
Encapsulation of Metal Nanoparticles within Metal–Organic Frameworks for the Reduction of Nitro Compounds



 and
and 
Abstract
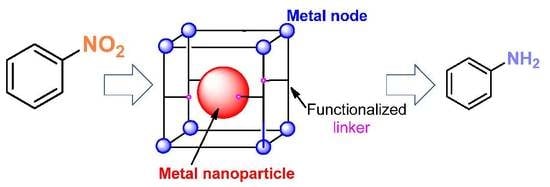
:
1. Introduction
2. Monometallic MNPs@MOFs
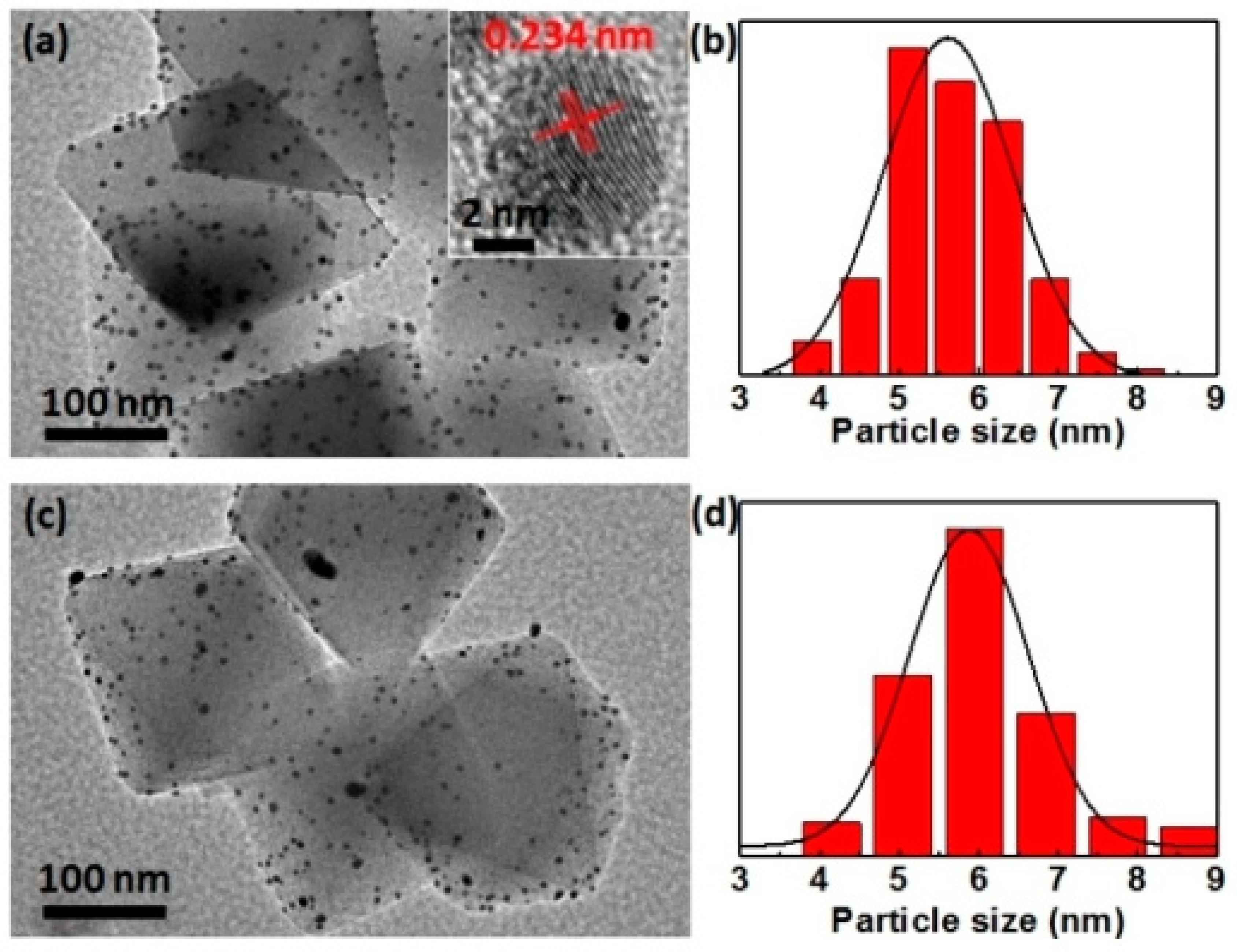
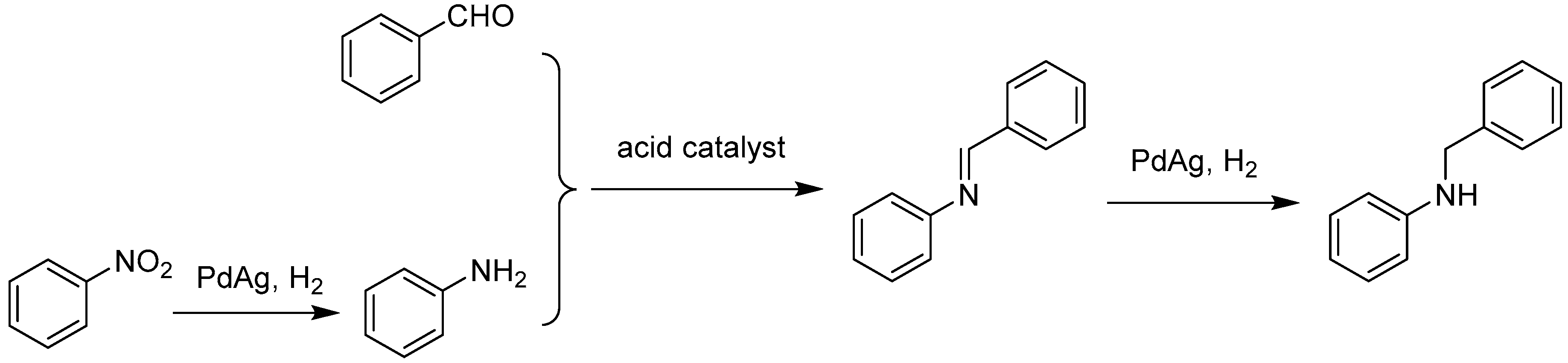
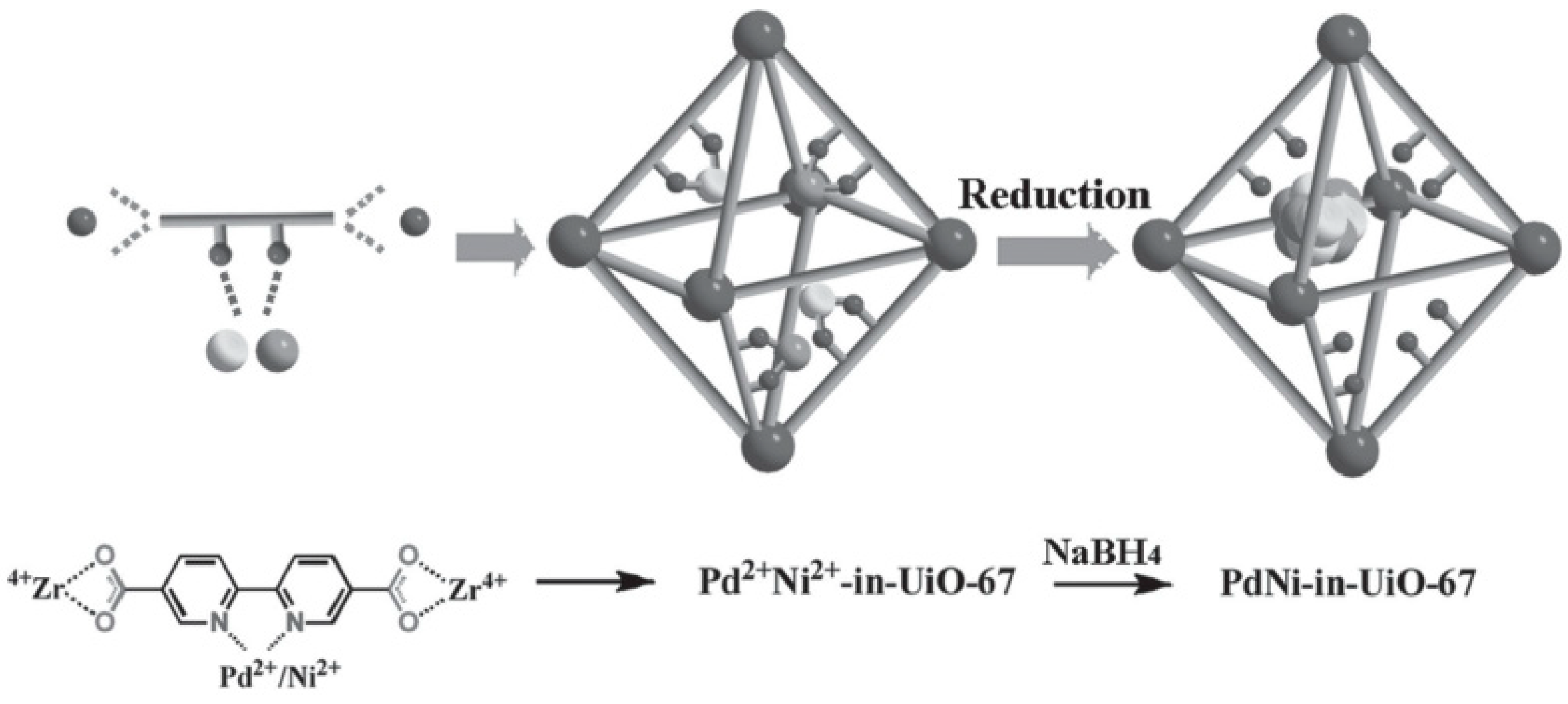
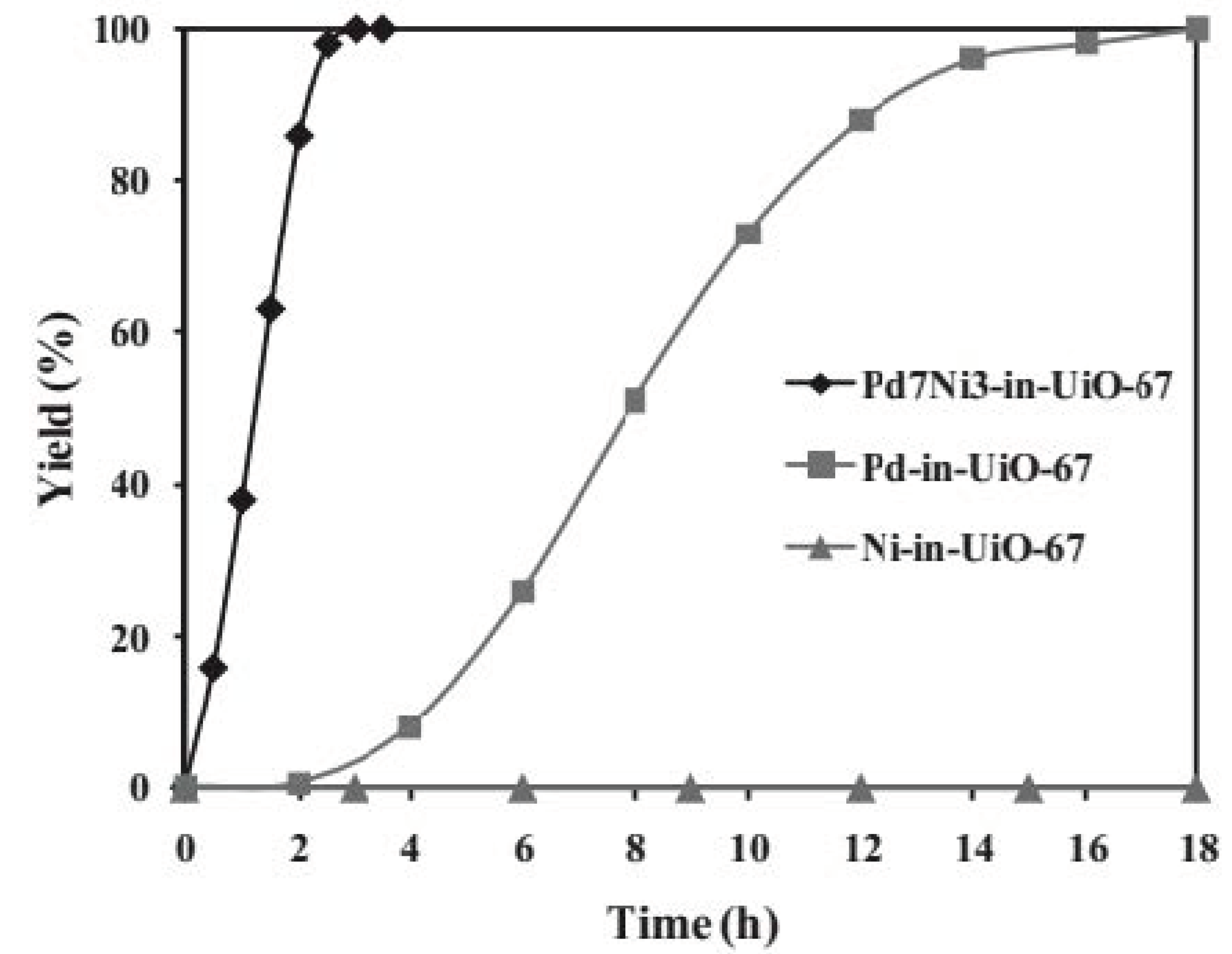
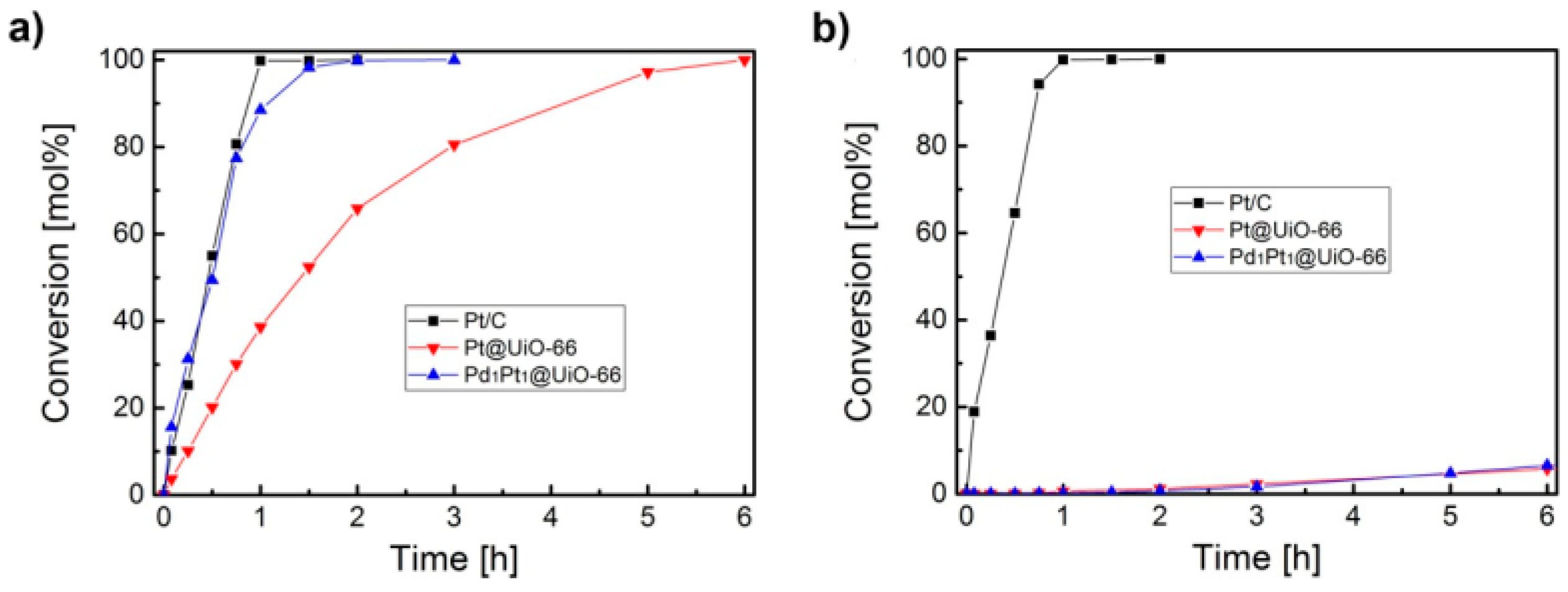
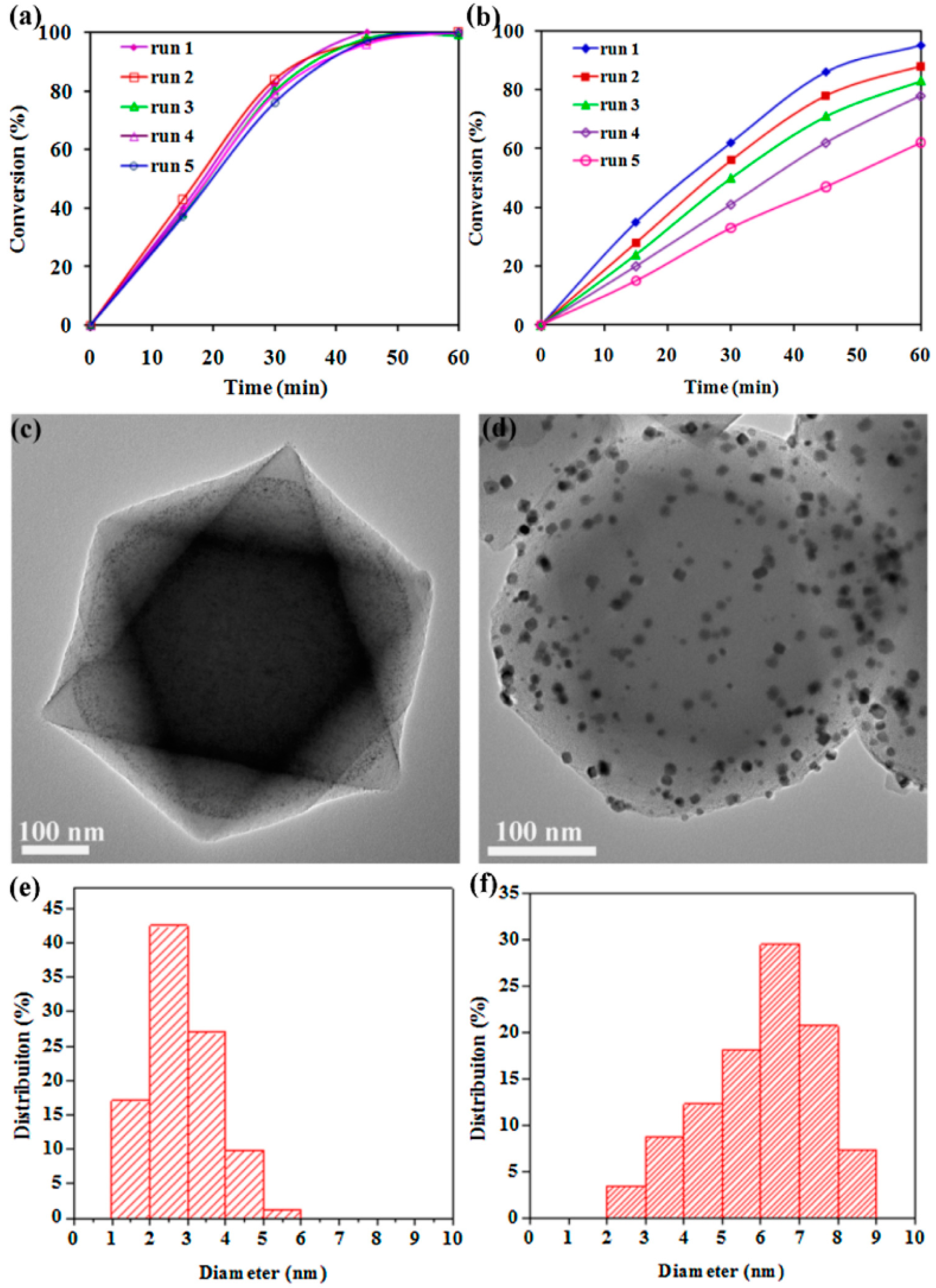
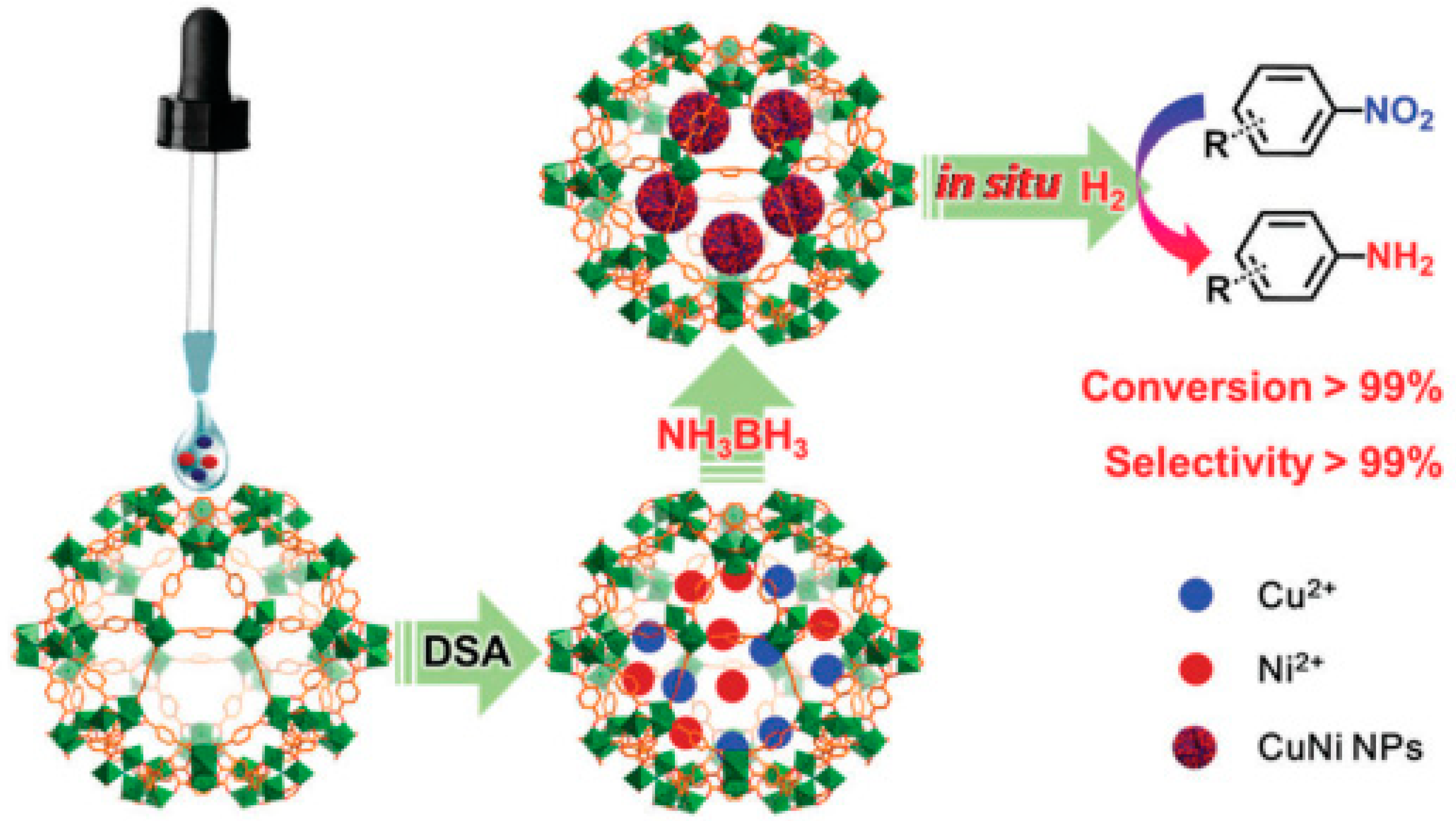
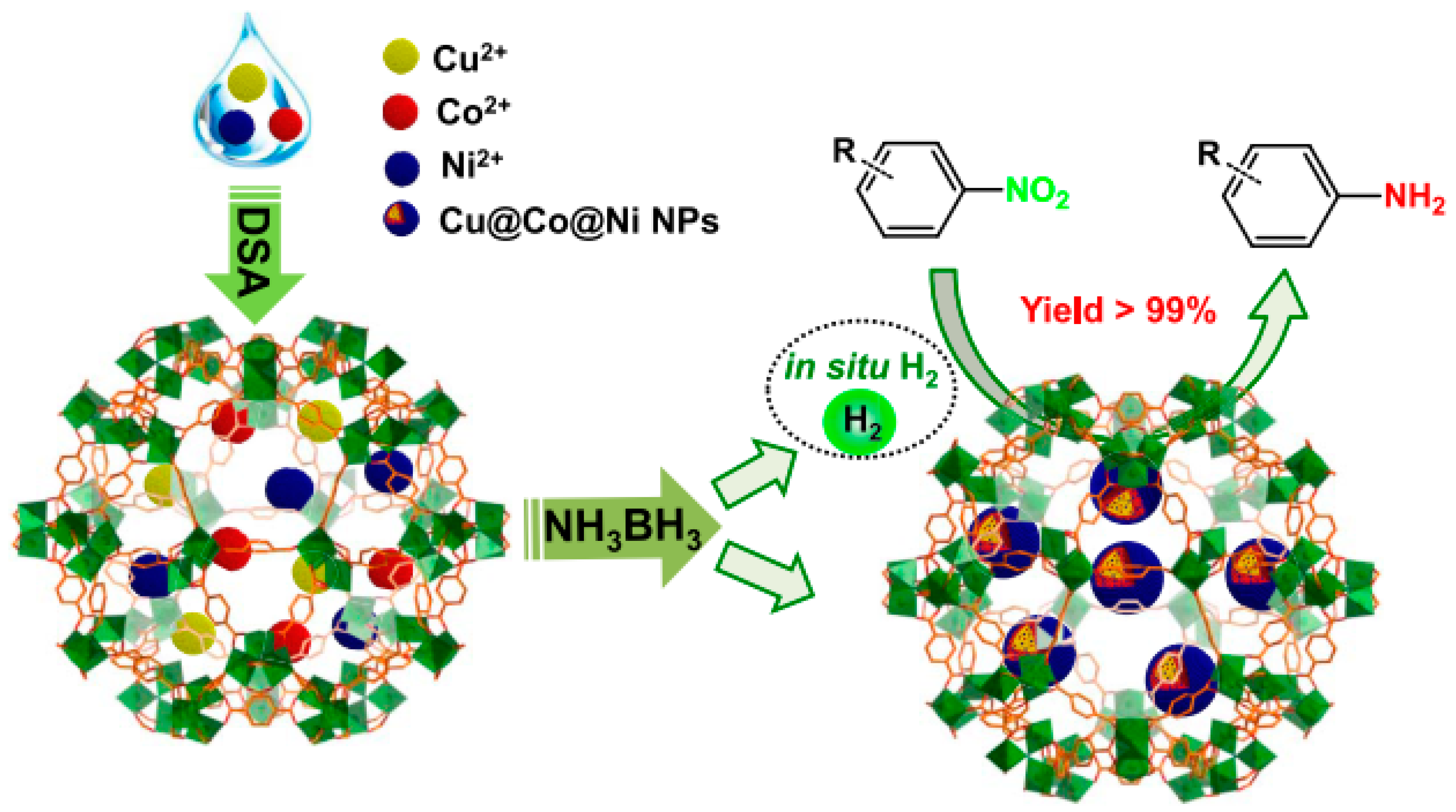
3. Bimetallic and Trimetallic MNPs@MOFs
4. Summary and Outlook
Funding
Acknowledgments
Conflicts of Interest
References
- Eddaoudi, M.; Kim, J.; Rosi, N.; Vodak, D.; Wachter, J.; O’Keeffe, M.; Yaghi, O.M. Systematic design of pore size and functionality in isoreticular MOFs and their application in methane storage. Science 2002, 295, 469–472. [Google Scholar] [CrossRef]
- Ferey, G.; Mellot-Draznieks, C.; Serre, C.; Millange, F.; Dutour, J.; Surble, S.; Margiolaki, I. A Chromium terephthalate-based solid with unusually large pore volumes and surface area. Science 2005, 309, 2040–2042. [Google Scholar] [CrossRef]
- Furukawa, H.; Cordova, K.E.; O’Keeffe, M.; Yaghi, O.M. The chemistry and applications of metal-organic frameworks. Science 2013, 341, 1230444. [Google Scholar] [CrossRef]
- Kitagawa, S.; Kitaura, R.; Noro, S.-I. Functional porous coordination polymers. Angew. Chem. Int. Ed. 2004, 43, 2334–2375. [Google Scholar] [CrossRef]
- Yaghi, O.M.; O’Keeffe, M.; Ockwig, N.W.; Chae, H.K.; Eddaoudi, M.; Kim, J. Reticular synthesis and the design of new materials. Nature 2003, 423, 705–714. [Google Scholar] [CrossRef]
- Silva, P.; Vilela, S.M.F.; Tomé, J.P.C.; Almeida Paz, F.A. Multifunctional metal-organic frameworks: From academia to industrial applications. Chem. Soc. Rev. 2015, 44, 6774–6803. [Google Scholar] [CrossRef]
- Stock, N.; Biswas, S. Synthesis of metal-organic frameworks (MOFs): Routes to various MOF topologies, morphologies, and composites. Chem. Rev. 2012, 112, 933–969. [Google Scholar] [CrossRef]
- Liu, F.-L.; Kozlevčar, B.; Strauch, P.; Zhuang, G.-L.; Guo, L.-Y.; Wang, Z.; Sun, D. Robust cluster building unit: Icosanuclear heteropolyoxocopperate templated by carbonate. Chem. Eur. J. 2015, 21, 18847–18854. [Google Scholar] [CrossRef]
- Wang, X.-P.; Chen, W.-M.; Qi, H.; Li, X.-Y.; Rajnák, C.; Feng, Z.-Y.; Kurmoo, M.; Boča, R.; Jia, C.-J.; Tung, C.-H.; et al. Solvent-controlled phase transition of a CoII-organic framework: From achiral to chiral and two to three dimensions. Chem. Eur. J. 2017, 23, 7990–7996. [Google Scholar] [CrossRef]
- Hu, Z.; Zhao, D. Metal–organic frameworks with Lewis acidity: Synthesis, characterization, and catalytic applications. CrystEngComm 2017, 19, 4066–4081. [Google Scholar] [CrossRef]
- Maksimchuk, N.V.; Zalomaeva, O.V.; Skobelev, I.Y.; Kovalenko, K.A.; Fedin, V.P.; Kholdeeva, O.A. Metal–organic frameworks of the MIL-101 family as heterogeneous single-site catalysts. Proc. R. Soc. A 2012, 468, 2017–2034. [Google Scholar] [CrossRef]
