Impact of the Position of the Chemically Modified 5-Furyl-2′-Deoxyuridine Nucleoside on the Thrombin DNA Aptamer–Protein Complex: Structural Insights into Aptamer Response from MD Simulations



Abstract
:1. Introduction
2. Results and Discussion
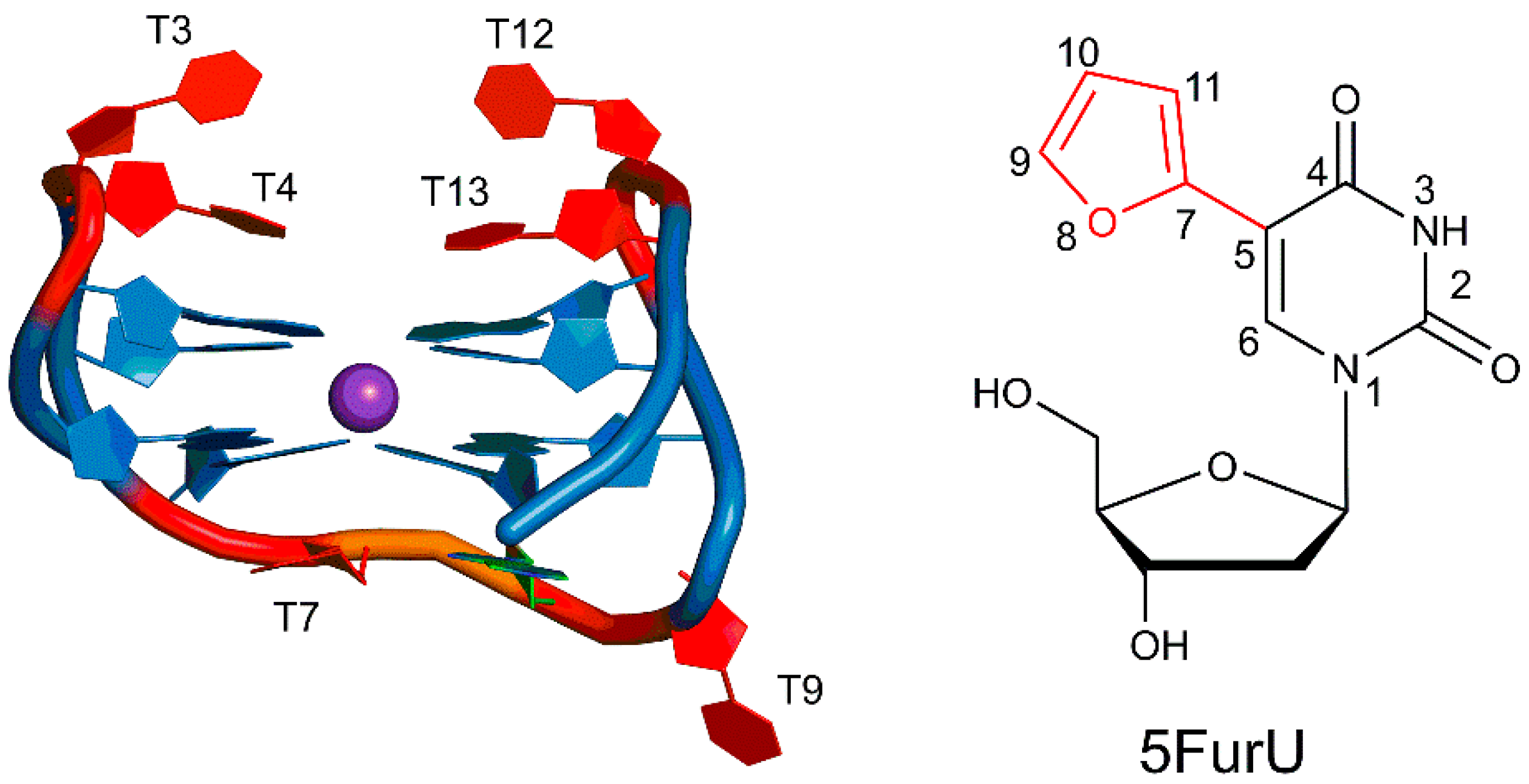
2.1. 5FurU Modified TBA Maintains the G-Quadruplex Structure of Native TBA
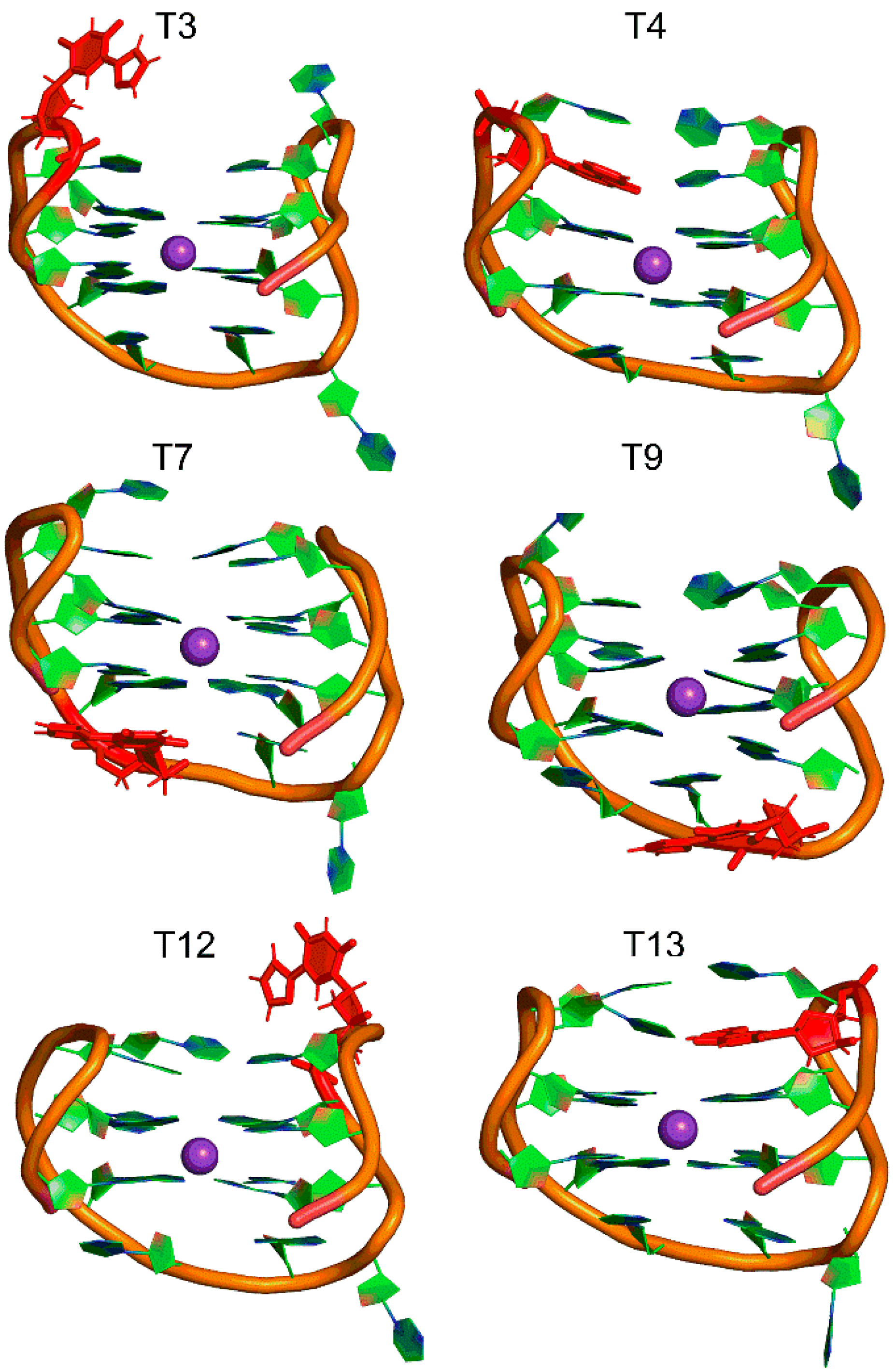
2.2. 5FurU Position Influences TBA Structural Dynamics and Intramolecular Interactions, Which Impact TBA Stability and Photophysical Properties
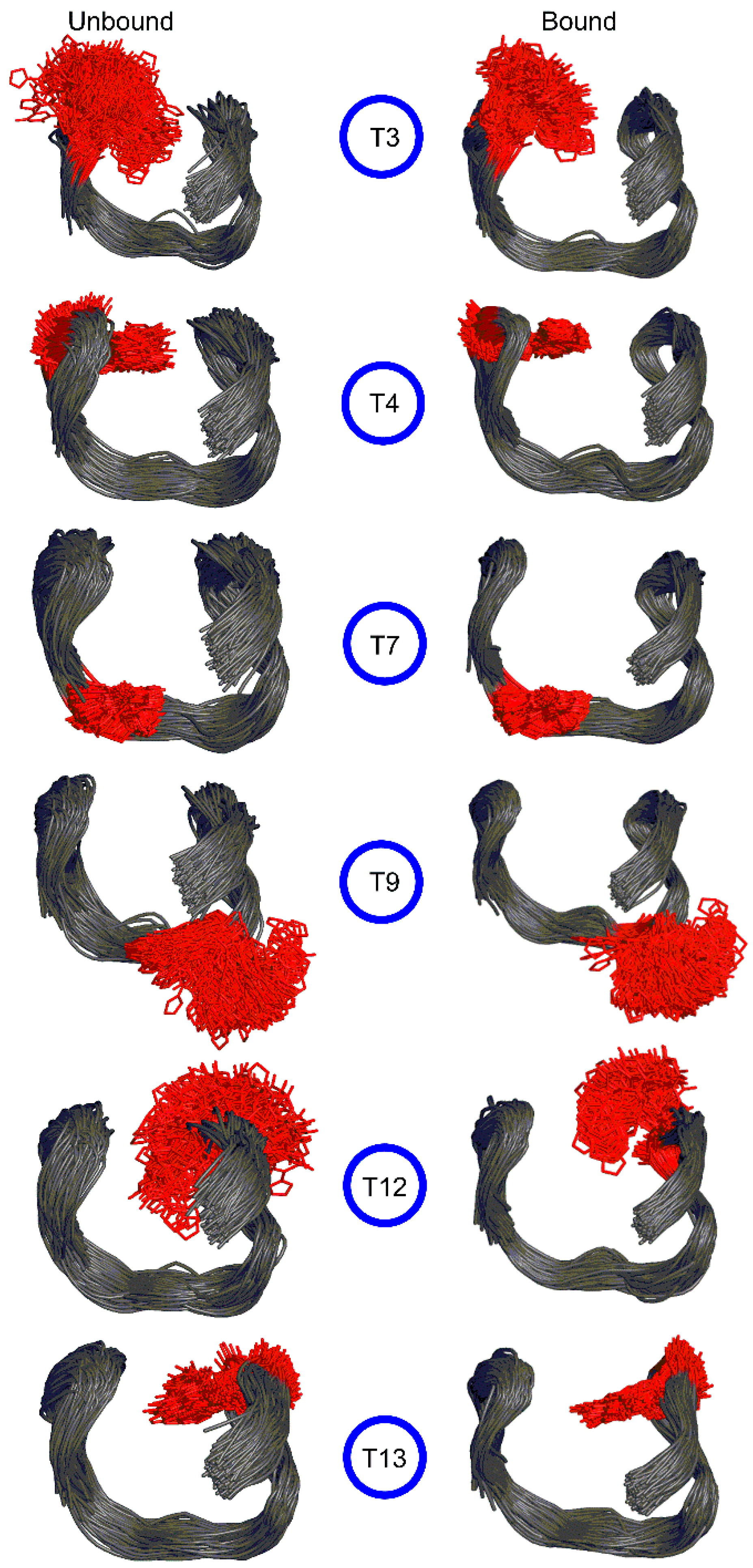
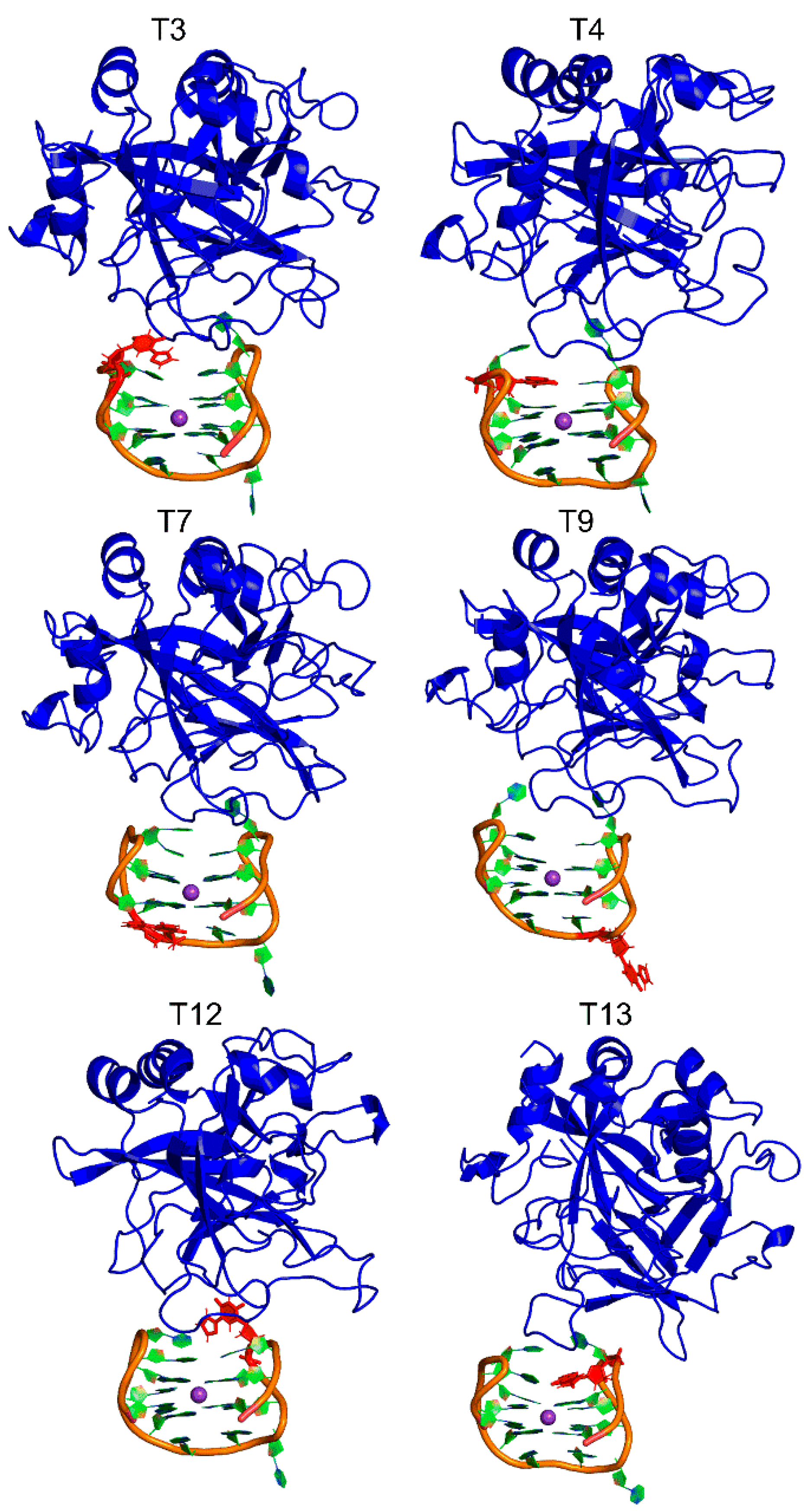
2.3. 5FurU Modification Does Not Affect the Overall Conformation of TBA Bound to Thrombin
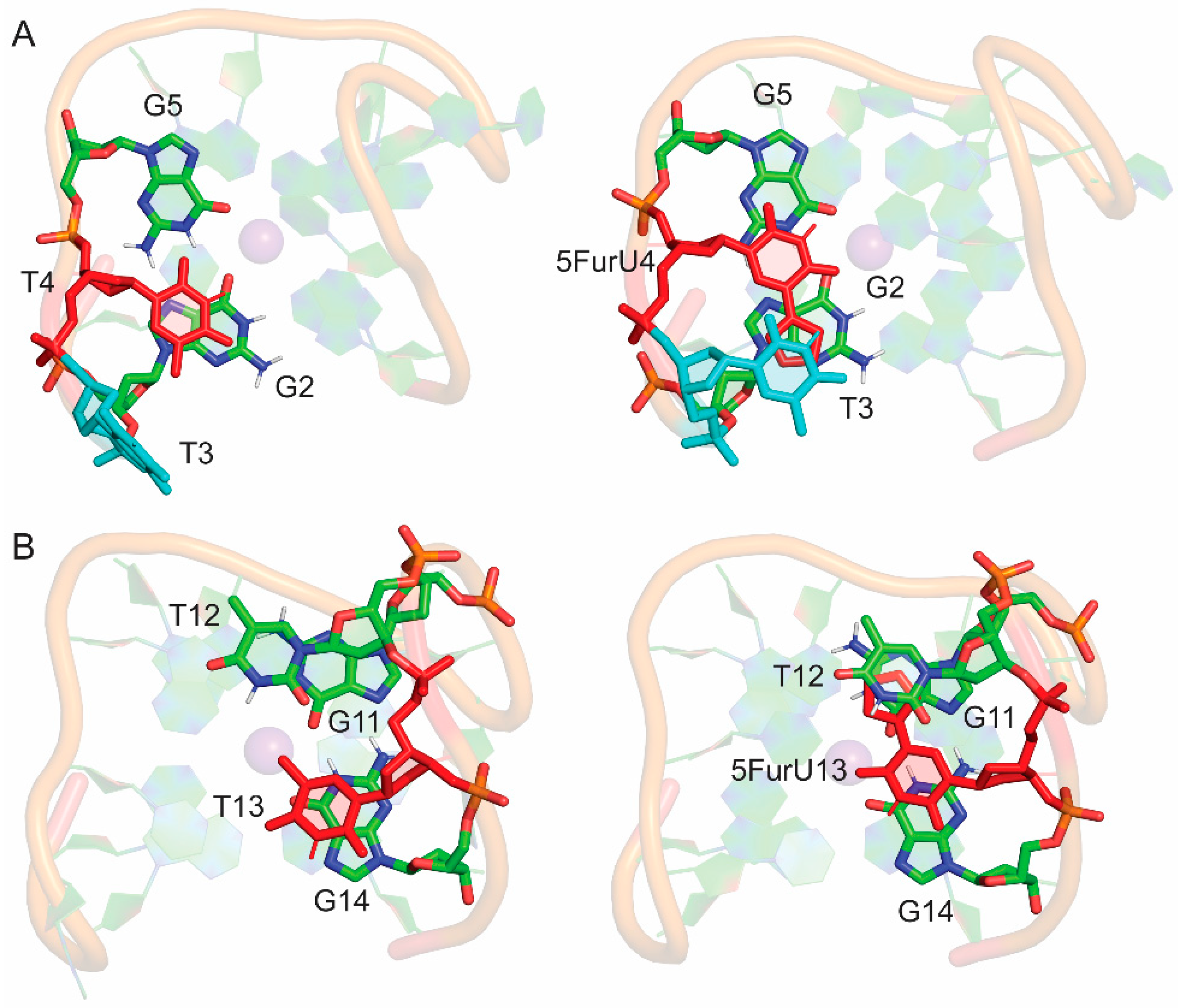
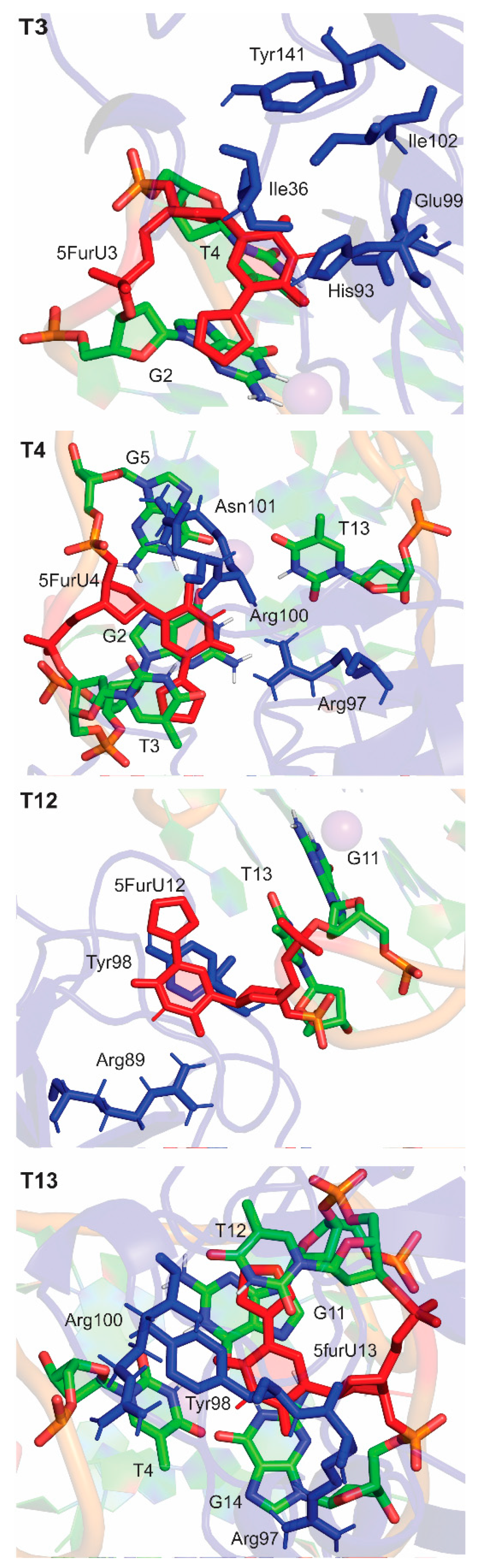
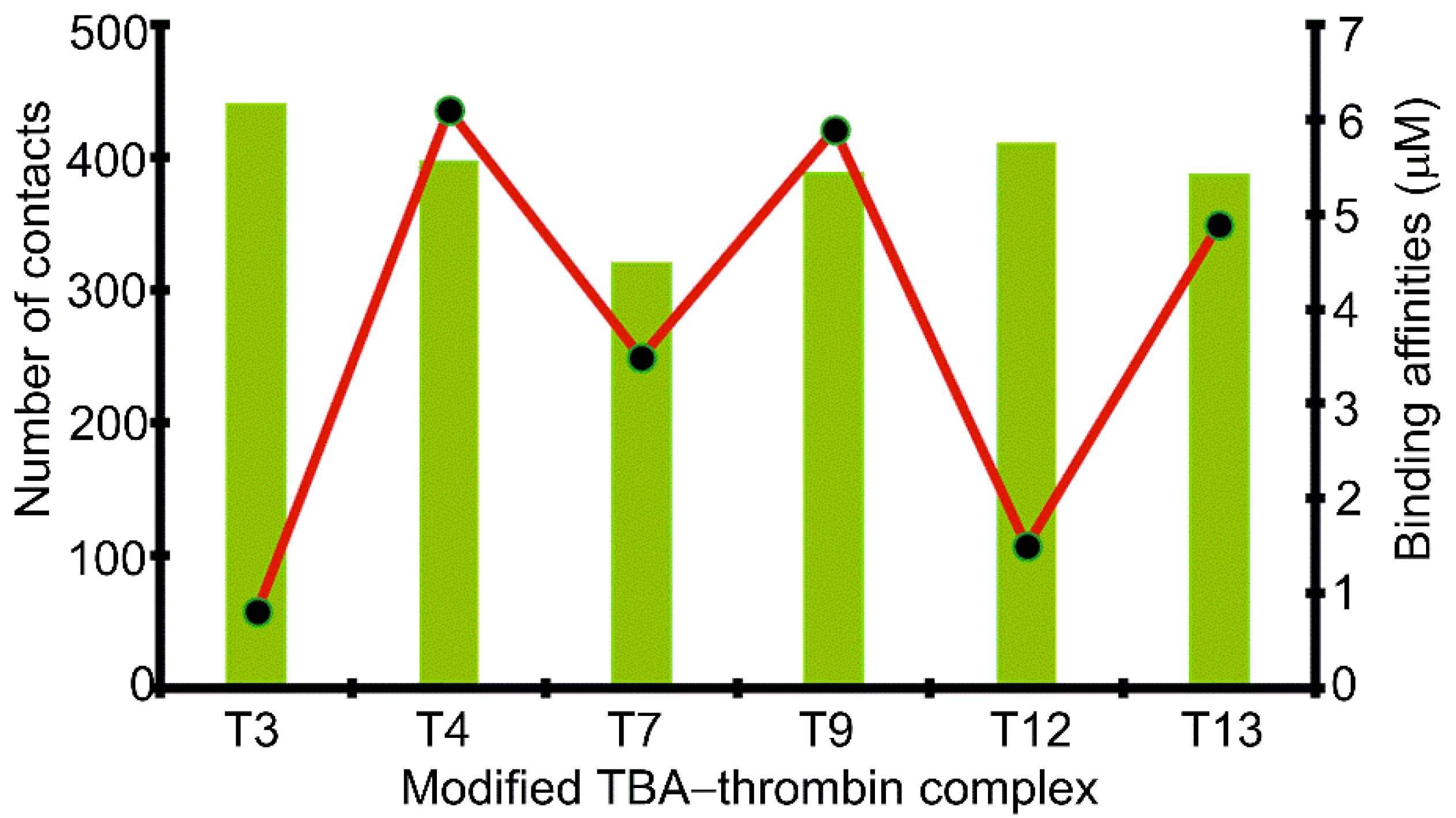
2.4. 5FurU Position Differentially Affects Interactions at the TBA–Thrombin Interface, which Rationalizes Aptamer Binding Affinity and Probe Response
3. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181. [Google Scholar] [CrossRef] [PubMed]
- Dunn, M.R.; Jimenez, R.M.; Chaput, J.C. Analysis of aptamer discovery and technology. Nat. Rev. Chem. 2017, 1, 0076. [Google Scholar] [CrossRef]
- Cai, S.; Yan, J.; Xiong, H.; Liu, Y.; Peng, D.; Liu, Z. Investigations on the interface of nucleic acid aptamers and binding targets. Analyst 2018, 143, 5317–5338. [Google Scholar] [CrossRef] [PubMed]
- Nimjee, S.M.; White, R.R.; Becker, R.C.; Sullenger, B.A. Aptamers as therapeutics. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Jayasena, S.D. Aptamers: An emerging class of molecules that rival antibodies in diagnostics. Clin. Chem 1999, 45, 1628–1650. [Google Scholar] [PubMed]
- Rhodes, D.; Lipps, H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef] [Green Version]
- Hänsel-Hertsch, R.; di Antonio, M.; Balasubramanian, S. DNA G-quadruplexes in the human genome: Detection, functions and therapeutic potential. Nat. Rev. Mol. Cell Biol. 2017, 18, 279. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Huang, R.; Zhou, Y.; Zhang, M.; Deng, M.; Wang, X.; Weng, X.; Zhou, X. Aptamer-based turn-on fluorescent four-branched quaternary ammonium pyrazine probe for selective thrombin detection. Chem. Comm. 2011, 47, 1273–1275. [Google Scholar] [CrossRef]
