
Self-Assembly of Covalently Linked Porphyrin Dimers at the Solid–Liquid Interface


Abstract
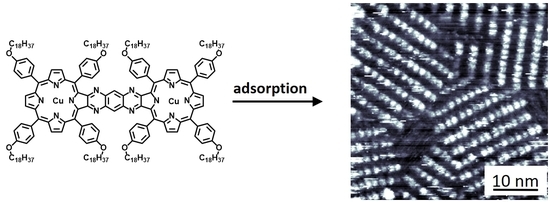
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. Self-Assembly of Conjugated Porphyrin Dimers Cu21 and Mn21 at a Solid–Liquid Interface
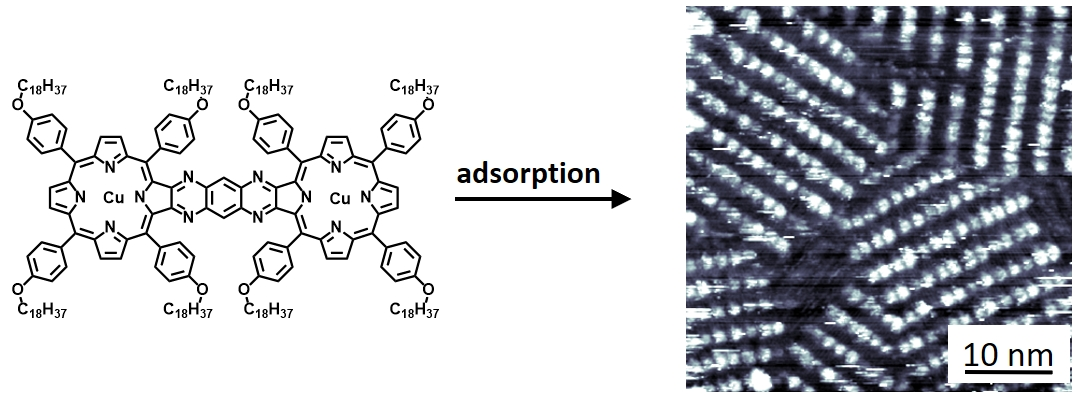
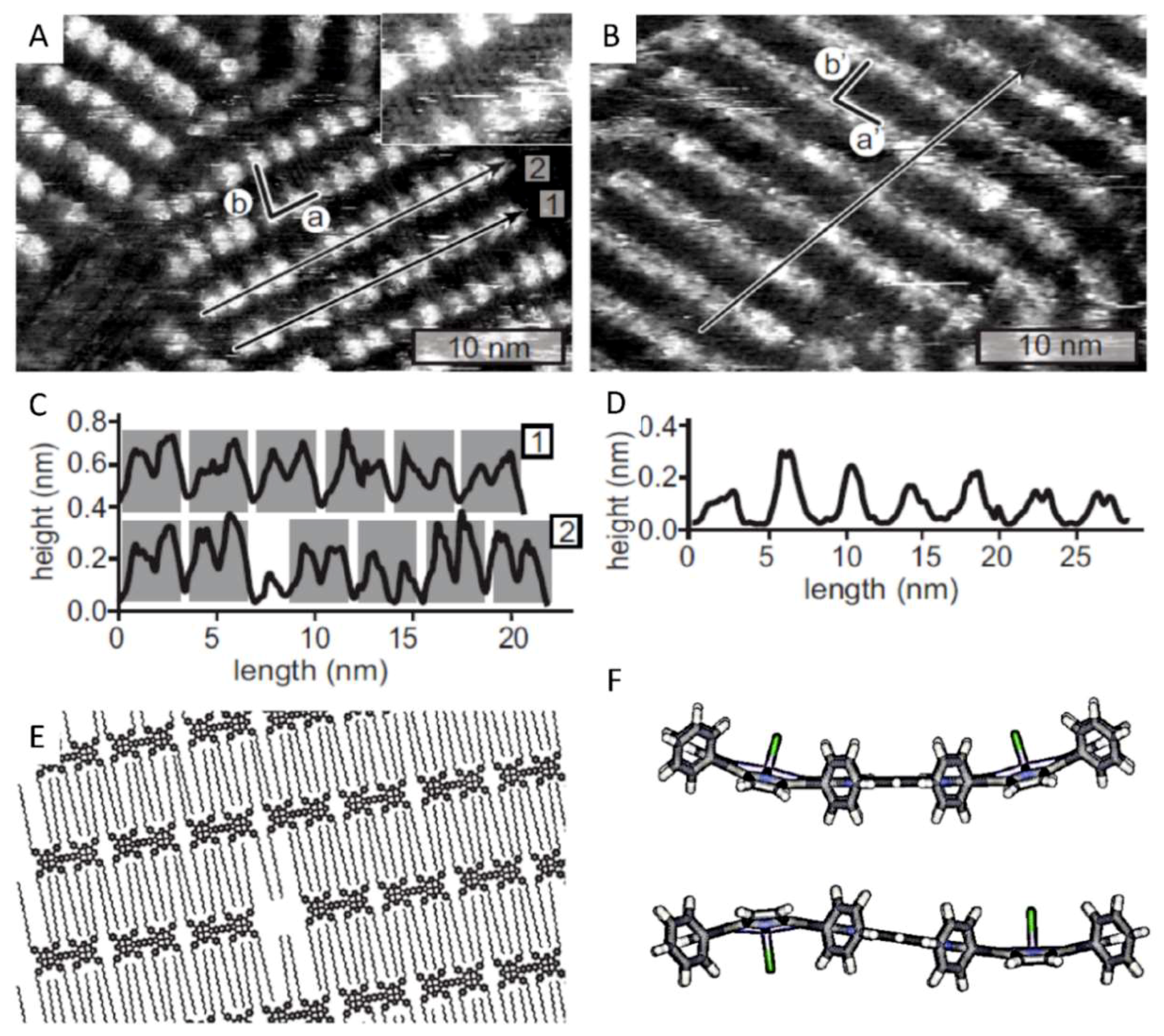
2.3. Self-Assembly of the Non-Conjugated Porphyrin Dimers Cu22, Mn22, and MnCu2 at a Solid–Liquid Interface
2.4. 2D Polymorphism of Porphyrin Dimer MnCu2
3. Materials and Methods
3.1. General Materials and Methods
3.2. Syntheses
3.3. STM Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- McDermott, G.; Prince, S.M.; Freer, A.A.; Hawthornthwaite-Lawless, A.M.; Papiz, M.Z.; Cogdell, R.J.; Isaacs, N.W. Crystal structure of an integral membrane light-harvesting complex from photosynthetic bacteria. Nature 1995, 374, 517. [Google Scholar] [CrossRef]
- Yu, L.-J.; Suga, M.; Wang-Otomo, Z.-Y.; Shen, J.-R. Structure of photosynthetic LH1–RC supercomplex at 1.9 Å resolution. Nature 2018, 556, 209. [Google Scholar] [CrossRef] [PubMed]
- Paoli, M.; Liddington, R.; Tame, J.; Wilkinson, A.; Dodson, G. Crystal structure of T state haemoglobin with oxygen bound at all four haems. J. Mol. Biol. 1996, 256, 775. [Google Scholar] [CrossRef] [PubMed]
- Feiters, M.C.; Rowan, A.E.; Nolte, R.J.M. From simple to supramolecular cytochrome P450 mimics. Chem. Soc. Rev. 2000, 29, 375. [Google Scholar] [CrossRef]
- Ortiz de Montellano, P.R. Hydrocarbon hydroxylation by cytochrome P450 enzymes. Chem. Rev. 2010, 110, 932. [Google Scholar] [CrossRef] [PubMed]
- Elemans, J.A.A.W.; van Hameren, R.; Nolte, R.J.M.; Rowan, A.E. Molecular materials by the self-assembly of porphyrins, phthalocyanines and perylenes. Adv. Mater. 2006, 18, 1251. [Google Scholar] [CrossRef]
- Jurow, M.; Schuckman, A.E.; Batteas, J.D.; Drain, C.M. Porphyrins as molecular electronic components of functional devices. Coord. Chem. Rev. 2010, 254, 2297. [Google Scholar] [CrossRef]
- Longevial, J.-F.; Clément, S.; Wytko, J.A.; Ruppert, R.; Weiss, J.; Richeter, S. Peripherally metalated porphyrins with applications in catalysis, molecular electronics and biomedicine. Chem. Eur. J. 2018, 24, 15442. [Google Scholar] [CrossRef]
- Meunier, B. Metalloporphyrins as versatile catalysts for oxidation reactions and oxidative DNA cleavage. Chem. Rev. 1992, 92, 1411. [Google Scholar] [CrossRef]