- Santiago-Portillo, A.; Blandez, J.F.; Navalón, S.; Álvaro, M.; García, H. Influence of the organic linker substituent on the catalytic activity of MIL-101(Cr) for the oxidative coupling of benzylamines to imines. Catal. Sci. Technol. 2017, 7, 1351–1362. [Google Scholar] [CrossRef]
- Santiago-Portillo, A.; Navalón, S.; Concepción, P.; Álvaro, M.; García, H. Influence of terephthalic acid substituents on the catalytic activity of MIL-101(Cr) in three Lewis acid catalyzed reactions. ChemCatChem 2017, 9, 2506–2511. [Google Scholar] [CrossRef]
- Ding, M.; Flaig, R.W.; Jiang, H.-L.; Yaghi, O.M. Carbon capture and conversion using metal-organic frameworks and MOF-based materials. Chem. Soc. Rev. 2019, 48, 2783–2828. [Google Scholar] [CrossRef]
- Yuan, S.; Deng, Y.-K.; Sun, D. Unprecedented second-timescale blue/green emissions and iodine-uptake-induced single-crystal-to-single-crystal transformation in ZnII/CdII metal–organic frameworks. Chem. Eur. J. 2014, 20, 10093–10098. [Google Scholar] [CrossRef]
- Jiang, J.; Yaghi, O.M. Brønsted Acidity in Metal-Organic Frameworks. Chem. Rev. 2015, 115, 6966–6997. [Google Scholar] [CrossRef]
- Zhu, L.; Liu, X.-Q.; Jiang, H.-L.; Sun, L.-B. Metal-Organic frameworks for heterogeneous basic catalysis. Chem. Rev. 2017, 117, 8129–8176. [Google Scholar] [CrossRef]
- Chen, L.; Luque, R.; Li, Y. Controllable design of tunable nanostructures inside metal-organic frameworks. Chem. Soc. Rev. 2017, 46, 4614–4630. [Google Scholar] [CrossRef]
- Chughtai, A.H.; Ahmad, N.; Younus, H.A.; Laypkov, A.; Verpoort, F. Metal-organic frameworks: Versatile heterogeneous catalysts for efficient catalytic organic transformations. Chem. Soc. Rev. 2015, 44, 6804–6849. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, A.; Asiri, A.M.; Garcia, H. Metal organic frameworks as versatile hosts of Au nanoparticles in heterogeneous catalysis. ACS Catal. 2017, 7, 2896–2919. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, A.; Garcia, H. Catalysis by metal nanoparticles embedded on metal-organic frameworks. Chem. Soc. Rev. 2012, 41, 5262–5284. [Google Scholar] [CrossRef]
- Falcaro, P.; Ricco, R.; Yazdi, A.; Imaz, I.; Furukawa, S.; Maspoch, D.; Ameloot, R.; Evans, J.D.; Doonan, C.J. Application of metal and metal oxide nanoparticles at MOFs. Coord. Chem. Rev. 2016, 307, 237–254. [Google Scholar] [CrossRef]
- Hu, P.; Morabito, J.V.; Tsung, C.-K. Core-shell catalysts of metal nanoparticle core and metal-organic framework shell. ACS Catal. 2014, 4, 4409–4419. [Google Scholar] [CrossRef]
- Huang, Y.-B.; Liang, J.; Wang, X.-S.; Cao, R. Multifunctional metal-organic framework catalysts: Synergistic catalysis and tandem reactions. Chem. Soc. Rev. 2017, 46, 126–157. [Google Scholar] [CrossRef]
- Liu, J.; Chen, L.; Cui, H.; Zhang, J.; Zhang, L.; Su, C.-Y. Applications of metal-organic frameworks in heterogeneous supramolecular catalysis. Chem. Soc. Rev. 2014, 43, 6011–6061. [Google Scholar] [CrossRef]
