef]
- Frau, J.; Glossman-Mitnik, D. Conceptual DFT Study of the Local Chemical Reactivity of the Colored BISARG Melanoidin and Its Protonated Derivative. Front. Chem. 2018, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Frau, J.; Glossman-Mitnik, D. Molecular Reactivity of some Maillard Reaction Products Studied through Conceptual DFT. Contemp. Chem. 2018, 1, 1–14. [Google Scholar]
- Frau, J.; Glossman-Mitnik, D. Computational Study of the Chemical Reactivity of the Blue-M1 Intermediate Melanoidin. Comput. Theor. Chem. 2018, 1134, 22–29. [Google Scholar] [CrossRef]
- Frau, J.; Glossman-Mitnik, D. Chemical Reactivity Theory Applied to the Calculation of the Local Reactivity Descriptors of a Colored Maillard Reaction Product. Chem. Sci. Int. J. 2018, 22, 1–14. [Google Scholar] [CrossRef]
- Frau, J.; Glossman-Mitnik, D. Blue M2: An Intermediate Melanoidin Studied via Conceptual DFT. J. Mol. Model. 2018, 24, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision E.01, 2016; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Peverati, R.; Truhlar, D.G. Screened-Exchange Density Functionals with Broad Accuracy for Chemistry and Solid-State Physics. Phys. Chem. Chem. Phys. 2012, 14, 16187–16191. [Google Scholar] [CrossRef]
- Borghi, G.; Ferretti, A.; Nguyen, N.L.; Dabo, I.; Marzari, N. Koopmans-compliant Functionals and Their Performance Against Reference Molecular Data. Phys. Rev. B 2014, 90, 1. [Google Scholar] [CrossRef]
- Dabo, I.; Ferretti, A.; Poilvert, N.; Li, Y.; Marzari, N.; Cococcioni, M. Koopmans’ Condition for Density-Functional Theory. Phys. Rev. B 2010, 82, 115121. [Google Scholar] [CrossRef]
- Kar, R.; Song, J.W.; Hirao, K. Long-Range Corrected Functionals Satisfy Koopmans’ Theorem: Calculation of Correlation and Relaxation Energies. J. Comput. Chem. 2013, 34, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Salzner, U.; Baer, R. Koopmans’ Springs to Life. J. Chem. Phys. 2009, 131, 231101. [Google Scholar] [CrossRef] [PubMed]
- Vanfleteren, D.; Van Neck, D.; Ayers, P.W.; Morrison, R.C.; Bultinck, P. Exact Ionization Potentials from Wavefunction Asymptotics: The Extended Koopmans’ Theorem, Revisited. J. Chem. Phys. 2009, 130, 194104. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting Basis Sets for H to R. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.; Cramer, C.; Truhlar, D. Universal Solvation Model Based on Solute Electron Density and a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Merck Molecular Force Field. I. Basis, Form, Scope, Parameterization, and Performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck Molecular Force Field. II. MMFF94 van der Waals and Electrostatic Parameters for Intermolecular Interactions. J. Comput. Chem. 1996, 17, 520–552. [Google Scholar] [CrossRef]
- Halgren, T.A. MMFF VI. MMFF94s Option for Energy Minimization Studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Halgren, T.A.; Nachbar, R.B. Merck Molecular Force Field. IV. Conformational Energies and Geometries for MMFF94. J. Comput. Chem. 1996, 17, 587–615. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck Molecular Force field. V. Extension of MMFF94 Using Experimental Data, Additional Computational Data, and Empirical Rules. J. Comput. Chem. 1996, 17, 616–641. [Google Scholar] [CrossRef]
- Becke, A.D. Vertical Excitation Energies From the Adiabatic Connection. J. Chem. Phys. 2016, 145, 194107. [Google Scholar] [CrossRef] [PubMed]
- Baerends, E.J.; Gritsenko, O.V.; van Meer, R. The Kohn-Sham Gap, the Fundamental Gap and the Optical Gap: The Physical Meaning of Occupied and Virtual Kohn-Sham Orbital Energies. Phys. Chem. Chem. Phys. 2013, 15, 16408–16425. [Google Scholar] [CrossRef] [PubMed]
- van Meer, R.; Gritsenko, O.V.; Baerends, E.J. Physical Meaning of Virtual Kohn-Sham Orbitals and Orbital Energies: An Ideal Basis for the Description of Molecular Excitations. J. Chem. Theory Comput. 2014, 10, 4432–4441. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1873. [Google Scholar] [CrossRef]
- Parr, R.; Szentpaly, L.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Gázquez, J.; Cedillo, A.; Vela, A. Electrodonating and Electroaccepting Powers. J. Phys. Chem. A 2007, 111, 1966–1970. [Google Scholar] [CrossRef]