- Pellissier, H.; Clavier, H. Enantioselective cobalt-catalyzed transformations. Chem. Rev. 2014, 114, 2775. [Google Scholar] [CrossRef]
- Barona-Castaño, J.C.; Carmona-Vargas, C.C.; Brocksom, T.J.; de Oliveira, K.T. Porphyrins as catalysts in scalable organic reactions. Molecules 2016, 21, 310. [Google Scholar] [CrossRef] [PubMed]
- Elemans, J.A.A.W.; Bijsterveld, E.J.A.; Rowan, A.E.; Nolte, R.J.M. A host-guest epoxidation catalyst with enhanced activity and stability. Chem. Commun. 2000, 2443. [Google Scholar]
- Elemans, J.A.A.W.; Bijsterveld, E.J.A.; Rowan, A.E.; Nolte, R.J.M. Manganese porphyrin hosts as epoxidation catalysts: Activity and stability control by axial ligand effects. Eur. J. Org. Chem. 2007, 751. [Google Scholar] [CrossRef]
- Monnereau, C.; Ramos, P.H.; Deutman, A.B.C.; Elemans, J.A.A.W.; Nolte, R.J.M.; Rowan, A.E. Porphyrin macrocyclic catalysts for the processive oxidation of polymer substrates. J. Am. Chem. Soc. 2010, 132, 1529. [Google Scholar] [CrossRef] [PubMed]
- Bernar, I.; Rutjes, F.P.J.T.; Elemans, J.A.A.W.; Nolte, R.J.M. Aerobic epoxidation of low molecular weight and polymeric olefins by a supramolecular manganese–porphyrin catalyst. Catalysts 2019, 9, 195. [Google Scholar] [CrossRef]
- Elemans, J.A.A.W.; Nolte, R.J.M. Porphyrin cage compounds based on glycoluril – from enzyme mimics to functional molecular machines. Chem. Commun. 2019, 55, 9590. [Google Scholar] [CrossRef]
- Elemans, J.A.A.W.; Lensen, M.C.; Gerritsen, J.W.; van Kempen, H.; Speller, S.; Nolte, R.J.M.; Rowan, A.E. Scanning probe studies of porphyrin assemblies and their supramolecular manipulation at a solid-liquid interface. Adv. Mater. 2003, 15, 2070. [Google Scholar] [CrossRef]
- Lensen, M.C.; van Dingenen, S.J.T.; Elemans, J.A.A.W.; Dijkstra, H.P.; van Klink, G.P.M.; van Koten, G.; Gerritsen, J.W.; Speller, S.; Nolte, R.J.M.; Rowan, A.E. Synthesis and self-assembly of giant porphyrin discs. Chem. Commun. 2004, 762. [Google Scholar] [CrossRef]
- Lensen, M.C.; Elemans, J.A.A.W.; van Dingenen, S.J.T.; Gerritsen, J.W.; Speller, S.; Rowan, A.E.; Nolte, R.J.M. Giant porphyrin disks. Control of their self-assembly at liquid-solid interfaces by metal-ligand interactions. Chem. Eur. J. 2007, 13, 7948. [Google Scholar] [CrossRef]
- den Boer, D.; Habets, T.; Coenen, M.J.J.; van der Maas, M.; Peters, T.P.J.; Crossley, M.J.; Khoury, T.; Rowan, A.E.; Nolte, R.J.M.; Speller, S.; et al. Controlled templating of porphyrins by a molecular command layer. Langmuir 2011, 27, 2644. [Google Scholar] [CrossRef]