- Moon, H.R.; Lim, D.-W.; Suh, M.P. Fabrication of metal nanoparticles in metal-organic frameworks. Chem. Soc. Rev. 2013, 42, 1807–1824. [Google Scholar] [CrossRef]
- Qiu, J.; He, M.; Jia, M.; Yao, J. Metal organic frameworks for bi- and multi-metallic catalyst and their applications. Prog. Chem. 2016, 28, 1016–1028. [Google Scholar]
- Rösler, C.; Fischer, R.A. Metal-organic frameworks as hosts for nanoparticles. CrystEngComm 2015, 17, 199–217. [Google Scholar] [CrossRef]
- Wang, N.; Sun, Q.; Yu, J. Ultrasmall metal nanoparticles confined within crystalline nanoporous materials: A fascinating class of nanocatalysts. Adv. Mater. 2019, 31, 1803966. [Google Scholar] [CrossRef]
- Xiang, W.; Zhang, Y.; Lin, H.; Liu, C.-J. Nanoparticle/metal-organic framework composites for catalytic applications: Current status and perspective. Molecules 2017, 22, 2103. [Google Scholar] [CrossRef]
- Yang, Q.; Xu, Q.; Jiang, H.-L. Metal-organic frameworks meet metal nanoparticles: Synergistic effect for enhanced catalysis. Chem. Soc. Rev. 2017, 46, 4774–4808. [Google Scholar] [CrossRef]
- Yang, Q.; Yang, C.-C.; Lin, C.-H.; Jiang, H.-L. Metal-organic-framework-derived hollow N-doped porous carbon with ultrahigh concentrations of single Zn atoms for efficient carbon dioxide conversion. Angew. Chem. Int. Ed. 2019, 58, 3511–3515. [Google Scholar] [CrossRef]
- Cui, Y.; Li, B.; He, H.; Zhou, W.; Chen, B.; Qian, G. Metal-Organic Frameworks as Platforms for Functional Materials. Acc. Chem. Res. 2016, 49, 483–493. [Google Scholar] [CrossRef]
- James, S.L. Metal-organic frameworks. Chem. Soc. Rev. 2003, 32, 276–288. [Google Scholar] [CrossRef]
- Li, H.; Eddaoudi, M.; O’Keeffe, M.; Yaghi, O.M. Design and synthesis of an exceptionally stable and highly porous metal-organic framework. Nature 1999, 402, 276–279. [Google Scholar] [CrossRef] [Green Version]
- Rowsell, J.L.C.; Yaghi, O.M. Metal-organic frameworks: A new class of porous materials. Microporous Mesoporous Mater. 2004, 73, 3–14. [Google Scholar] [CrossRef]
- Zhou, H.-C.; Long, J.R.; Yaghi, O.M. Introduction to metal-organic frameworks. Chem. Rev. 2012, 112, 673–674. [Google Scholar] [CrossRef]
- Meilikhov, M.; Yusenko, K.; Esken, D.; Turner, S.; Van Tendeloo, G.; Fischer, R.A. Metals@MOFs—Loading MOFs with metal nanoparticles for hybrid functions. Eur. J. Inorg. Chem. 2010, 2010, 3701–3714. [Google Scholar] [CrossRef]
- Santiago-Portillo, A.; Cabrero-Antonino, M.; Álvaro, M.; Navalón, S.; García, H. Tuning the Microenvironment of Gold Nanoparticles Encapsulated within MIL-101(Cr) for the Selective Oxidation of Alcohols with O2: Influence of the Amino Terephthalate Linker. Chem. Eur. J. 2019, 25, 9280–9286. [Google Scholar] [CrossRef]
- Zanon, A.; Verpoort, F. Metals@ZIFs: Catalytic applications and size selective catalysis. Coord. Chem. Rev. 2017, 353, 201–222. [Google Scholar] [CrossRef]
- Aditya, T.; Pal, A.; Pal, T. Nitroarene reduction: A trusted model reaction to test nanoparticle catalysts. Chem. Commun. 2015, 51, 9410–9431. [Google Scholar] [CrossRef] [PubMed]
