CA-170 – A Potent Small-Molecule PD-L1 Inhibitor or Not?

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
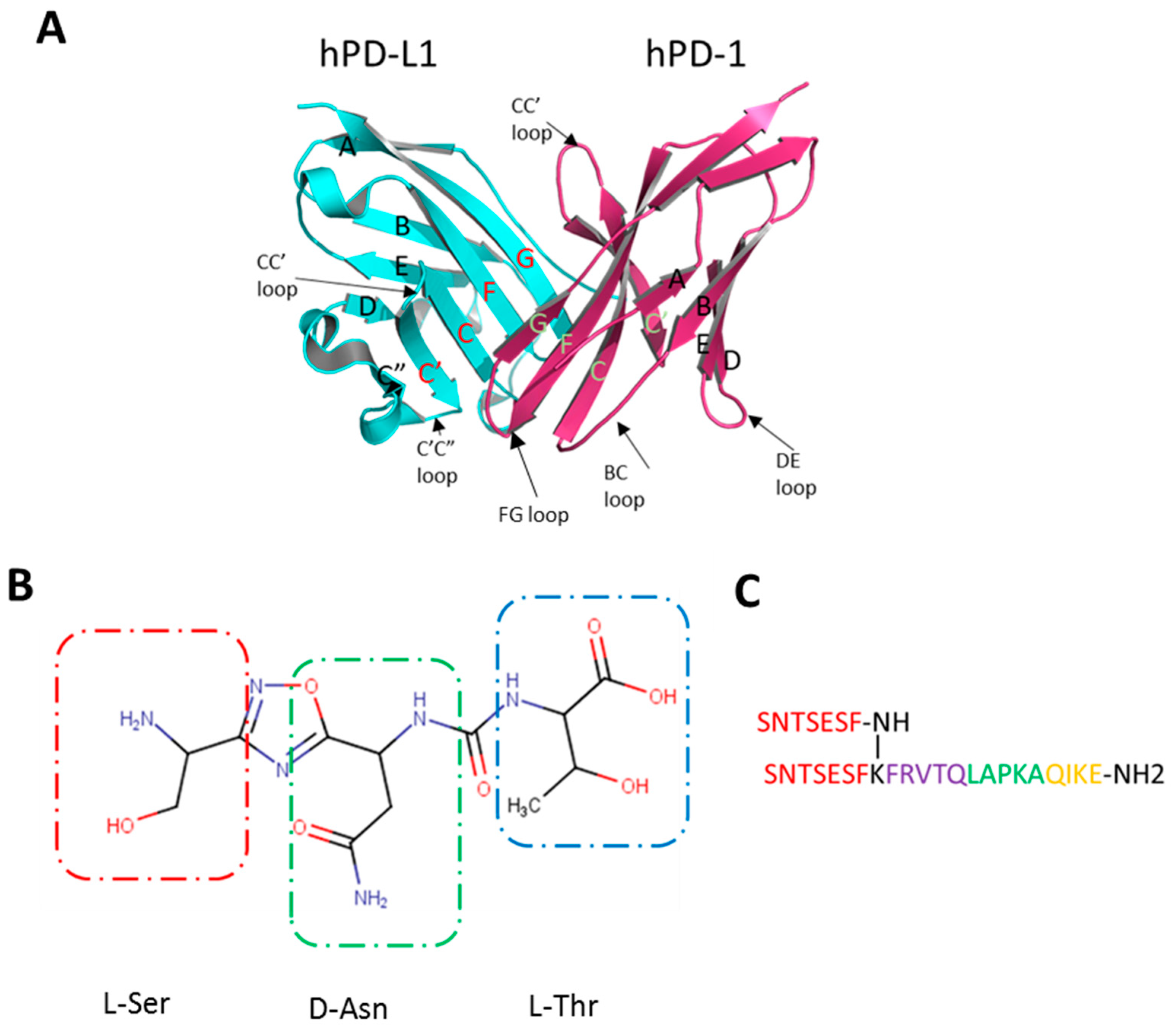
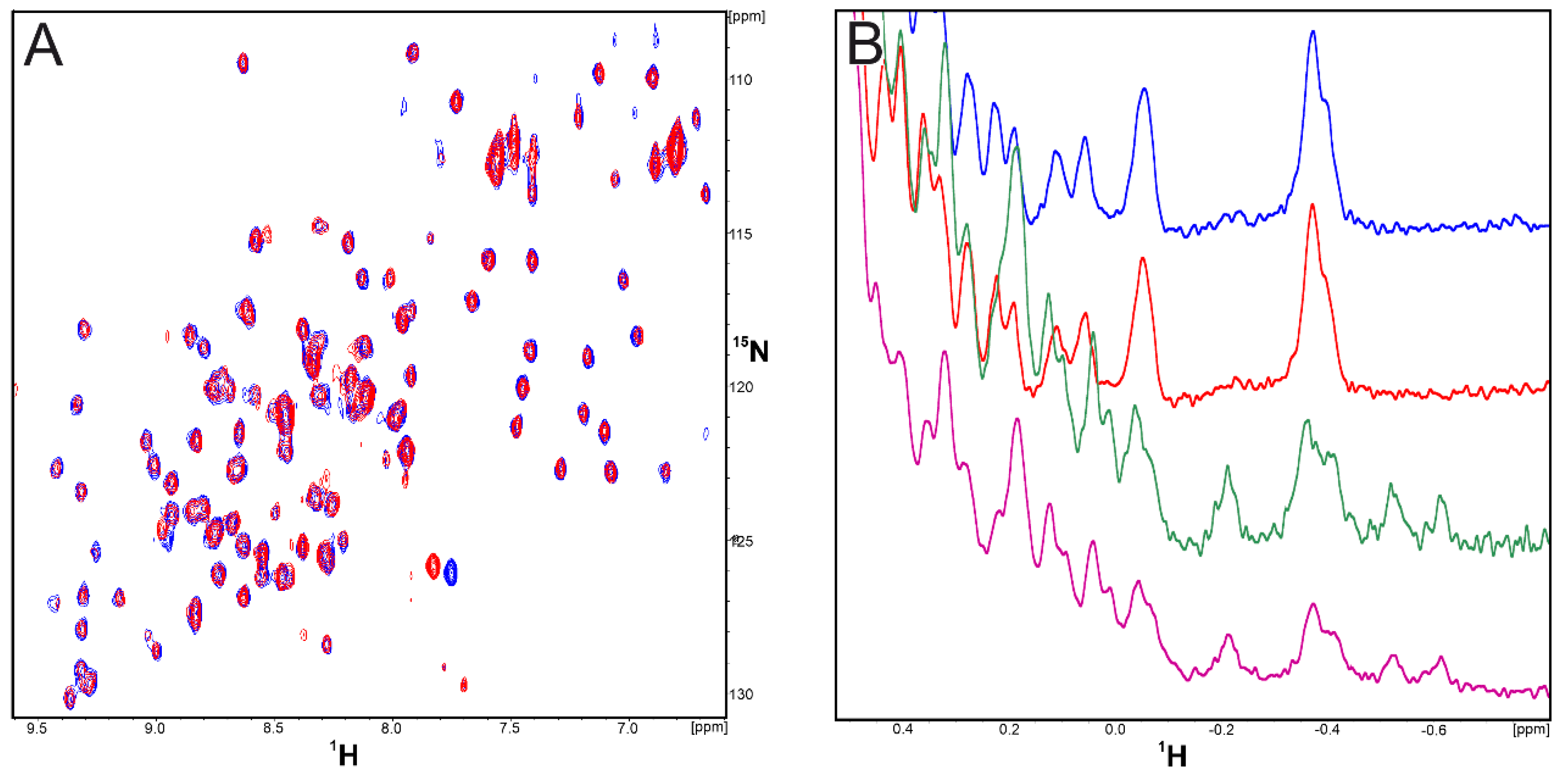
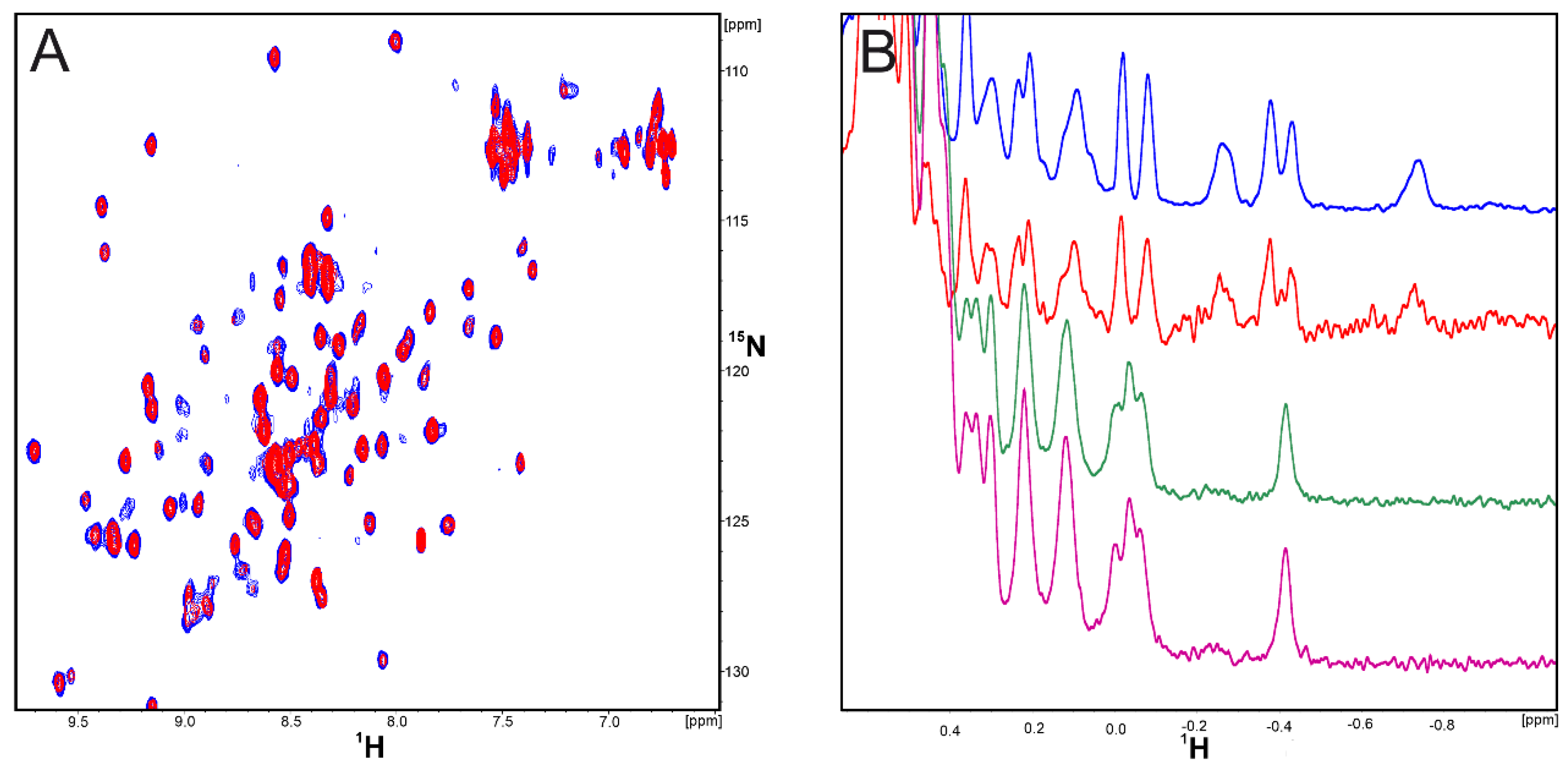
2.1. CA-170 Does Not Bind to hPD-L1 According to the NMR Binding Assay
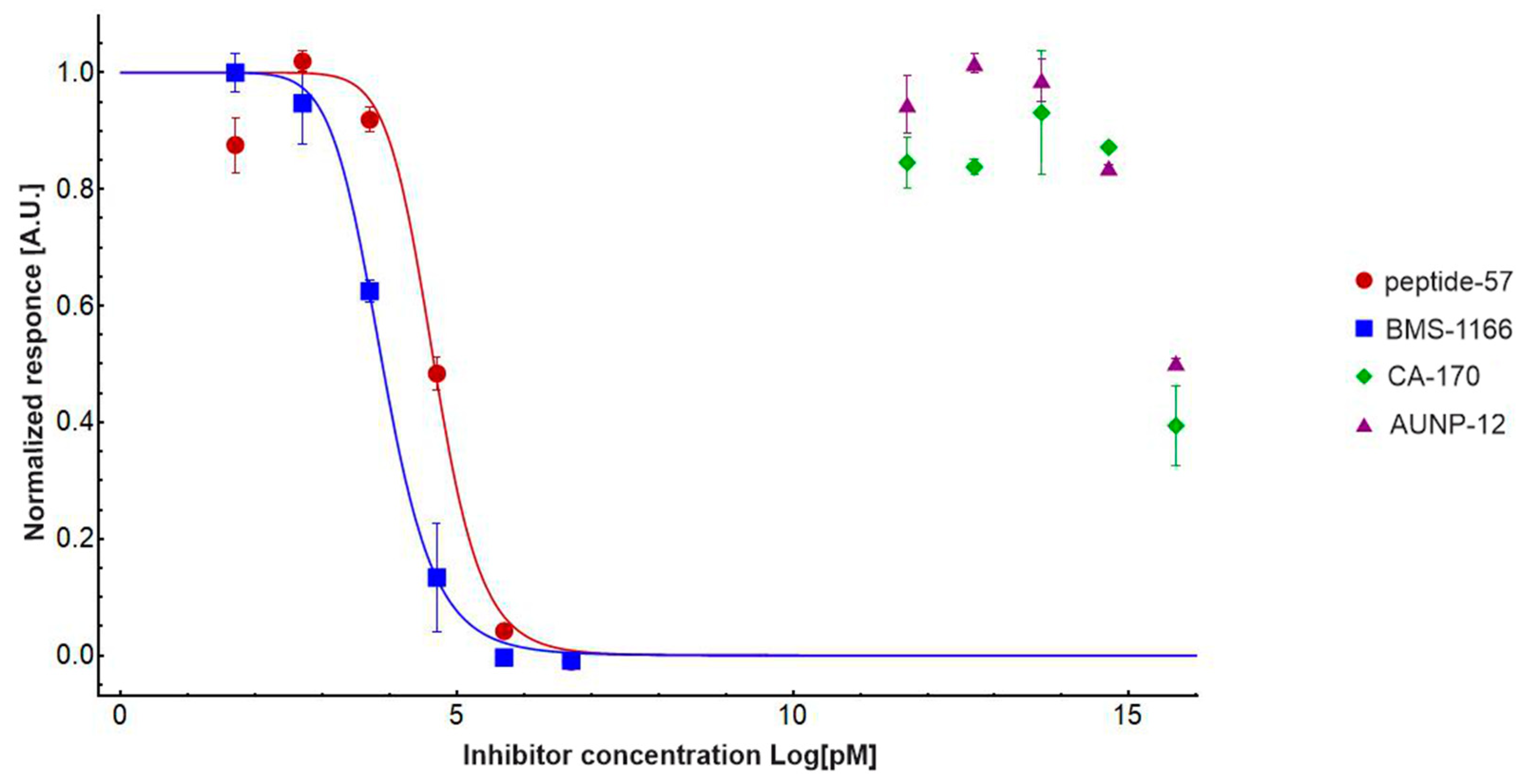