- Chattaraj, P.; Chakraborty, A.; Giri, S. Net Electrophilicity. J. Phys. Chem. A 2009, 113, 10068–10074. [Google Scholar] [CrossRef]
- Frau, J.; Hernández-Haro, N.; Glossman-Mitnik, D. Computational Prediction of the pKas of Small Peptides through Conceptual DFT Descriptors. Chem. Phys. Lett. 2017, 671, 138–141. [Google Scholar] [CrossRef]
- Chermette, H. Chemical Reactivity Indexes in Density Functional Theory. J. Comput. Chem. 1999, 20, 129–154. [Google Scholar] [CrossRef]
- Morell, C.; Grand, A.; Toro-Labbé, A. New Dual Descriptor for Chemical Reactivity. J. Phys. Chem. A 2005, 109, 205–212. [Google Scholar] [CrossRef]
- Morell, C.; Grand, A.; Toro-Labbé, A. Theoretical Support for Using the Δf(r) Descriptor. Chem. Phys. Lett. 2006, 425, 342–346. [Google Scholar] [CrossRef]
- Martínez-Araya, J.I. Revisiting Caffeate’s Capabilities as a Complexation Agent to Silver Cation in Mining Processes by means of the Dual Descriptor—A Conceptual DFT Approach. J. Mol. Model. 2012, 18, 4299–4307. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Araya, J.I. Explaining Reaction Mechanisms Using the Dual Descriptor: A Complementary Tool to the Molecular Electrostatic Potential. J. Mol. Model. 2012, 19, 2715–2722. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Araya, J.I. Why is the Dual Descriptor a More Accurate Local Reactivity Descriptor than Fukui Functions? J. Math. Chem. 2015, 53, 451–465. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P.; Sáez, J. Understanding the Local Reactivity in Polar Organic Reactions through Electrophilic and Nucleophilic Parr Functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]
- Chamorro, E.; Pérez, P.; Domingo, L.R. On the Nature of Parr Functions to Predict the Most Reactive Sites along Organic Polar Reactions. Chem. Phys. Lett. 2013, 582, 141–143. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| HOMO | LUMO | HOMO-LUMO Gap | ||
| Alaviroidin | −6.072 | −1.700 | 4.372 | 284 |
| Deoxoviroidin | −5.929 | −1.467 | 4.462 | 278 |
| Deoxovirosin | −5.681 | −1.228 | 4.453 | 278 |
| Viroidin | −6.056 | −1.622 | 4.434 | 280 |
| Viroisin | −5.888 | −1.593 | 4.295 | 289 |
| Molecule | Electronegativity | Global Hardness | Electrophilicity |
| Alaviroidin | 3.886 | 4.372 | 1.727 |
| Deoxoviroidin | 3.698 | 4.462 | 1.533 |
| Deoxoviroisin | 3.454 | 4.453 | 1.340 |
| Viroidin | 3.839 | 4.434 | 1.662 |
| Viroisin | 3.740 | 4.295 | 1.629 |
| Molecule | Electrodonating Power | Electroaccepting Power | Net Electrophilicity |
| Alaviroidin | 5.670 | 1.784 | 7.454 |
| Deoxoviroidin | 5.193 | 1.495 | 6.688 |
| Deoxoviroisin | 4.685 | 1.231 | 5.916 |
| Viroidin | 5.520 | 1.682 | 7.202 |
| Viroisin | 5.396 | 1.655 | 7.051 |
| Molecule | pKa |
| FAR | 12.69 |
| FAY | 12.62 |
| FVY | 12.63 |
| FWC | 12.64 |
| FWY | 12.76 |
| Alaviroidin | |||
| Atom | |||
| 50 C | 8.71 | 0.227 | 0.007 |
| 24 N | −13.59 | 0.048 | 0.275 |
| Deoxoviroidin | |||
| Atom | |||
| 1 S | 6.43 | 0.071 | 0.010 |
| 48 C | −11.44 | 0.103 | 0.358 |
| Deoxoviroisin | |||
| Atom | |||
| 59 C | 12.91 | 0.259 | 0.067 |
| 49 C | −15.01 | 0.058 | 0.386 |
| Viroidin | |||
| Atom | |||
| 50 C | 7.07 | 0.268 | 0.045 |
| 24 N | −10.04 | 0.050 | 0.225 |
| Viroisin | |||
| Atom | |||
| 1 S | 7.83 | 0.086 | 0.005 |
| 25 N | −9.77 | 0.044 | 0.206 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. CDFT-Based Reactivity Descriptors as a Useful MEDT Chemoinformatics Tool for the Study of the Virotoxin Family of Fungal Peptides. Molecules 2019, 24, 2707. https://doi.org/10.3390/molecules24152707
Flores-Holguín N, Frau J, Glossman-Mitnik D. CDFT-Based Reactivity Descriptors as a Useful MEDT Chemoinformatics Tool for the Study of the Virotoxin Family of Fungal Peptides. Molecules. 2019; 24(15):2707. https://doi.org/10.3390/molecules24152707
Chicago/Turabian StyleFlores-Holguín, Norma, Juan Frau, and Daniel Glossman-Mitnik. 2019. "CDFT-Based Reactivity Descriptors as a Useful MEDT Chemoinformatics Tool for the Study of the Virotoxin Family of Fungal Peptides" Molecules 24, no. 15: 2707. https://doi.org/10.3390/molecules24152707
APA StyleFlores-Holguín, N., Frau, J., & Glossman-Mitnik, D. (2019). CDFT-Based Reactivity Descriptors as a Useful MEDT Chemoinformatics Tool for the Study of the Virotoxin Family of Fungal Peptides. Molecules, 24(15), 2707. https://doi.org/10.3390/molecules24152707