- He, H.Z.; Chan, D.S.H.; Leung, C.H.; Ma, D.L. G-quadruplexes for luminescent sensing and logic gates. Nucleic Acids Res. 2013, 41, 4345–4359. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Kaplan, A.; Grant, G.; Toole, J.; Leung, L. A novel nucleotide-based thrombin inhibitor inhibits clot-bound thrombin and reduces arterial platelet thrombus formation. Blood 1994, 83, 677–682. [Google Scholar] [Green Version]
- Russo Krauss, I.; Merlino, A.; Randazzo, A.; Novellino, E.; Mazzarella, L.; Sica, F. High-resolution structures of two complexes between thrombin and thrombin-binding aptamer shed light on the role of cations in the aptamer inhibitory activity. Nucleic Acids Res. 2012, 40, 8119–8128. [Google Scholar] [CrossRef] [Green Version]
- Pagano, B.; Martino, L.; Randazzo, A.; Giancola, C. Stability and binding properties of a modified thrombin binding aptamer. Biophys. J. 2008, 94, 562–569. [Google Scholar] [CrossRef]
- Yang, C.; Kulkarni, M.; Lim, M.; Pak, Y. In silico direct folding of thrombin-binding aptamer G-quadruplex at all-atom level. Nucleic Acids Res. 2017, 45, 12648–12656. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Jin, H.; Zhao, X.; Liu, Z.; Guan, Y.; Yang, Z.; Zhang, L.; Zhang, L. Unfolding and conformational variations of thrombin-binding DNA aptamers: Synthesis, circular dichroism and molecular dynamics simulations. ChemMedChem 2014, 9, 993–1001. [Google Scholar] [CrossRef]
- Bian, Y.; Song, F.; Cao, Z.; Zhao, L.; Yu, J.; Guo, X.; Wang, J. Fast-folding pathways of the thrombin-binding aptamer G-quadruplex revealed by a markov state model. Biophys. J. 2018, 114, 1529–1538. [Google Scholar] [CrossRef]
- Reshetnikov, R.; Golovin, A.; Spiridonova, V.; Kopylov, A.; Sponer, J. Structural dynamics of thrombin-binding DNA aptamer d (GGTTGGTGTGGTTGG) quadruplex DNA studied by large-scale explicit solvent simulations. J. Chem. Theory Comput. 2010, 6, 3003–3014. [Google Scholar] [CrossRef]
- Kim, E.; Yang, C.; Pak, Y. Free-energy landscape of a thrombin-binding DNA aptamer in aqueous environment. J. Chem. Theory Comput. 2012, 8, 4845–4851. [Google Scholar] [CrossRef]