2.2. CA-170 Cannot Disrupt hPD-1/hPD-L1 Complex as Determined with HTRF Assay
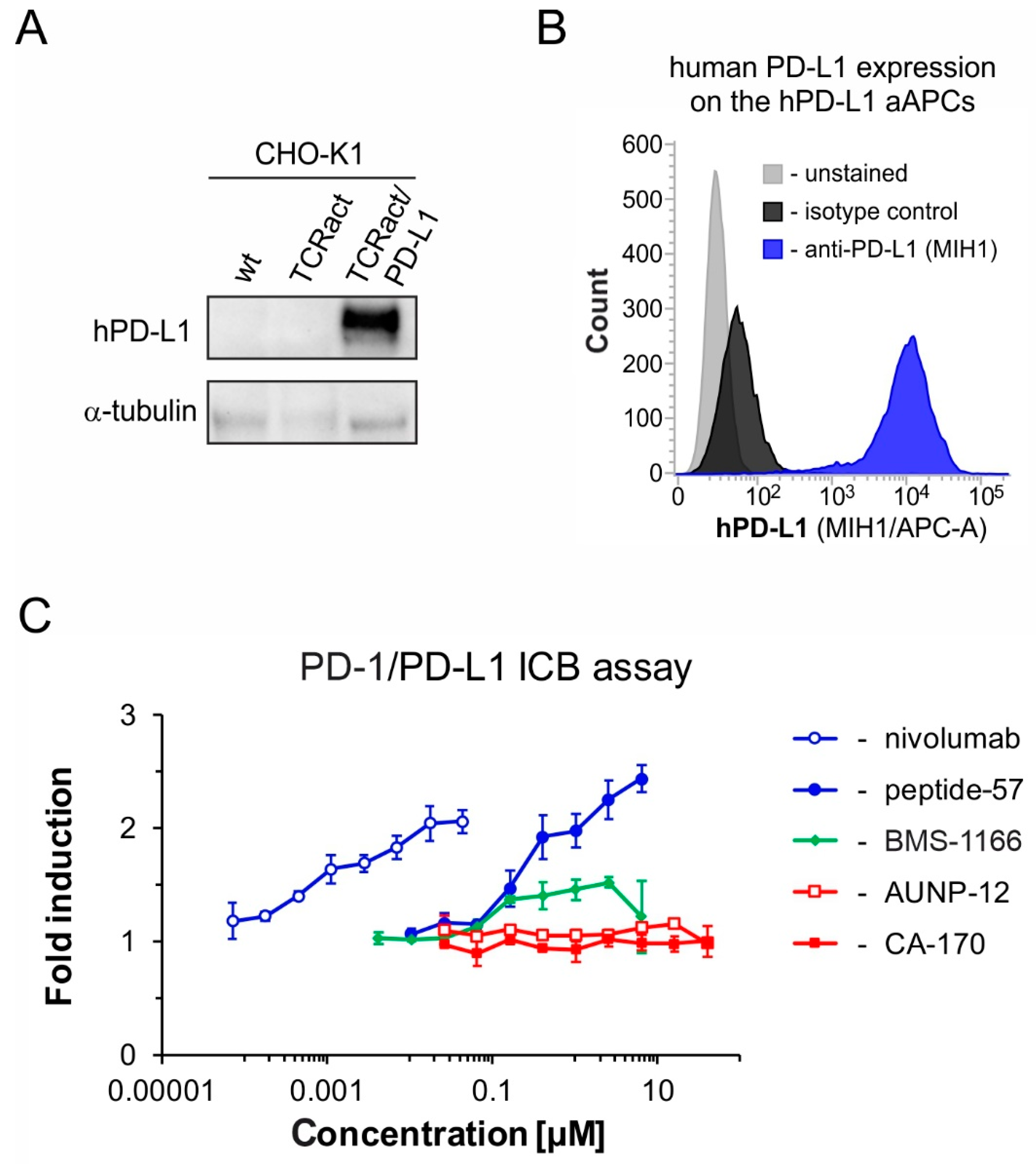
2.3. CA-170 Fails to Restore the Activation of hPD-1/hPD-L1-Blocked Effector Jurkat T Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Protein Expression and Purification
4.3. NMR Binding Assay
4.4. Homogenous Time Resolved FRET
4.5. Cell Culture
4.6. hPD-1/hPD-L1 Immune Checkpoint Blockade Assay
4.7. Western Blot Analysis
4.8. Flow Cytometry Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, P.N. The Cancer Immunotherapy Revolution. Science 2018, 359, 1344–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dömling, A.; Holak, T.A. Programmed Death-1: Therapeutic Success after More than 100 Years of Cancer Immunotherapy. Angew. Chem. Int. Ed. 2014, 53, 2286–2288. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H.; Else, H.; Warren, M. Cancer immunologists scoop medicine Nobel prize. Nature 2018, 562, 20–21. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, J.; Couzin-Frankel, J. Cancer immunotherapy sweeps Nobel for medicine. Science 2018, 362, 13. [Google Scholar] [CrossRef] [PubMed]
- Global $4.92 Billion Programmed Death-1 (PD-1) & amp; Programmed Death Ligand-1 (PD-L1) Inhibitors Pipeline Analysis 2017-2025-Research and Markets. Available online: https://www.prnewswire.com/news-releases/global-492-billion-programmed-death-1-pd-1--programmed-death-ligand-1-pd-l1-inhibitors-pipeline-analysis-2017-2025---research-and-markets-300422553.html (accessed on 4 June 2019).