- Münninghoff, J.A.W.; Elemans, J.A.A.W. Chemistry at the square nanometer: Reactivity at liquid/solid interfaces revealed with STM. Chem. Commun. 2017, 53, 1769. [Google Scholar] [CrossRef] [PubMed]
- Coenen, M.J.J.; Cremers, M.; den Boer, D.; van den Bruele, F.J.; Khoury, T.; Sintic, M.; Crossley, M.J.; van Enckevort, W.J.P.; Hendriksen, B.L.M.; Elemans, J.A.A.W.; et al. Little exchange at the liquid/solid interface: Defect-mediated equilibration of physisorbed porphyrin monolayers. Chem. Commun. 2011, 47, 9666. [Google Scholar] [CrossRef] [PubMed]
- Coenen, M.J.J.; den Boer, D.; van den Bruele, F.J.; Habets, T.; Timmers, K.A.A.M.; van der Maas, M.; Khoury, T.; Panduwinata, D.; Crossley, M.J.; Reimers, J.R.; et al. Polymorphism in porphyrin monolayers: The relation between adsorption configuration and molecular conformation. Phys. Chem. Chem. Phys. 2013, 15, 12451. [Google Scholar] [CrossRef] [PubMed]
- Coenen, M.J.J.; Khoury, T.; Crossley, M.J.; Hendriksen, B.L.M.; Elemans, J.A.A.W.; Speller, S. Nanostructuring of self-assembled porphyrin networks at a solid/liquid interface: Local manipulation under global control. ChemPhysChem 2014, 15, 3484. [Google Scholar] [CrossRef] [PubMed]
- Hulsken, B.; van Hameren, R.; Gerritsen, J.W.; Khoury, T.; Thordarson, P.; Crossley, M.J.; Rowan, A.E.; Nolte, R.J.M.; Elemans, J.A.A.W.; Speller, S. Real-time single molecule imaging of alkene oxidation by manganese porphyrins at a liquid-solid interface. Nat. Nanotechnol. 2007, 2, 285. [Google Scholar] [CrossRef] [PubMed]
- den Boer, D.; Li, M.; Habets, T.; Iavicoli, P.; Rowan, A.E.; Nolte, R.J.M.; Speller, S.; Amabilino, D.B.; De Feyter, S.; Elemans, J.A.A.W. Detection of different oxidation states of individual manganese porphyrins during their reaction with oxygen at a solid/liquid interface. Nat. Chem. 2013, 5, 621. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; den Boer, D.; Iavicoli, P.; Adisoejoso, J.; Uji-i, H.; Van der Auweraer, M.; Amabilino, D.B.; Elemans, J.A.A.W.; De Feyter, S. Tip-induced chemical manipulation of metal porphyrins at the liquid-solid interface. J. Am. Chem. Soc. 2014, 136, 17418. [Google Scholar] [CrossRef] [PubMed]
- Pawlicki, M.; Morisue, M.; Davis, N.K.S.; McLean, D.G.; Haley, J.E.; Beuerman, E.; Drobizhev, M.; Rebane, A.; Thompson, A.L.; Pascu, S.I.; et al. Engineering conjugation in para-phenylene-bridged porphyrin tapes. Chem. Sci. 2012, 3, 1541. [Google Scholar] [CrossRef]