- Reshetnikov, R.V.; Sponer, J.; Rassokhina, O.I.; Kopylov, A.M.; Tsvetkov, P.O.; Makarov, A.A.; Golovin, A.V. Cation binding to 15-TBA quadruplex DNA is a multiple-pathway cation-dependent process. Nucleic Acids Res. 2011, 39, 9789–9802. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.W.; Rhee, Y.M.; Shin, S.K. Charge–dipole interactions in G-quadruplex thrombin-binding aptamer. Phys. Chem. Chem. Phys. 2018, 20, 21068–21074. [Google Scholar] [CrossRef]
- Xiao, J.; Salsbury, F.R. Molecular dynamics simulations of aptamer-binding reveal generalized allostery in thrombin. J. Biomol. Struct. Dyn. 2017, 35, 3354–3369. [Google Scholar] [CrossRef]
- Dolot, R.; Lam, C.H.; Sierant, M.; Zhao, Q.; Liu, F.W.; Nawrot, B.; Egli, M.; Yang, X. Crystal structures of thrombin in complex with chemically modified thrombin DNA aptamers reveal the origins of enhanced affinity. Nucleic Acids Res. 2018, 46, 4819–4830. [Google Scholar] [CrossRef]
- Aaldering, L.J.; Poongavanam, V.; Langkjær, N.; Murugan, N.A.; Jørgensen, P.T.; Wengel, J.; Veedu, R.N. Development of an efficient G-quadruplex-stabilised thrombin-binding aptamer containing a three-carbon spacer molecule. ChemBioChem 2017, 18, 755–763. [Google Scholar] [CrossRef]
- Kotkowiak, W.; Czapik, T.; Pasternak, A. Novel isoguanine derivative of unlocked nucleic acid—Investigations of thermodynamics and biological potential of modified thrombin binding aptamer. PLoS ONE 2018, 13, e0197835. [Google Scholar] [CrossRef]
- E Wang, R.; Zhang, Y.; Cai, J.; Cai, W.; Gao, T. Aptamer-based fluorescent biosensors. Curr. Med. Chem. 2011, 18, 4175–4184. [Google Scholar] [CrossRef]
- Musumeci, D.; Platella, C.; Riccardi, C.; Moccia, F.; Montesarchio, D. Fluorescence sensing using DNA aptamers in cancer research and clinical diagnostics. Cancers 2017, 9, 174. [Google Scholar] [CrossRef]
- Jhaveri, S.D.; Kirby, R.; Conrad, R.; Maglott, E.J.; Bowser, M.; Kennedy, R.T.; Glick, G.; Ellington, A.D. Designed signaling aptamers that transduce molecular recognition to changes in fluorescence intensity. J. Am. Chem. Soc. 2000, 122, 2469–2473. [Google Scholar] [CrossRef]