- Tang, J.; Shalabi, A.; Hubbard-Lucey, V.M. Comprehensive analysis of the clinical immuno-oncology landscape. Ann. Oncol. 2018, 29, 84–91. [Google Scholar] [CrossRef]
- Zak, K.M.; Kitel, R.; Przetocka, S.; Golik, P.; Guzik, K.; Musielak, B.; Dömling, A.; Dubin, G.; Holak, T.A. Structure of the Complex of Human Programmed Death 1, PD-1, and Its Ligand PD-L1. Structure 2015, 23, 2341–2348. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.Y.-w.; Tanaka, Y.; Iwasaki, M.; Gittis, A.G.; Su, H.P.; Mikami, B.; Okazaki, T.; Honjo, T.; Minato, N.; Garboczi, D.N. The PD-1/PD-L1 complex resembles the antigen-binding Fv domains of antibodies and T cell receptors. Proc. Natl. Acad. Sci. USA 2008, 105, 3011–3016. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Riella, L.V.; Paterson, A.M.; Sharpe, A.H.; Chandraker, A. Role of the PD-1 Pathway in the Immune Response. Am. J. Transplant. 2012, 12, 2575–2587. [Google Scholar] [CrossRef] [PubMed]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, E.A.; Grigg, A.; Chong, G. Programmed cell death-1 inhibition in lymphoma. Lancet Oncol. 2015, 16, e234–e245. [Google Scholar] [CrossRef]
- Farid, S.S. Process economics of industrial monoclonal antibody manufacture. J. Chromatogr. B 2007, 848, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Baldo, B. Adverse events to monoclonal antibodies used for cancer therapy: Focus on hypersensitivity responses. Oncoimmunology 2013, 2, e26333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michot, J.M.; Bigenwald, C.; Champiat, S.; Collins, M.; Carbonnel, F.; Postel-Vinay, S.; Berdelou, A.; Varga, A.; Bahleda, R.; Hollebecque, A.; et al. Immune-related adverse events with immune checkpoint blockade: A comprehensive review. Eur. J. Cancer 2016, 54, 139–148. [Google Scholar] [CrossRef]
- Adams, J.L.; Smothers, J.; Srinivasan, R.; Hoos, A. Big opportunities for small molecules in immuno-oncology. Nat. Rev. Drug Discov. 2015, 14, 603–622. [Google Scholar] [CrossRef]
- Guzik, K.; Zak, K.M.; Grudnik, P.; Magiera, K.; Musielak, B.; Törner, R.; Skalniak, L.; Dömling, A.; Dubin, G.; Holak, T.A. Small-Molecule Inhibitors of the Programmed Cell Death-1/Programmed Death-Ligand 1 (PD-1/PD-L1) Interaction via Transiently Induced Protein States and Dimerization of PD-L1. J. Med. Chem. 2017, 60, 5857–5867. [Google Scholar] [CrossRef] [PubMed]
- Acúrcio, R.C.; Scomparin, A.; Conniot, J.; Salvador, J.A.R.; Satchi-Fainaro, R.; Florindo, H.F.; Guedes, R.C. Structure-Function Analysis of Immune Checkpoint Receptors to Guide Emerging Anticancer Immunotherapy. J. Med. Chem. 2018, 61, 10957–10975. [Google Scholar] [CrossRef] [PubMed]
- Huck, B.R.; Kötzner, L.; Urbahns, K. Small Molecules Drive Big Improvements in Immuno-Oncology Therapies. Angew. Chem. Int. Ed. 2018, 57, 4412–4428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasikumar, P.G.; Ramachandra, M. Small-Molecule Immune Checkpoint Inhibitors Targeting PD-1/PD-L1 and Other Emerging Checkpoint Pathways. BioDrugs 2018, 32, 481–497. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hu, L. Immunomodulators targeting the PD-1/PD-L1 protein-protein interaction: From antibodies to small molecules. Med. Res. Rev. 2019, 39, 265–301. [Google Scholar] [CrossRef]