- Blaser, H.-U.; Malan, C.; Pugin, B.; Spindler, F.; Steiner, H.; Studer, M. Selective Hydrogenation for fine chemicals: Recent trends and new developments. Adv. Synth. Catal. 2003, 345, 103–151. [Google Scholar] [CrossRef]
- Tafesh, A.M.; Weiguny, J. A review of the selective catalytic reduction of aromatic nitro compounds into aromatic amines, isocyanates, carbamates, and ureas using CO. Chem. Rev. 1996, 96, 2035–2052. [Google Scholar] [CrossRef] [PubMed]
- Dan-Hardi, M.; Serre, C.; Frot, T.; Rozes, L.; Maurin, G.; Sanchez, C.; Férey, G. A new photoactive crystalline highly porous titanium(IV) dicarboxylate. J. Am. Chem. Soc. 2009, 131, 10857–10859. [Google Scholar] [CrossRef] [PubMed]
- Jhung, S.H.; Lee, J.-H.; Yoon, J.W.; Serre, C.; Férey, G.; Chang, J.-S. Microwave synthesis of chromium terephthalate MIL-101 and its benzene sorption ability. Adv. Mater. 2007, 19, 121–124. [Google Scholar] [CrossRef]
- Cavka, J.H.; Jakobsen, S.; Olsbye, U.; Guillou, N.; Lamberti, C.; Bordiga, S.; Lillerud, K.P. A new zirconium inorganic building brick forming metal organic frameworks with exceptional stability. J. Am. Chem. Soc. 2008, 130, 13850–13851. [Google Scholar] [CrossRef]
- Kandiah, M.; Nilsen, M.H.; Usseglio, S.; Jakobsen, S.; Olsbye, U.; Tilset, M.; Larabi, C.; Quadrelli, E.A.; Bonino, F.; Lillerud, K.P. Synthesis and stability of tagged UiO-66 Zr-MOFs. Chem. Mater. 2010, 22, 6632–6640. [Google Scholar]
- Aijaz, A.; Karkamkar, A.; Choi, Y.J.; Tsumori, N.; Rönnebro, E.; Autrey, T.; Shioyama, H.; Xu, Q. Immobilizing highly catalytically active Pt nanoparticles inside the pores of metal-organic framework: A double solvents approach. J. Am. Chem. Soc. 2012, 134, 13926–13929. [Google Scholar] [CrossRef]
- Zhu, Q.-L.; Li, J.; Xu, Q. Immobilizing metal nanoparticles to metal-organic frameworks with size and location control for optimizing catalytic performance. J. Am. Chem. Soc. 2013, 135, 10210–10213. [Google Scholar] [CrossRef]
- Li, G.; Zhao, S.; Zhang, Y.; Tang, Z. Metal–organic frameworks encapsulating active nanoparticles as emerging composites for catalysis: Recent progress and perspectives. Adv. Mater. 2018, 30, 1800702. [Google Scholar] [CrossRef]
- Esken, D.; Turner, S.; Lebedev, O.I.; Van Tendeloo, G.; Fischer, R.A. Au@ZIFs: Stabilization and encapsulation of cavity-size matching gold clusters inside functionalized zeolite imidazolate frameworks, ZIFs. Chem. Mater. 2010, 22, 6393–6401. [Google Scholar] [CrossRef]
- Chen, Y.-Z.; Zhou, Y.-X.; Wang, H.; Lu, J.; Uchida, T.; Xu, Q.; Yu, S.-H.; Jiang, H.-L. Multifunctional PdAg@MIL-101 for one-pot cascade reactions: Combination of host–guest cooperation and bimetallic synergy in catalysis. ACS Catal. 2015, 5, 2062–2069. [Google Scholar] [CrossRef]
- Zhuang, G.-L.; Bai, J.-Q.; Tan, L.; Huang, H.-L.; Gao, Y.-F.; Zhong, X.; Zhong, C.-L.; Wang, J.-G. Preparation and catalytic properties of Pd nanoparticles supported on micro-crystal DUT-67 MOFs. RSC Adv. 2015, 5, 32714–32719. [Google Scholar] [CrossRef]