- Sproviero, M.; Fadock, K.L.; Witham, A.A.; Manderville, R.A.; Sharma, P.; Wetmore, S.D. Electronic tuning of fluorescent 8-aryl-guanine probes for monitoring DNA duplex–quadruplex exchange. Chem. Sci. 2014, 5, 788–796. [Google Scholar] [CrossRef]
- Cservenyi, T.Z.; van Riesen, A.J.; Berger, F.D.; Desoky, A.; Manderville, R.A. A simple molecular rotor for defining nucleoside environment within a DNA aptamer–protein complex. ACS Chem. Biol. 2016, 11, 2576–2582. [Google Scholar] [CrossRef]
- Lietard, J.; Abou Assi, H.; Gomez-Pinto, I.; González, C.; Somoza, M.M.; Damha, M.J. Mapping the affinity landscape of thrombin-binding aptamers on 2’ F-ANA/DNA chimeric G-quadruplex microarrays. Nucleic Acids Res. 2017, 45, 1619–1632. [Google Scholar] [CrossRef]
- Van Riesen, A.J.; Fadock, K.L.; Deore, P.S.; Desoky, A.; Manderville, R.A.; Sowlati-Hashjin, S.; Wetmore, S.D. Manipulation of a DNA aptamer–protein binding site through arylation of internal guanine residues. Org. Biomol. Chem. 2018, 16, 3831–3840. [Google Scholar] [CrossRef]
- Fadock, K.L.; Manderville, R.A.; Sharma, P.; Wetmore, S.D. Optimization of fluorescent 8-heteroaryl-guanine probes for monitoring protein-mediated duplex→G-quadruplex exchange. Org. Biomol. Chem. 2016, 14, 4409–4419. [Google Scholar] [CrossRef]
- Mo, M.; Kong, D.; Ji, H.; Lin, D.; Tang, X.; Yang, Z.; He, Y.; Wu, L. Reversible photocontrol of thrombin activity by replacing loops of thrombin binding aptamer using azobenzene derivatives. Bioconjug. Chem. 2018, 30, 231–241. [Google Scholar] [CrossRef]
- Kolganova, N.A.; Tsvetkov, V.B.; Smirnov, I.P.; Timofeev, E.N. Probing the nitroindole-modified central loop of thrombin aptamer HD1 as a recognition site. Nucleic Acid Ther. 2019, 29, 4. [Google Scholar] [CrossRef]
- Coppola, T.; Varra, M.; Oliviero, G.; Galeone, A.; D’Isa, G.; Mayol, L.; Morelli, E.; Bucci, M.R.; Vellecco, V.; Cirino, G. Synthesis, structural studies and biological properties of new TBA analogues containing an acyclic nucleotide. Bioorg. Med. Chem. 2008, 16, 8244–8253. [Google Scholar] [CrossRef]
- Martino, L.; Virno, A.; Randazzo, A.; Virgilio, A.; Esposito, V.; Giancola, C.; Bucci, M.; Cirino, G.; Mayol, L. A new modified thrombin binding aptamer containing a 5′–5′ inversion of polarity site. Nucleic Acids Res. 2006, 34, 6653–6662. [Google Scholar] [CrossRef]