- Matano, Y.; Matsumoto, K.; Hayashi, H.; Nakao, Y.; Kumpulainen, T.; Chukharev, V.; Tkachenko, N.V.; Lemmetyinen, H.; Shimizu, S.; Kobayashi, N.; et al. Effects of carbon−metal−carbon linkages on the optical, photophysical, and electrochemical properties of phosphametallacycle-linked coplanar porphyrin dimers. J. Am. Chem. Soc. 2012, 134, 1825. [Google Scholar] [CrossRef] [PubMed]
- Gehrold, A.C.; Bruhn, T.; Bringmann, G. Axial, helical, and planar chirality in directly linked basket-handle porphyrin arrays. J. Org. Chem. 2016, 81, 1075. [Google Scholar] [CrossRef] [PubMed]
- Gao, K.; Fukui, N.; Jung, S.I.; Yorimitsu, H.; Kim, D.; Osuka, A. Pictet–Spengler synthesis of quinoline-fused porphyrins and phenanthroline-fused diporphyrins. Angew. Chem. Int. Ed. 2016, 55, 13038. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Osuka, A. Meta- and para-phenylenediamine-fused porphyrin dimers: Synthesis and magnetic interactions of their dication diradicals. Angew. Chem. Int. Ed. 2019, 58, 8546. [Google Scholar] [CrossRef] [PubMed]
- Bonifazi, D.; Spillmann, H.; Kiebele, A.; de Wild, M.; Seiler, P.; Chen, F.; Güntherodt, H.-J.; Jung, T.; Diederich, F. Supramolecular patterned surfaces driven by cooperative assembly of C60 and porphyrins on metal substrates. Angew. Chem. Int. Ed. 2004, 43, 4759. [Google Scholar] [CrossRef] [PubMed]
- Bennett, N.; Xu, G.; Esdaile, L.J.; Anderson, H.L.; Macdonald, J.E.; Elliott, M. Transition voltage spectroscopy of porphyrin molecular wires. Small 2010, 6, 2604. [Google Scholar] [CrossRef] [PubMed]
- Krasnikov, S.A.; Sergeeva, N.N.; Sergeeva, Y.N.; Sengeband, M.O.; Cafolla, A.A. Self-assembled rows of Ni porphyrin dimers on the Ag(111) surface. Phys. Chem. Chem. Phys. 2010, 12, 6666. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, M.-A.; Dekkiche, H.; Karmazin, L.; Sanchez, F.; Vincent, B.; Kanesato, M.; Kikkawa, Y.; Ruppert, R. Synthesis and study at a solid/liquid interface of porphyrin dimers linked by metal ions. Inorg. Chem. 2017, 56, 15081. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, M.-A.; Dekkiche, H.; Nagasaki, M.; Kikkawa, Y.; Ruppert, R. Coordination-driven construction of porphyrin nanoribbons at a highly oriented pyrolytic graphite (HOPG)/liquid interface. J. Am. Chem. Soc. 2019, 141, 10137. [Google Scholar] [CrossRef]
- Qiu, X.; Wang, C.; Zeng, Q.; Xu, B.; Yin, S.; Wang, H.; Xu, S.; Bai, C. Alkane-assisted adsorption and assembly of phthalocyanines and porphyrins. J. Am. Chem. Soc. 2000, 122, 5550. [Google Scholar] [CrossRef]