- Zheng, D.-Y.; Zhou, X.-M.; Mutyala, S.; Huang, X.-C. High catalytic activity of C60Pdn encapsulated in metal–organic framework UiO-67, for tandem hydrogenation reaction. Chem. Eur. J. 2018, 24, 19141–19145. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhang, H.-Y.; Wang, L.; Zhang, Y.; Zhao, J. Ru/UiO-66 catalyst for the reduction of nitroarenes and tandem reaction of alcohol oxidation/Knoevenagel condensation. ACS Omega 2018, 3, 4199–4212. [Google Scholar] [CrossRef]
- Gole, B.; Sanyal, U.; Mukherjee, P.S. A smart approach to achieve an exceptionally high loading of metal nanoparticles supported by functionalized extended frameworks for efficient catalysis. Chem. Commun. 2015, 51, 4872–4875. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Xi, Q.; Zhang, Y.; Jin, M.; Zhao, Y.; Wu, C.; Zhou, H.; Guo, P.; Xu, J. Atomic-scale engineering of MOF array confined Au nanoclusters for enhanced heterogeneous catalysis. Nanoscale 2019, 11, 1169–1176. [Google Scholar] [CrossRef]
- Zhang, H.-J.; Qi, S.-D.; Niu, X.-Y.; Hu, J.; Ren, C.-L.; Chen, H.-L.; Chen, X.-G. Metallic nanoparticles immobilized in magnetic metal–organic frameworks: Preparation and application as highly active, magnetically isolable and reusable catalysts. Catal. Sci. Technol. 2014, 4, 3013–3024. [Google Scholar] [CrossRef]
- Park, Y.K.; Choi, S.B.; Nam, H.J.; Jung, D.-Y.; Choon Ahn, H.C.; Choi, K.; Furukawa, H.; Kim, J. Catalytic nickel nanoparticles embedded in a mesoporous metal-organic frameworkw. Chem. Commun. 2010, 46, 3086–3088. [Google Scholar] [CrossRef]
- Yin, D.; Li, C.; Ren, H.; Liu, J.; Liang, C. Gold-palladium-alloy-catalyst loaded UiO-66-NH2 for reductive amination with nitroarenes exhibiting high selectivity. ChemistrySelect 2018, 3, 5092–5097. [Google Scholar] [CrossRef]
- Chen, C.; Chen, X.; Liu, H.; Li, Y. Encapsulation of mono or bimetal nanoparticles inside metal-organic-frameworks via in situ incorporation of metal precursors. Small 2015, 11, 2642–2648. [Google Scholar] [CrossRef] [PubMed]
- Rçsler, C.; Dissegna, S.; Rechac, V.L.; Kauer, M.; Guo, P.; Turner, S.; Ollegott, K.; Kobayashi, H.; Yamamoto, T.; Peeters, D.; et al. Encapsulation of bimetallic metal nanoparticles into robust zirconium-based metal–organic frameworks: evaluation of the catalytic potential for size-selective hydrogenation. Chem. Eur. J. 2017, 23, 3583–3594. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Li, Y. One-step encapsulation of Pt-Co bimetallic nanoparticles within MOFs for advanced room temperature nanocatalysis. Mol. Catal. 2017, 433, 77–83. [Google Scholar] [CrossRef]
- Zhou, Y.-H.; Yang, Q.; Chen, Y.-Z.; Jiang, H.-L. Low-cost CuNi@MIL-101 as an excellent catalyst toward cascade reaction: Integration of ammonia borane dehydrogenation with nitroarene hydrogenation. Chem. Commun. 2017, 53, 12361–12364. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.-L.; Chen, Y.-Z.; Ge, B.-D.; Li, J.-H.; Wang, G.-M. Three-shell Cu@Co@Ni nanoparticles stabilized with a metal–organic framework for enhanced tandem catalysis. ACS Appl. Mater. Interfaces 2019, 11, 940–947. [Google Scholar] [CrossRef] [PubMed]