- Tan, S.Y.; Acquah, C.; Sidhu, A.; Ongkudon, C.M.; Yon, L.; Danquah, M.K. SELEX modifications and bioanalytical techniques for aptamer–target binding characterization. Crit. Rev. Anal. Chem. 2016, 46, 521–537. [Google Scholar] [CrossRef]
- Borbone, N.; Bucci, M.; Oliviero, G.; Morelli, E.; Amato, J.; D’Atri, V.; D’Errico, S.; Vellecco, V.; Cirino, G.; Piccialli, G. Investigating the role of T7 and T12 residues on the biological properties of thrombin-binding aptamer: Enhancement of anticoagulant activity by a single nucleobase modification. J. Med. Chem. 2012, 55, 10716–10728. [Google Scholar] [CrossRef]
- Zaitseva, M.; Kaluzhny, D.; Shchyolkina, A.; Borisova, O.; Smirnov, I.; Pozmogova, G. Conformation and thermostability of oligonucleotide d(GGTTGGTGTGGTTGG) containing thiophosphoryl internucleotide bonds at different positions. Biophys. Chem. 2010, 146, 1–6. [Google Scholar] [CrossRef]
- Pozmogova, G.; Zaitseva, M.; Smirnov, I.; Shvachko, A.; Murina, M.; Sergeenko, V. Anticoagulant effects of thioanalogs of thrombin-binding DNA-aptamer and their stability in the plasma. Bull. Exp. Biol. Med. 2010, 150, 180–184. [Google Scholar] [CrossRef]
- Liu, B.; Li, D. Structural transformation induced by locked nucleic acid or 2′–O-methyl nucleic acid site-specific modifications on thrombin binding aptamer. Chem. Cent. J. 2014, 8, 19. [Google Scholar] [CrossRef]
- Aviñó, A.; Mazzini, S.; Fàbrega, C.; Peñalver, P.; Gargallo, R.; Morales, J.C.; Eritja, R. The effect of l-thymidine, acyclic thymine and 8-bromoguanine on the stability of model G-quadruplex structures. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1205–1212. [Google Scholar] [CrossRef]
- Virgilio, A.; Petraccone, L.; Scuotto, M.; Vellecco, V.; Bucci, M.; Mayol, L.; Varra, M.; Esposito, V.; Galeone, A. 5-Hydroxymethyl-2′-deoxyuridine residues in the thrombin binding aptamer: Investigating anticoagulant activity by making a tiny chemical modification. ChemBioChem 2014, 15, 2427–2434. [Google Scholar] [CrossRef]