- Zarganes-Tzitzikas, T.; Konstantinidou, M.; Gao, Y.; Krzemien, D.; Zak, K.; Dubin, G.; Holak, T.A.; Dömling, A. Inhibitors of programmed cell death 1 (PD-1): A patent review (2010–2015). Expert Opin. Ther. Pat. 2016, 26, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidou, M.; Zarganes-Tzitzikas, T.; Magiera-Mularz, K.; Holak, T.A.; Dömling, A. Immune Checkpoint PD-1/PD-L1: Is There Life Beyond Antibodies? Angew. Chem. Int. Ed. 2018, 57, 4840–4848. [Google Scholar] [CrossRef]
- Shaabani, S.; Huizinga, H.P.S.; Butera, R.; Kouchi, A.; Guzik, K.; Magiera-Mularz, K.; Holak, T.A.; Dömling, A. A patent review on PD-1/PD-L1 antagonists: Small molecules, peptides, and macrocycles (2015–2018). Expert Opin. Ther. Pat. 2018, 28, 665–678. [Google Scholar] [CrossRef]
- Lazorchak, A.S.; Patterson, T.; Ding, Y.; Sasikumar, P.G.; Sudarshan, N.S.; Gowda, N.M.; Ramachandra, R.K.; Samiulla, D.S.; Giri, S.; Eswarappa, R.; et al. Abstract A36: CA-170, an oral small molecule PD-L1 and VISTA immune checkpoint antagonist, promotes T cell immune activation and inhibits tumor growth in pre-clinical models of cancer. In Proceedings of the AACR Special Conference on Tumor Immunology and Immunotherapy, Boston, MA, USA, 20–23 October 2016; p. A36. [Google Scholar]
- Sasikumar, P.; Sudarshan, N.S.; Gowda, N.; Samiulla, D.S.; Ramachandra, R.; Chandrasekhar, T.; Adurthi, S.; Mani, J.; Nair, R.; Singh, S.; et al. Abstract 4861: Oral immune checkpoint antagonists targeting PD-L1/VISTA or PD-L1/Tim3 for cancer therapy. In Proceedings of the AACR 107th Annual Meeting 2016, New Orleans, LA, USA, 16–20 April 2016; p. 4861. [Google Scholar]
- Available online: http://www.curis.com/images/stories/pdfs/posters/SITC2018CA-170RPD962.pdf (accessed on 5 June 2019).
- Available online: http://www.curis.com/images/stories/pdfs/posters/SITC2018CA-170RPD961.pdf (accessed on 5 June 2019).
- Available online: http://www.curis.com/images/stories/pdfs/posters/SITC2018CA-170P714ASIAD.pdf (accessed on 5 June 2019).
- Sasikumar, P.G.; Ramachandra, R.K.; Adurthi, S.; Dhudashiya, A.A.; Vadlamani, S.; Vemula, K.; Vunnum, S.; Satyam, L.K.; Samiulla, D.S.; Subbarao, K.; et al. A Rationally Designed Peptide Antagonist of the PD-1 Signaling Pathway as an Immunomodulatory Agent for Cancer Therapy. Mol. Cancer Ther. 2019, 18, 1081–1091. [Google Scholar] [CrossRef] [Green Version]
- Sasikumar, P.G.N.; Ramachandra, M.; Naremadde-palli, S.S.S. WO 2015/033301 Al 2015. Available online: http://www.eapatis.com/getdocument.asp?Document=//patstorage/EapatisStorage/wo/wo215010/wo2015033301a1.pdf (accessed on 5 June 2019).
- CA-170 MedKoo. Available online: https://medkoo.com/products/18283 (accessed on 5 June 2019).
- CA-170 - InvivoChem. Available online: https://www.invivochem.com/ca-170/ (accessed on 5 June 2019).
- CA-170 Glixxlabs. Available online: https://www.glixxlabs.com/chemical-products/bioactive-screen-leads-p6/GLXC-15291 (accessed on 5 June 2019).
- CA-170. Available online: http://www.dcchemicals.com/product_show-PD_1_IN_1.html (accessed on 5 June 2019).