- Bellina, F.; Calandri, C.; Cauteruccio, S.; Rossi, R. Efficient and highly regioselective direct C-2 arylation of azoles, including free (NH)-imidazole, -benzimidazole and -indole, with aryl halides. Tetrahedron 2007, 63, 1970–1980. [Google Scholar] [CrossRef]
- Nagashima, H.; Kato, Y.; Yamaguchi, H.; Kimura, E.; Kawamishi, T.; Kato, M.; Saito, Y.; Haga, M.; Itoh, K. Synthesis and reactions of organoplatinum compounds of C60, C60Ptn. Chem. Lett. 1994, 23, 1207–1210. [Google Scholar] [CrossRef]
- Nagashima, H.; Nakaoka, A.; Saito, Y.; Kato, M.; Kawanishi, T.; Itoh, K. C60Pdn: The first organometallic polymer of buckminsterfullerene. J. Chem. Soc. Chem. Commun. 1992, 377–379. [Google Scholar] [CrossRef]
- Nagashima, H.; Nakaoka, A.; Tajima, S.; Saito, Y.; Itoh, K. Synthesis of optically clear molecular organogels comprising phenol and surfactants of sulfosuccinic acid derivatives. Chem. Lett. 2017, 46, 1361–1364. [Google Scholar]
- Li, B.; Xu, Z. A nonmetal catalyst for molecular hydrogen activation with comparable catalytic hydrogenation capability to noble metal catalyst. J. Am. Chem. Soc. 2009, 131, 16380–16382. [Google Scholar] [CrossRef]
- Han, Y.; Liu, M.; Li, K.; Zuo, Y.; Wei, Y.; Xu, S.; Zhang, G.; Song, C.; Zhang, Z.; Guo, X. Facile synthesis of morphology and sizecontrolled zirconium metal–organic framework UiO-66: The role of hydrofluoric acid in crystallization. CrystEngComm 2015, 17, 6434–6440. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Z.; Huang, D.; Cheng, M.; Zeng, G.; Lai, C.; Zhang, C.; Zhou, C.; Wang, W.; Jiang, D.; et al. Metal or metal-containing nanoparticle@MOF nanocomposites as a promising type of photocatalyst. Coord. Chem. Rev. 2019, 388, 63–78. [Google Scholar] [CrossRef]
- Layek, K.; Lakshmi Kantam, M.L.; Shirai, M.; Nishio-Hamane, D.; Sasaki, T.; Maheswarana, H. Gold nanoparticles stabilized on nanocrystalline magnesium oxide as an active catalyst for reduction of nitroarenes in aqueous medium at room temperature. Green Chem. 2012, 14, 3164–3174. [Google Scholar] [CrossRef]
- Hayakawa, K.; Yoshimura, T.; Esumi, K. Preparation of gold−dendrimer nanocomposites by laser irradiation and their catalytic reduction of 4-nitrophenol. Langmuir 2003, 19, 5517–5521. [Google Scholar] [CrossRef]
- Lee, J.; Park, J.C.; Song, H. A nanoreactor framework of a Au@SiO2 yolk/shell structure for catalytic reduction of p-nitrophenol. Adv. Mater. 2008, 20, 1523–1528. [Google Scholar] [CrossRef]
- Fazzini, S.; Cassani, M.C.; Ballarin, B.; Boanini, E.; Girardon, J.S.; Mamede, A.-S.; Mignani, A.; Nanni, D. Novel synthesis of gold nanoparticles supported on alkyne-functionalized nanosilica. J. Phys. Chem. C 2014, 118, 24538–24547. [Google Scholar] [CrossRef]
- Ballarin, B.; Barreca, D.; Boanini, E.; Bonansegna, E.; Cristina Cassani, M.; Carraro, G.; Fazzini, S.; Mignani, A.; Nanni, D.; Pinelli, D. Functionalization of silica through thiol-yne radical chemistry: A catalytic system based on gold nanoparticles supported on amino-sulfide-branched silica. RSC Adv. 2016, 6, 25780–25788. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, G.; Chaker, M.; Rosei, F.; Ma, D. Gold nanoparticle decorated ceria nanotubes with significantly high catalytic activity for the reduction of nitrophenol and mechanism study. Appl. Catal. B Environ. 2013, 132–133, 107–115. [Google Scholar] [CrossRef]