- Varizhuk, A.M.; Tsvetkov, V.B.; Tatarinova, O.N.; Kaluzhny, D.N.; Florentiev, V.L.; Timofeev, E.N.; Shchyolkina, A.K.; Borisova, O.F.; Smirnov, I.P.; Grokhovsky, S.L. Synthesis, characterization and in vitro activity of thrombin-binding DNA aptamers with triazole internucleotide linkages. Eur. J. Med. Chem. 2013, 67, 90–97. [Google Scholar] [CrossRef]
- Saneyoshi, H.; Mazzini, S.; Avino, A.; Portella, G.; Gonzalez, C.; Orozco, M.; Marquez, V.E.; Eritja, R. Conformationally rigid nucleoside probes help understand the role of sugar pucker and nucleobase orientation in the thrombin-binding aptamer. Nucleic Acids Res. 2009, 37, 5589–5601. [Google Scholar] [CrossRef] [Green Version]
- Pasternak, A.; Hernandez, F.J.; Rasmussen, L.M.; Vester, B.; Wengel, J. Improved thrombin binding aptamer by incorporation of a single unlocked nucleic acid monomer. Nucleic Acids Res. 2010, 39, 1155–1164. [Google Scholar] [CrossRef] [Green Version]
- Jensen, T.B.; Henriksen, J.R.; Rasmussen, B.E.; Rasmussen, L.M.; Andresen, T.L.; Wengel, J.; Pasternak, A. Thermodynamic and biological evaluation of a thrombin binding aptamer modified with several unlocked nucleic acid (UNA) monomers and a 2′-C-piperazino-UNA monomer. Bioorg. Med. Chem. 2011, 19, 4739–4745. [Google Scholar] [CrossRef]
- Esposito, V.; Russo, A.; Amato, T.; Vellecco, V.; Bucci, M.; Mayol, L.; Russo, G.; Virgilio, A.; Galeone, A. The “Janus face” of the thrombin binding aptamer: Investigating the anticoagulant and antiproliferative properties through straightforward chemical modifications. Bioorg. Chem. 2018, 76, 202–209. [Google Scholar] [CrossRef]
- Aviñó, A.; Mazzini, S.; Ferreira, R.; Gargallo, R.; Marquez, V.E.; Eritja, R. The effect on quadruplex stability of north-nucleoside derivatives in the loops of the thrombin-binding aptamer. Bioorg. Med. Chem. 2012, 20, 4186–4193. [Google Scholar] [CrossRef]
- Gunjal, A.D.; Fernandes, M.; Erande, N.; Rajamohanan, P.R.; Kumar, V.A. Functional isoDNA aptamers: Modified thrombin binding aptamers with a 2′-5′-linked sugar-phosphate backbone (isoTBA). Chem. Comm. 2014, 50, 605–607. [Google Scholar] [CrossRef]