- Lensen, D.; Habets, T.; Elemans, J.A.A.W. Dynamic rearrangement of bilayers of porphyrin hetero-dimers at a solid/liquid interface. Chem. Commun. 2014, 50, 7291. [Google Scholar] [CrossRef]
- Brückner, C.; Rettig, S.J.; Dolphin, D. Formation of a meso-tetraphenylsecochlorin and a homoporphyrin with a twist. J. Org. Chem. 1998, 63, 2094. [Google Scholar] [CrossRef]
- Mazur, U.; Hipps, K.W. Kinetic and thermodynamic processes of organic species at the solution-solid interface: The view through an STM. Chem. Commun. 2015, 51, 4737. [Google Scholar] [CrossRef] [PubMed]
- Reimers, J.R.; Panduwinata, D.; Visser, J.; Chin, Y.; Tang, C.; Goerigk, L.; Ford, M.J.; Sintic, M.; Sum, T.J.; Coenen, M.J.J.; et al. A-priori calculations of the free energy of formation from solution of polymorphic self-assembled monolayers. Proc. Natl. Acad. Sci. USA 2015, 112, E6101. [Google Scholar] [CrossRef] [PubMed]
- Reimers, J.R.; Panduwinata, D.; Visser, J.; Chin, Y.; Tang, C.; Goerigk, L.; Ford, M.; Baker, M.; Sum, T.J.; Coenen, M.J.J.; et al. From chaos to order: Chain-length dependence of the free energy of formation of meso-tetraalkylporphyrin self-assembled monolayer polymorphs. J. Phys. Chem. C 2016, 120, 1739. [Google Scholar] [CrossRef]
- Marie, C.; Silly, F.; Tortech, L.; Müllen, K.; Fichou, D. Tuning the packing density of 2D supramolecular self-assemblies at the solid−liquid interface using variable temperature. ACS Nano 2010, 4, 1288. [Google Scholar] [CrossRef] [PubMed]
- Blunt, M.O.; Adisoejoso, J.; Tahara, K.; Katayama, K.; Van der Auweraer, M.; Tobe, Y.; De Feyter, S. Temperature-induced structural phase transitions in a two-dimensional self-assembled network. J. Am. Chem. Soc. 2013, 135, 12068. [Google Scholar] [CrossRef] [PubMed]
- Mali, K.S.; Wu, D.; Feng, X.; Müllen, K.; Van der Auweraer, M.; De Feyter, S. Scanning tunneling microscopy-induced reversible phase transformation in the two-dimensional crystal of a positively charged discotic polycyclic aromatic hydrocarbon. J. Am. Chem. Soc. 2011, 133, 5686. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-L.; Hsu, Y.-J.; Wu, H.-J.; Lin, H.-A.; Hsu, H.-F.; Chen, C.-H. Electrical pulse triggered reversible assembly of molecular adlayers. Chem. Commun. 2012, 48, 11748. [Google Scholar] [CrossRef] [PubMed]
- Samorì, P.; Engelkamp, H.; de Witte, P.; Rowan, A.E.; Nolte, R.J.M.; Rabe, J.P. Self-assembly and manipulation of crown ether phthalocyanines at the gel-graphite interface. Angew. Chem. Int. Ed. 2001, 40, 2348. [Google Scholar] [CrossRef]