- Sasikumar, P.G.N.; Ramachandra, M.; Vadlamani, S.K.; Vemula, K.R.; Satyam, L.K.; Subbarao, K.; Shrimali, K.R.; Kandepu, S. Immunosuppression Modulating Compounds. U.S. Patent 2011/0318373 A1 2011, 29 December 2011. [Google Scholar]
- Skalniak, L.; Zak, K.M.; Guzik, K.; Magiera, K.; Musielak, B.; Pachota, M.; Szelazek, B.; Kocik, J.; Grudnik, P.; Tomala, M.; et al. Small-molecule inhibitors of PD-1/PD-L1 immune checkpoint alleviate the PD-L1-induced exhaustion of T-cells. Oncotarget 2017, 8, 72167–72181. [Google Scholar] [CrossRef]
- Rehm, T.; Huber, R.; Holak, T.A. Application of NMR in Structural Proteomics. Structure 2002, 10, 1613–1618. [Google Scholar] [CrossRef]
- Shuker, S.B.; Hajduk, P.J.; Meadows, R.P.; Fesik, S.W. Discovering High-Affinity Ligands for Proteins: SAR by NMR. Science 1996, 274, 1531–1534. [Google Scholar] [CrossRef] [PubMed]
- Degorce, F. HTRF: A Technology Tailored for Drug Discovery-A Review of Theoretical Aspects and Recent Applications. Curr. Chem. Genom. 2009, 3, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.J.; Karassina, N.; Grailer, J.; Hartnett, J.; Fan, F.; Cong, M. Abstract 5440: Novel PD-1 blockade bioassay to assess therapeutic antibodies in PD-1 and PD-L1 immunotherapy programs. In Proceedings of the AACR 106th Annual Meeting 2015, Philadelphia, PA, USA, 18–22 April 2015; p. 5440. [Google Scholar]
- Wang, C.; Thudium, K.B.; Han, M.; Wang, X.T.; Huang, H.; Feingersh, D.; Garcia, C.; Wu, Y.; Kuhne, M.; Srinivasan, M.; et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol. Res. 2014, 2, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Blevins, D.; Hanley, R.; Bolduc, T.A.; Powell, D.; Gignac, M.; Walker, K.D.; Carr, M.; Hof, F.E.; Wulff, J. In Vitro Assessment of Putative PD-1/PD-L1 Inhibitors: Suggestions of an Alternative Mode of Action. ACS Med. Chem. Lett. 2019. [Google Scholar] [CrossRef]
- Magiera-Mularz, K.; Skalniak, L.; Zak, K.M.; Musielak, B.; Rudzinska-Szostak, E.; Berlicki, Ł.; Kocik, J.; Grudnik, P.; Sala, D.; Zarganes-Tzitzikas, T.; et al. Bioactive Macrocyclic Inhibitors of the PD-1/PD-L1 Immune Checkpoint. Angew. Chem. Int. Ed. 2017, 56, 13732–13735. [Google Scholar] [CrossRef]
- Van Kuppeveld, F.J.M.; Van der Logt, J.T.M.; Angulo, A.F.; Van Zoest, M.J.; Quint, W.G.V.; Niesters, H.G.M.; Galama, J.M.D.; Melchers, W.J.G. Genus- and species-specific identification of mycoplasmas by 16S rRNA amplification. Appl. Environ. Microbiol. 1992, 58, 2606–2615. [Google Scholar] [PubMed]
Sample Availability: Samples of the compounds are not available. |






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musielak, B.; Kocik, J.; Skalniak, L.; Magiera-Mularz, K.; Sala, D.; Czub, M.; Stec, M.; Siedlar, M.; Holak, T.A.; Plewka, J. CA-170 – A Potent Small-Molecule PD-L1 Inhibitor or Not? Molecules 2019, 24, 2804. https://doi.org/10.3390/molecules24152804
Musielak B, Kocik J, Skalniak L, Magiera-Mularz K, Sala D, Czub M, Stec M, Siedlar M, Holak TA, Plewka J. CA-170 – A Potent Small-Molecule PD-L1 Inhibitor or Not? Molecules. 2019; 24(15):2804. https://doi.org/10.3390/molecules24152804
Chicago/Turabian StyleMusielak, Bogdan, Justyna Kocik, Lukasz Skalniak, Katarzyna Magiera-Mularz, Dominik Sala, Miroslawa Czub, Malgorzata Stec, Maciej Siedlar, Tad A. Holak, and Jacek Plewka. 2019. "CA-170 – A Potent Small-Molecule PD-L1 Inhibitor or Not?" Molecules 24, no. 15: 2804. https://doi.org/10.3390/molecules24152804
APA StyleMusielak, B., Kocik, J., Skalniak, L., Magiera-Mularz, K., Sala, D., Czub, M., Stec, M., Siedlar, M., Holak, T. A., & Plewka, J. (2019). CA-170 – A Potent Small-Molecule PD-L1 Inhibitor or Not? Molecules, 24(15), 2804. https://doi.org/10.3390/molecules24152804