- Qiu, L.; Peng, Y.; Liu, B.; Lin, B.; Peng, Y.; Malik, M.J.; Yan, F. Polypyrrole nanotube-supported gold nanoparticles: An efficient electrocatalyst for oxygen reduction and catalytic reduction of 4-nitrophenol. Appl. Catal. A Gen. 2012, 413–414, 230–237. [Google Scholar] [CrossRef]
- Ke, F.; Zhu, J.; Qiu, L.-G.; Jiang, X. Controlled synthesis of novel Au@MIL-100(Fe) core–shell nanoparticles with enhanced catalytic performance. Chem. Commun. 2013, 49, 1267–1269. [Google Scholar] [CrossRef]
- Huang, X.; Guo, C.; Zuo, J.; Zheng, N.; Stucky, G.D. An assembly route to inorganic catalytic nanoreactors containing sub-10-nm gold nanoparticles with anti-aggregation properties. Small 2009, 5, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.-L.; Akita, T.; Ishida, T.; Haruta, M.; Xu, Q. Synergistic catalysis of Au@Ag core-shell nanoparticles stabilized on metal-organic framework. J. Am. Chem. Soc. 2011, 133, 1304–1306. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Liu, B.; Wang, Q.; Liu, Y.; Liu, Y.; Jing, P.; Yu, S.; Liua, L.; Zhang, J. A magnetic double-shell microsphere as a highly efficient reusable catalyst for catalytic applications. Chem. Commun. 2013, 49, 7596–7598. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Guo, Z.; Xiao, C.; Goh, T.W.; Tesfagaber, D.; Huang, W. Tandem catalysis by palladium nanoclusters encapsulated in metal-organic frameworks. ACS Catal. 2014, 4, 3490–3497. [Google Scholar] [CrossRef]
- Zhao, M.; Deng, K.; He, L.; Liu, Y.; Li, G.; Zhao, H.; Tang, Z. Core-shell palladium nanoparticle@metal-organic frameworks as multifunctional catalysts for cascade reactions. J. Am. Chem. Soc. 2014, 136, 1738–1741. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yu, R.; Huang, J.; Shi, Y.; Zhang, D.; Zhong, X.; Wang, D.; Wu, Y.; Li, Y. Platinum–nickel frame within metal-organic framework fabricated in situ for hydrogen enrichment and molecular sieving. Nat. Commun. 2015, 6, 8248. [Google Scholar] [CrossRef] [PubMed]
- Metin, O.; Özkar, S.; Sun, S. Monodisperse nickel nanoparticles supported on SiO2 as an effective catalyst for the hydrolysis of ammonia-borane. Nano Res. 2010, 3, 676–684. [Google Scholar] [CrossRef]
- Chen, Y.-Z.; Xu, Q.; Yu, S.-H.; Jiang, H.-L. Tiny Pd@Co core-shell nanoparticles confined inside a metal-organic framework for highly efficient catalysis. Small 2015, 11, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhu, Q.-L.; Xu, Q. Non-noble bimetallic CuCo nanoparticles encapsulated in the pores of metal-organic frameworks: Synergetic catalysis in the hydrolysis of ammonia borane for hydrogen generation. Catal. Sci. Technol. 2015, 5, 525–530. [Google Scholar] [CrossRef]