- France, C.B.; Schroeder, P.G.; Parkinson, B.A. Direct observation of a widely spaced periodic row structure at the pentacene/Au(111) interface using scanning tunneling microscopy. Nano Lett. 2002, 2, 693. [Google Scholar] [CrossRef]
- Huber, V.; Lysetska, M.; Würthner, F. Self-assembled single- and double-stack π-aggregates of chlorophyll derivatives on highly ordered pyrolytic graphite. Small 2007, 3, 1007. [Google Scholar] [CrossRef]
- Kowalzik, P.; Rathgeber, S.; Karthäuser, S.; Waser, R.; Schnaebele, N.; Raimundoc, J.-M.; Gingrasz, M. Columnar self-assembly of a 3D-persulfurated coronene asterisk. The dominant role of aryl-sulfur bonds. New J. Chem. 2012, 36, 477. [Google Scholar] [CrossRef]
- Yano, M.; Endo, M.; Hasegawa, Y.; Okada, R.; Yamada, Y.; Sasaki, M. Well-ordered monolayers of alkali-doped coronene and picene: Molecular arrangements and electronic structures. J. Chem. Phys. 2014, 141, 034708. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Guo, Z.; Tahara, K.; Khosrowabadi Kotyk, J.F.; Nguyen, H.; Gotoda, J.; Iritani, K.; Rubin, Y.; Tobe, Y. Self-assembled dehydro [24]annulene monolayers at the liquid/solid interface: Toward on-surface synthesis of tubular π-conjugated nanowires. Langmuir 2016, 32, 5532. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xie, H.; Hu, Z.; Liu, C. The impact of the electric field on surface condensation of water vapor: Insight from molecular dynamics simulation. Nanomaterials 2019, 9, 64. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Habets, T.; Lensen, D.; Speller, S.; Elemans, J.A.A.W. Self-Assembly of Covalently Linked Porphyrin Dimers at the Solid–Liquid Interface. Molecules 2019, 24, 3018. https://doi.org/10.3390/molecules24163018
Habets T, Lensen D, Speller S, Elemans JAAW. Self-Assembly of Covalently Linked Porphyrin Dimers at the Solid–Liquid Interface. Molecules. 2019; 24(16):3018. https://doi.org/10.3390/molecules24163018
Chicago/Turabian StyleHabets, Thomas, Dennis Lensen, Sylvia Speller, and Johannes A.A.W. Elemans. 2019. "Self-Assembly of Covalently Linked Porphyrin Dimers at the Solid–Liquid Interface" Molecules 24, no. 16: 3018. https://doi.org/10.3390/molecules24163018
APA StyleHabets, T., Lensen, D., Speller, S., & Elemans, J. A. A. W. (2019). Self-Assembly of Covalently Linked Porphyrin Dimers at the Solid–Liquid Interface. Molecules, 24(16), 3018. https://doi.org/10.3390/molecules24163018