- Cai, B.; Yang, X.; Sun, L.; Fan, X.; Li, L.; Jin, H.; Wu, Y.; Guan, Z.; Zhang, L.; Zhang, L. Stability and bioactivity of thrombin binding aptamers modified with D-/L-isothymidine in the loop regions. Org. Biomol. Chem. 2014, 12, 8866–8876. [Google Scholar] [CrossRef]
- Jayapal, P.; Mayer, G.; Heckel, A.; Wennmohs, F. Structure–activity relationships of a caged thrombin binding DNA aptamer: Insight gained from molecular dynamics simulation studies. J. Struct. Biol. 2009, 166, 241–250. [Google Scholar] [CrossRef]
- Tsvetkov, V.B.; Varizhuk, A.M.; Pozmogova, G.E.; Smirnov, I.P.; Kolganova, N.A.; Timofeev, E.N. A universal base in a specific role: Tuning up a thrombin aptamer with 5-nitroindole. Sci. Rep. 2015, 5, 16337. [Google Scholar] [CrossRef]
- Aviñó, A.; Portella, G.; Ferreira, R.; Gargallo, R.; Mazzini, S.; Gabelica, V.; Orozco, M.; Eritja, R. Specific loop modifications of the thrombin-binding aptamer trigger the formation of parallel structures. FEBS J. 2014, 281, 1085–1099. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Xie, X.; Weng, W.; Jin, H. Structural and mechanistic insights into modified G-quadruplex thrombin-binding DNA aptamers. Biochem. Biophys. Res. Commun. 2019, 513, 753–759. [Google Scholar] [CrossRef]
- Wang, K.Y.; McCurdy, S.; Shea, R.G.; Swaminathan, S.; Bolton, P.H. A DNA aptamer which binds to and inhibits thrombin exhibits a new structural motif for DNA. Biochemistry 1993, 32, 1899–1904. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView; Version 6; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Ivani, I.; Dans, P.D.; Noy, A.; Pérez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goñi, R.; Balaceanu, A.; et al. Parmbsc1: A refined force field for DNA simulations. Nat. Methods 2015, 13, 55. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Dupradeau, F.Y.; Pigache, A.; Zaffran, T.; Savineau, C.; Lelong, R.; Grivel, N.; Lelong, D.; Rosanski, W.; Cieplak, P. The RED Tools: Advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys. 2010, 12, 7821–7839. [Google Scholar] [CrossRef]
- Case, D.A.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; Gohlke, H.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TBA | Sequence a |
|---|---|
| Native | 5′–GGTTGGTGTGGTTGG–3′ |
| T3 | 5′–GGXTGGTGTGGTTGG–3′ |
| T4 | 5′–GGTXGGTGTGGTTGG–3′ |