- Chen, Y.-Z.; Liang, L.; Yang, Q.; Hong, M.; Xu, Q.; Yu, S.-H.; Jiang, H.-L. A seed-mediated approach to the general and mild synthesis of non-noble metal nanoparticles stabilized by a metal-organic framework for highly efficient catalysis. Mater. Horiz. 2015, 2, 606–612. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
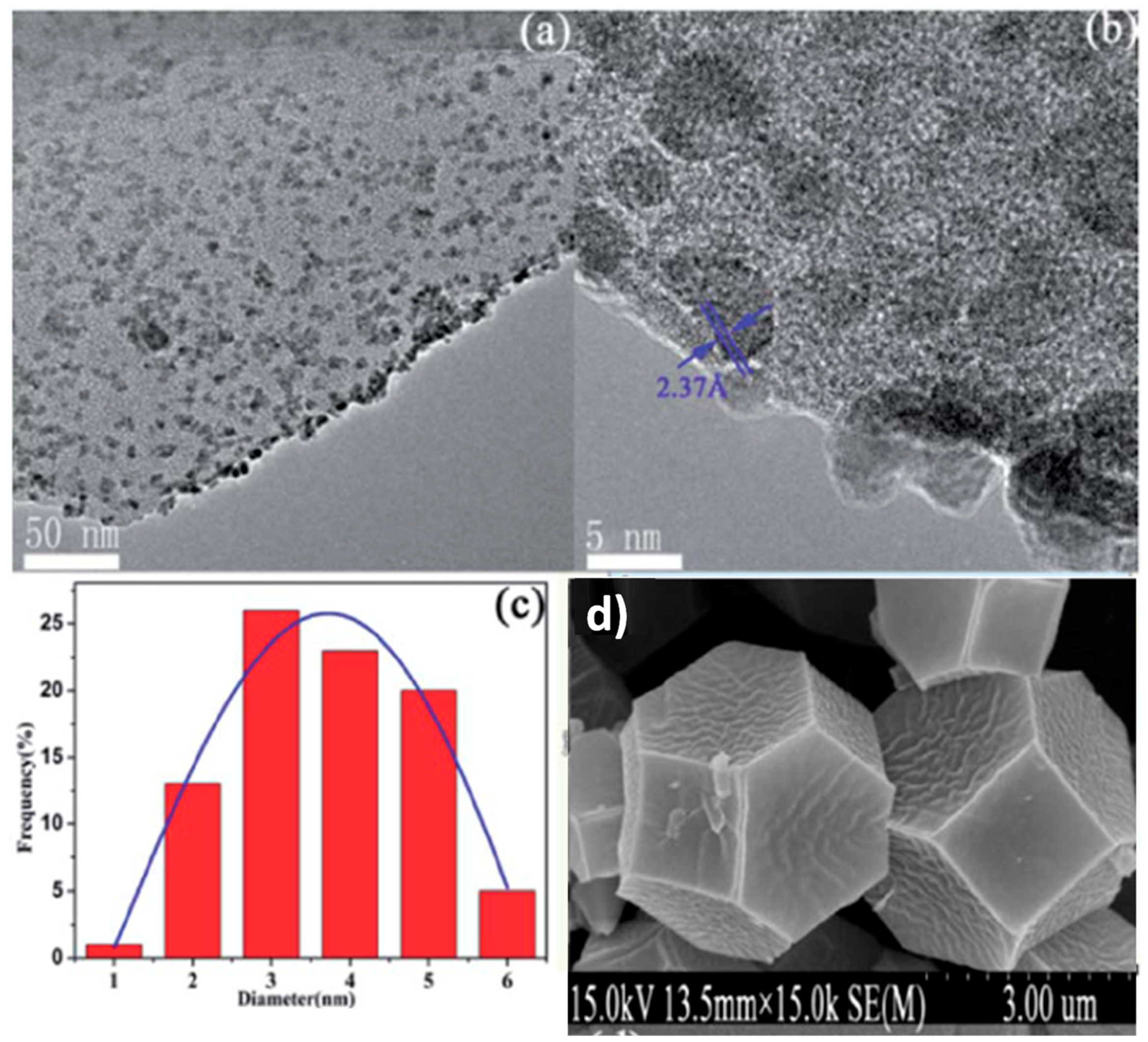{kind=link}
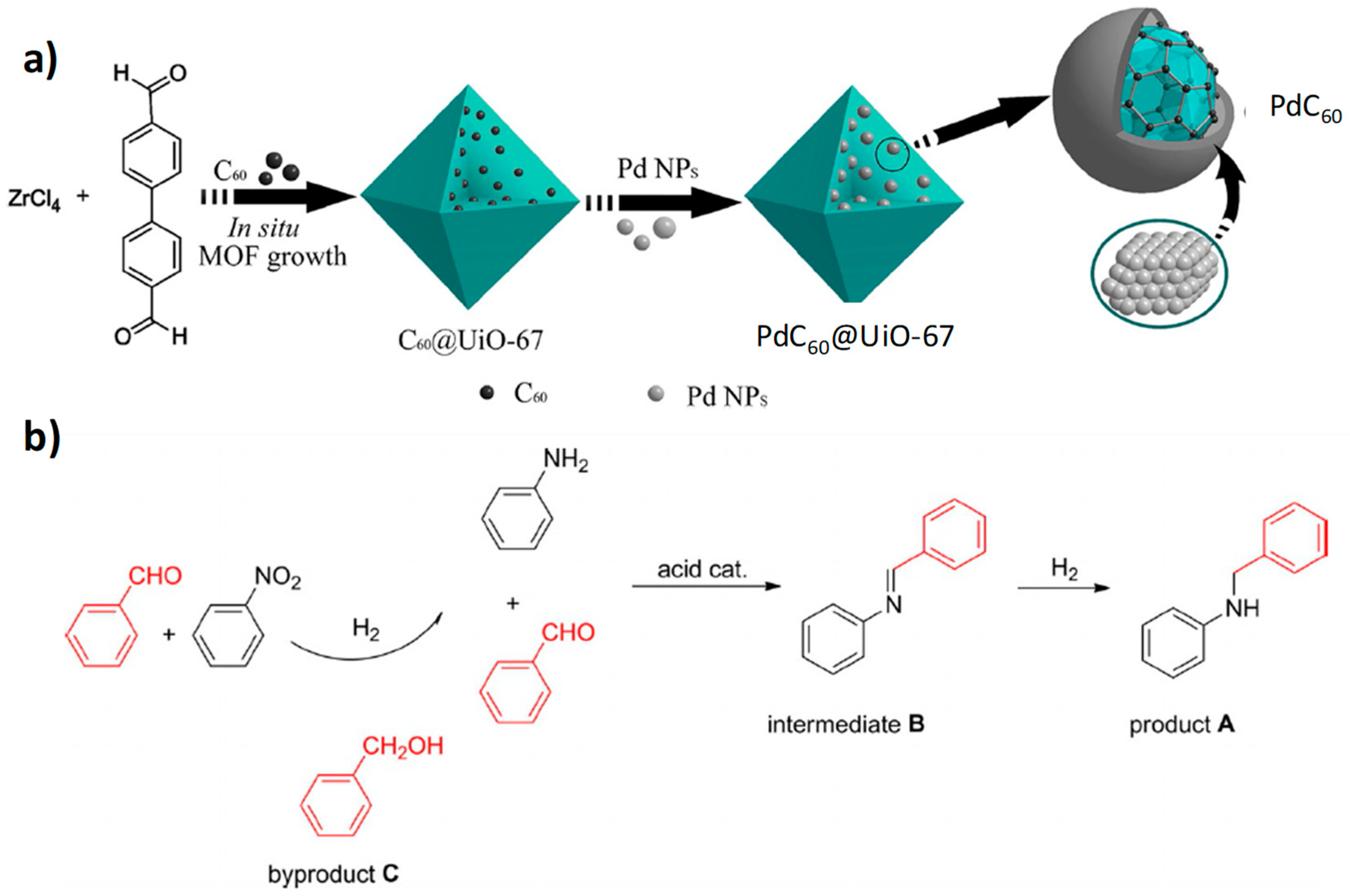{kind=link}
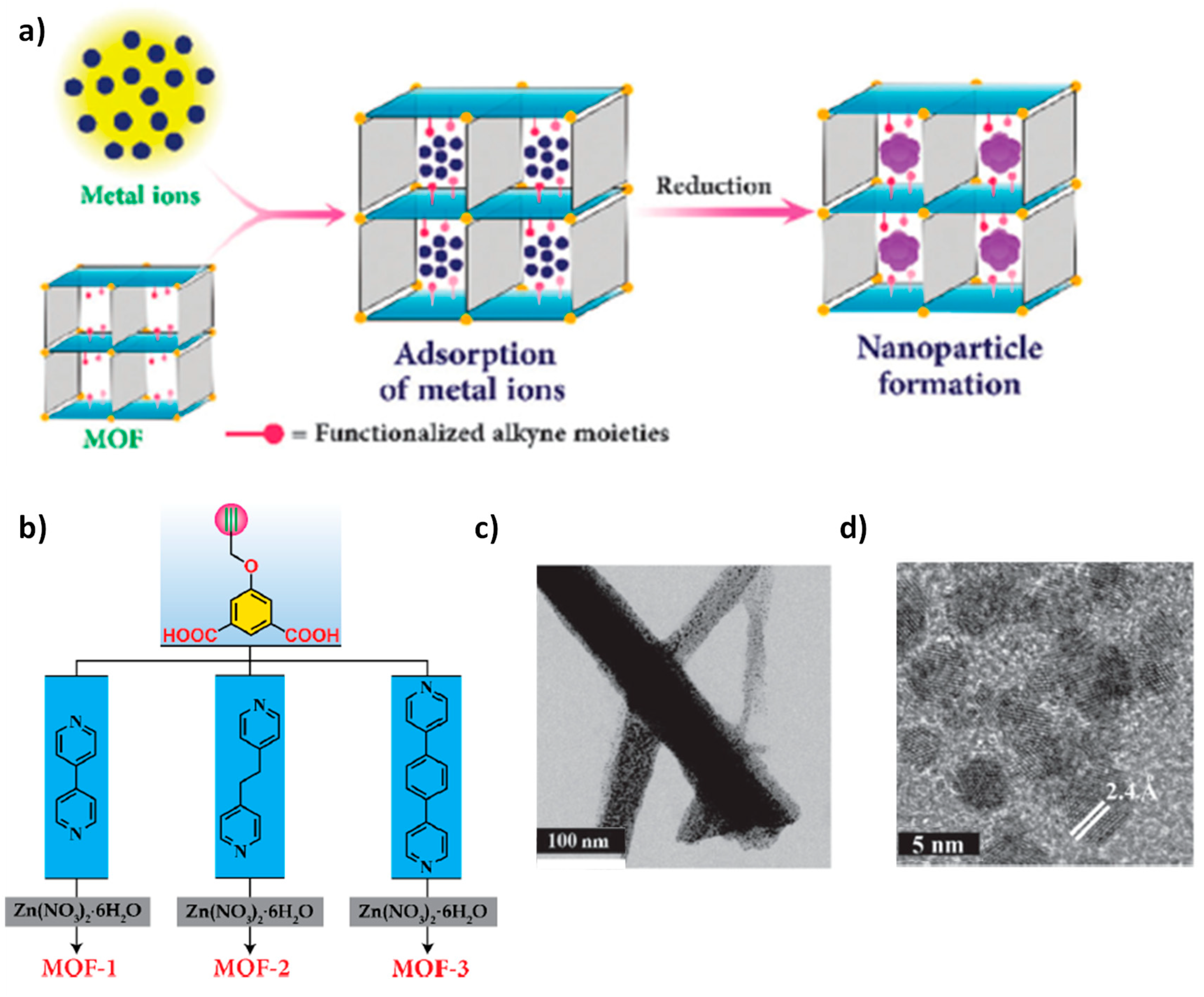{kind=link}
{kind=link}
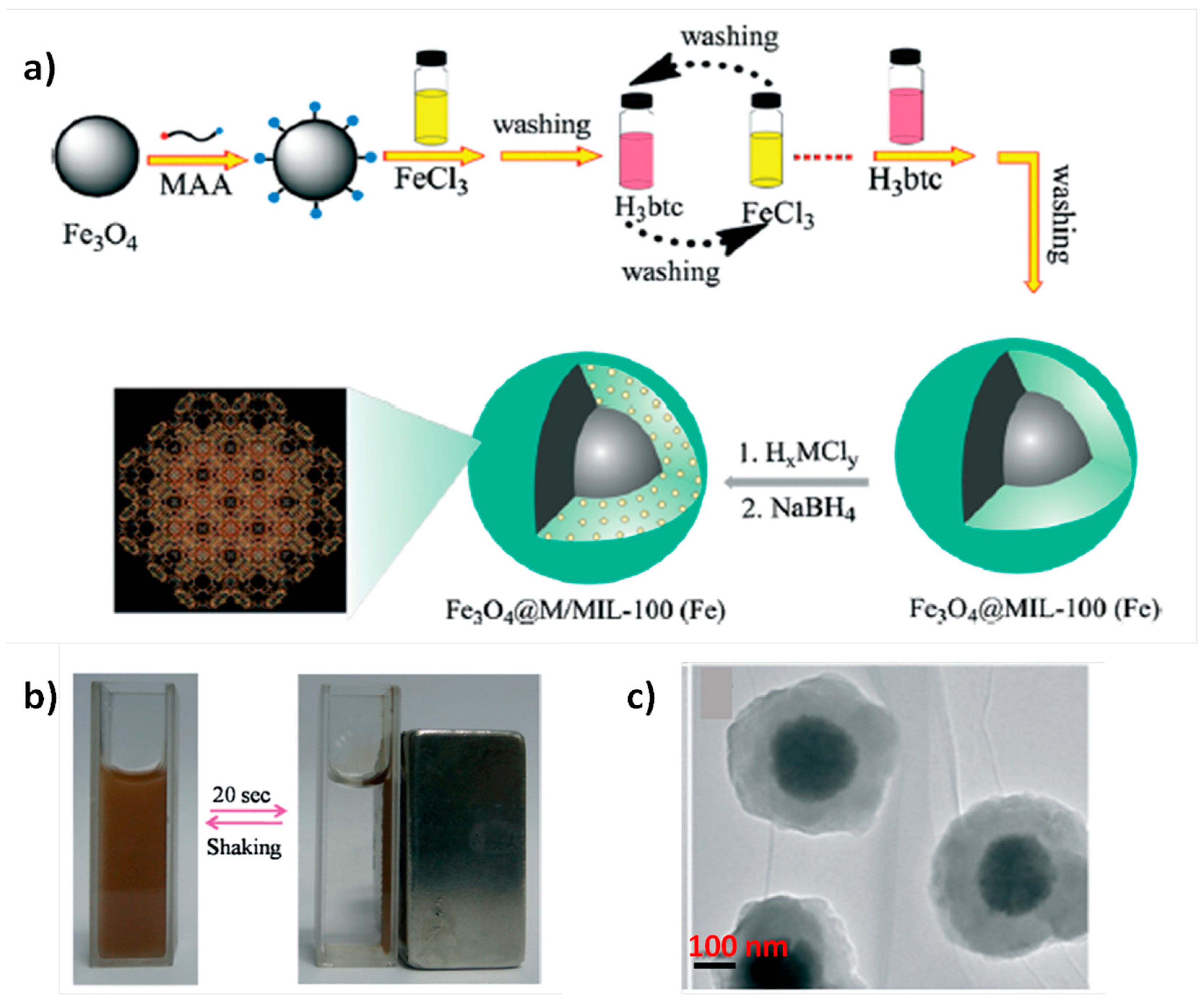{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | MNPs (Size, nm) | Reducing Agent | Catalytic Reaction | No. Reuses | Ref. |
|---|---|---|---|---|---|
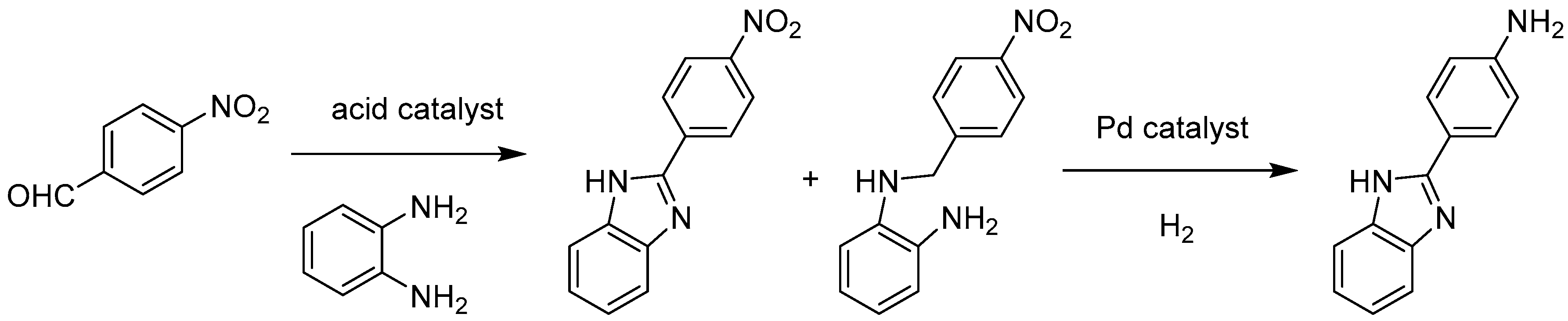
| Pd@MIL-101(Cr) | Pd (2.5 ± 0.3) | H2 | Synthesis of 2-(4-aminophenyl)-1H-benzimidazole from 4-nitrobenzaldehyde | 3 | [52] |
| Pd-DUT-67 | Pd (3.5) | H2 | Hydrogenation of nitrobenzene | - | [53] |
| PdC60@UiO-67(Zr) | Pd (5 ± 2) | H2 | Synthesis of N-benzylaniline from nitrobenzene | 5 | [54] |
| Ru-UiO-66(Zr) | Ru (1.07) | HCOOH | Hydrogenation of nitrobenzene | 6 | [55] |
| Au@MOF-3 | Au (1.85 ± 0.83) | NaBH4 | 4-Nitrophenol reduction | 5 | [56] |
| Au@ZIF-8(Zn,Cu) | Au nanoclusters (<2) | NaBH4 | 4-Nitrophenol reduction | 10 | [57] |
| Fe3O4@MIL-100(Fe)-Pt | Pt (1.9 ± 0.2) | NaBH4 | 4-Nitrophenol reduction | 10 | [58] |
| Ni@MesMOF-1 | Ni (1.4) | NaBH4 | Hydrogenation of nitrobenzene | 3 | [59] |
| AuPd@UiO-66(Zr)-NH2 | Au-Pd0.03 (5.3) | H2 | Reductive amination of nitrobenzene | 5 | [60] |
| PdAg@MIL-101(Cr) | PdAg (1.5 ± 0.3) | H2 | Synthesis of secondary arylamines by hydrogenation of nitrobenzene | 3 | [52] |
| Pd7Ni3@UiO-67(Zr) | PdNi (3–4) | H2 | Hydrogenation of nitrobenzene | 5 | [61] |
| Pd1Pt1@UiO-66(Zr) | PdPt (4.2 ± 0.8) | H2 | Hydrogenation of nitrobenzene | 3 | [62] |
| Pt8Co1@UiO-66(Zr) | PtCo (2) | H2 | Hydrogenation of nitrobenzene | 5 | [63] |
| CuNi@MIL-101(Cr) | CuNi (3) | H2 | Hydrogenation of nitrobenzene | 7 | [64] |
| Cu@Co@Ni/MIL-101(Cr) | Cu@Co@Ni (3.3) | H2 | Hydrogenation of nitrobenzene | 5 | [65] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Navalón, S.; Álvaro, M.; Dhakshinamoorthy, A.; García, H. Encapsulation of Metal Nanoparticles within Metal–Organic Frameworks for the Reduction of Nitro Compounds. Molecules 2019, 24, 3050. https://doi.org/10.3390/molecules24173050
Navalón S, Álvaro M, Dhakshinamoorthy A, García H. Encapsulation of Metal Nanoparticles within Metal–Organic Frameworks for the Reduction of Nitro Compounds. Molecules. 2019; 24(17):3050. https://doi.org/10.3390/molecules24173050
Chicago/Turabian StyleNavalón, Sergio, Mercedes Álvaro, Amarajothi Dhakshinamoorthy, and Hermenegildo García. 2019. "Encapsulation of Metal Nanoparticles within Metal–Organic Frameworks for the Reduction of Nitro Compounds" Molecules 24, no. 17: 3050. https://doi.org/10.3390/molecules24173050
APA StyleNavalón, S., Álvaro, M., Dhakshinamoorthy, A., & García, H. (2019). Encapsulation of Metal Nanoparticles within Metal–Organic Frameworks for the Reduction of Nitro Compounds. Molecules, 24(17), 3050. https://doi.org/10.3390/molecules24173050