| T7 | 5′–GGTTGGXGTGGTTGG–3′ |
| T9 | 5′–GGTTGGTGXGGTTGG–3′ |
| T12 | 5′–GGTTGGTGTGGXTGG–3′ |
| T13 | 5′–GGTTGGTGTGGTXGG–3′ |
| Position | Unbound | Bound | ||||
|---|---|---|---|---|---|---|
| Native | Modified | Δa | Native | Modified | Δa | |
| T3 | 266.5 | 314.8 | 48.4 | 194.2 | 182.5 | −11.7 |
| (20.7) | (19.6) | (34.9) | (27.1) | |||
| T4 | 167.7 | 151.6 | −16.2 | 126.9 | 129.0 | 2.1 |
| (26.6) | (17.3) | (18.5) | (12.9) | |||
| T7 | 230.8 | 261.3 | 30.5 | 233.3 | 263.9 | 30.6 |
| (7.1) | (8.4) | (6.8) | (8.1) | |||
| T9 | 290.8 | 293.4 | 2.6 | 290.7 | 332.8 | 42.1 |
| (6.8) | (36.9) | (7.2) | (11.2) | |||
| T12 | 236.6 | 313.2 | 76.6 | 207.5 | 265.1 | 57.6 |
| (32.5) | (20.7) | (23.0) | (21.0) | |||
| T13 | 171.1 | 147.5 | −23.6 | 80.8 | 76.6 | −4.2 |
| (22.3) | (18.2) | (17.1) | (15.6) | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seelam Prabhakar, P.; A. Manderville, R.; D. Wetmore, S. Impact of the Position of the Chemically Modified 5-Furyl-2′-Deoxyuridine Nucleoside on the Thrombin DNA Aptamer–Protein Complex: Structural Insights into Aptamer Response from MD Simulations. Molecules 2019, 24, 2908. https://doi.org/10.3390/molecules24162908
Seelam Prabhakar P, A. Manderville R, D. Wetmore S. Impact of the Position of the Chemically Modified 5-Furyl-2′-Deoxyuridine Nucleoside on the Thrombin DNA Aptamer–Protein Complex: Structural Insights into Aptamer Response from MD Simulations. Molecules. 2019; 24(16):2908. https://doi.org/10.3390/molecules24162908
Chicago/Turabian StyleSeelam Prabhakar, Preethi, Richard A. Manderville, and Stacey D. Wetmore. 2019. "Impact of the Position of the Chemically Modified 5-Furyl-2′-Deoxyuridine Nucleoside on the Thrombin DNA Aptamer–Protein Complex: Structural Insights into Aptamer Response from MD Simulations" Molecules 24, no. 16: 2908. https://doi.org/10.3390/molecules24162908
APA StyleSeelam Prabhakar, P., A. Manderville, R., & D. Wetmore, S. (2019). Impact of the Position of the Chemically Modified 5-Furyl-2′-Deoxyuridine Nucleoside on the Thrombin DNA Aptamer–Protein Complex: Structural Insights into Aptamer Response from MD Simulations. Molecules, 24(16), 2908. https://doi.org/10.3390/molecules24162908